
BMAL1 in Ischemic Heart Disease: A Narrative Review from Molecular Clock to Myocardial Pathology

 , , and
, , and
Abstract
1. Introduction
2. Literature Search Strategy
3. Cardiac Regulation by BMAL1
3.1. Regulation of the Cardiac Vasculature by BMAL1
3.2. BMAL1 Regulation of Cardiac Function Circadian Rhythm
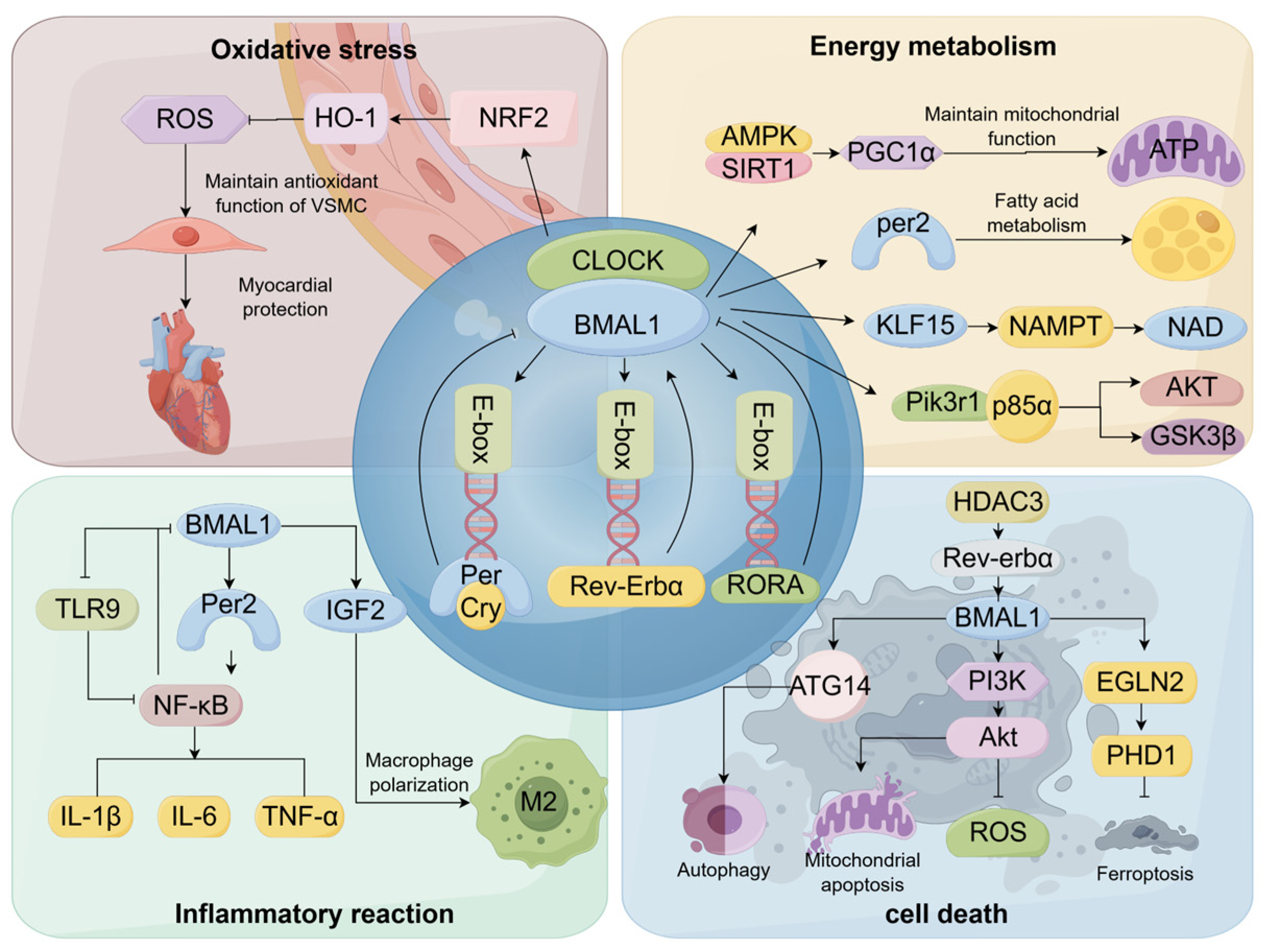
4. Regulatory Mechanisms of Myocardial Ischemia by BMAL1
4.1. BMAL1 Is Involved in the Regulation of Oxidative Stress Responses
4.2. BMAL1 Is Implicated in the Regulation of Energy Metabolism
4.3. BMAL1 Contributes to the Modulation of Immune-Inflammatory Responses
4.4. BMAL1 Is Involved in Apoptosis and Autophagy Regulation
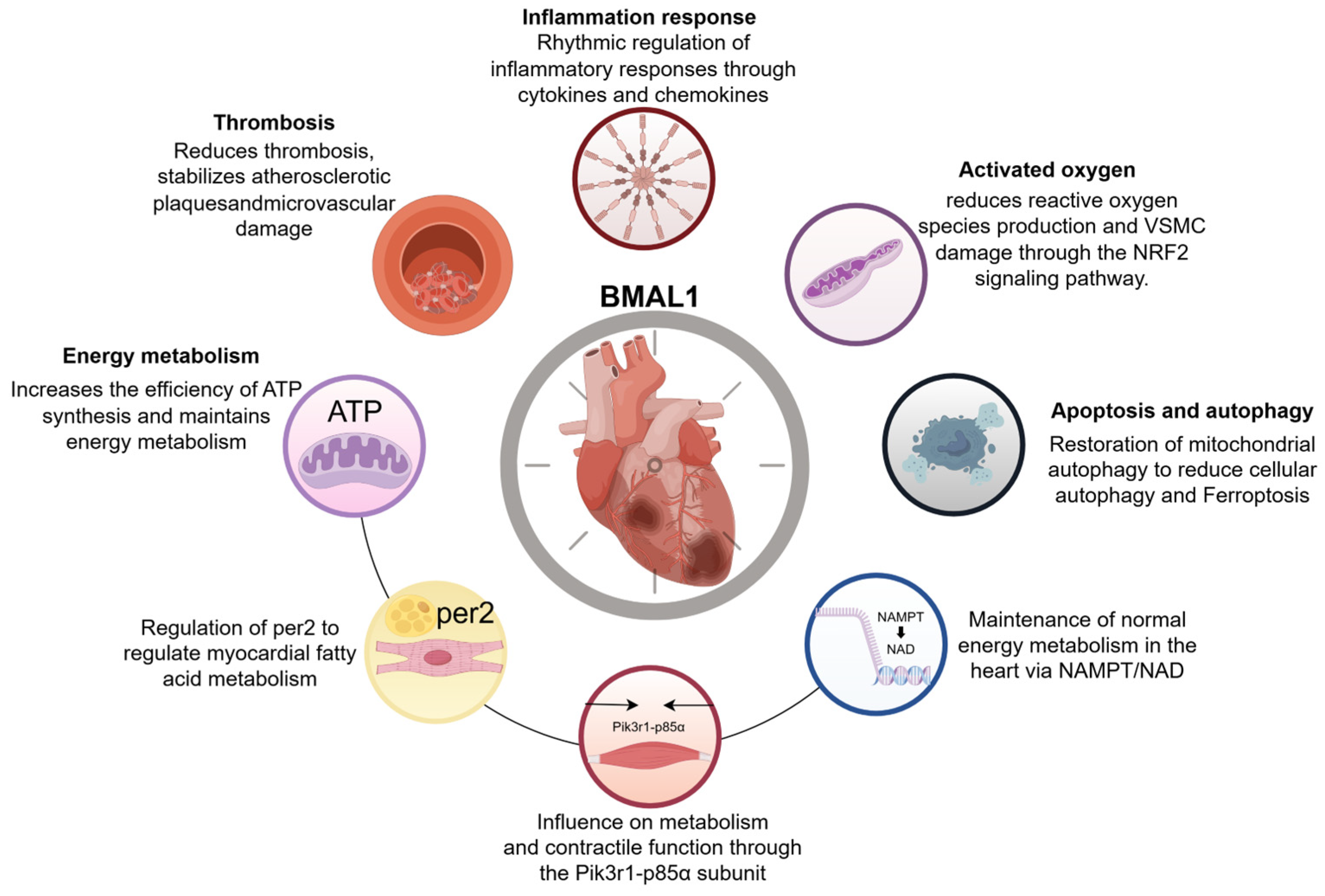
5. Discussion
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef]
- Falk, E.; Nakano, M.; Bentzon, J.F.; Finn, A.V.; Virmani, R. Update on acute coronary syndromes: The pathologists view. Eur. Heart J. 2013, 34, 719–728. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- López-Candales, A.; Sawalha, K. Improving diagnostic assessments in the ever-changing landscape of atherosclerosis. J. Cardiovasc. Med. 2023, 24, 221–229. [Google Scholar] [CrossRef]
- NCD Countdown 2030 Collaborators. NCD Countdown 2030: Worldwide trends in non-communicable disease mortality and progress towards Sustainable Development Goal target 3.4. Lancet 2018, 392, 1072–1088. [Google Scholar] [CrossRef]
- Moran, A.E.; Forouzanfar, M.H.; Roth, G.A.; Mensah, G.A.; Ezzati, M.; Murray, C.J.; Naghavi, M. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: The Global Burden of Disease 2010 study. Circulation 2014, 129, 1483–1492. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Xu, L.L.; Yao, S.Y.; Men, J.; Wang, Y.M.; Xia, L.D.; Cui, C.L.; Li, H.; Liu, Z.Y.; Zhang, S.Q. Bioinformatics Analysis of BMAL1 Protein in Mice. Heilongjiang Med. J. 2023, 47, 645–652. [Google Scholar] [CrossRef]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Shearman, L.P.; Sriram, S.; Weaver, D.R.; Maywood, E.S.; Chaves, I.; Zheng, B.; Kume, K.; Lee, C.C.; van der Horst, G.T.; Hastings, M.H.; et al. Interacting molecular loops in the mammalian circadian clock. Science 2000, 288, 1013–1019. [Google Scholar] [CrossRef]
- Erdem, J.S.; Das, M.K.; De Ryck, E.; Skare, Ø.; Lie, J.S.; Bugge, M.; Harding, B.; Jorgensen, I.L.; Mehlum, I.S.; Kogevinas, M.; et al. Night shift work and indicators of cardiovascular risk: A systematic review and meta-analysis. Environ. Res. 2025, 276, 121503. [Google Scholar] [CrossRef]
- Black, N.; D’Souza, A.; Wang, Y.; Piggins, H.; Dobrzynski, H.; Morris, G.; Boyett, M.R. Circadian rhythm of cardiac electrophysiology, arrhythmogenesis, and the underlying mechanisms. Heart Rhythm 2019, 16, 298–307. [Google Scholar] [CrossRef]
- Begemann, K.; Neumann, A.M.; Oster, H. Regulation and function of extra-SCN circadian oscillators in the brain. Acta Physiol. 2020, 229, e13446. [Google Scholar] [CrossRef]
- Fagiani, F.; Di Marino, D.; Romagnoli, A.; Travelli, C.; Voltan, D.; Di Cesare Mannelli, L.; Racchi, M.; Govoni, S.; Lanni, C. Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduct. Target. Ther. 2022, 7, 41. [Google Scholar] [CrossRef]
- Westgate, E.J.; Cheng, Y.; Reilly, D.F.; Price, T.S.; Walisser, J.A.; Bradfield, C.A.; FitzGerald, G.A. Genetic components of the circadian clock regulate thrombogenesis in vivo. Circulation 2008, 117, 2087–2095. [Google Scholar] [CrossRef]
- De Juan, A.; Ince, L.M.; Pick, R.; Chen, C.S.; Molica, F.; Zuchtriegel, G.; Wang, C.; Zhang, D.; Druzd, D.; Hessenauer, M.E.T.; et al. Artery-associated sympathetic innervation drives rhythmic vascular inflammation of arteries and veins. Circulation 2019, 140, 1100–1114. [Google Scholar] [CrossRef]
- Mastrullo, V.; Matos, R.S.; McVey, J.H.; Gupta, P.; Madeddu, P.; Johnston, J.D.; Van Der Veen, D.R.; Velliou, E.G.; Campagnolo, P. The vascular clock: A new insight into endothelial cells and pericytes crosstalk. Eur. Heart J. 2021, 42, 3382. [Google Scholar] [CrossRef]
- Anea, C.B.; Cheng, B.; Sharma, S.; Kumar, S.; Caldwell, R.W.; Yao, L.; Ali, M.I.; Merloiu, A.M.; Stepp, D.W.; Black, S.M.; et al. Increased superoxide and endothelial NO synthase uncoupling in blood vessels of Bmal1-knockout mice. Circ. Res. 2012, 111, 1157–1165. [Google Scholar] [CrossRef]
- Bhatwadekar, A.D.; Beli, E.; Diao, Y.; Chen, J.; Luo, Q.; Alex, A.; Caballero, S.; Dominguez, J.M., 2nd; Salazar, T.E.; Busik, J.V.; et al. Conditional Deletion of Bmal1 Accentuates Microvascular and Macrovascular Injury. Am. J. Pathol. 2017, 187, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xu, L.R.; Yan, D.; Zhou, M.; Han, T.L.; Lu, C.; Tang, X.; Lin, C.P.; Qian, R.Z.; Guo, D.Q. BMAL1 modulates smooth muscle cells phenotypic switch towards fibroblast-like cells and stabilizes atherosclerotic plaques by upregulating YAP1. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.K.; Ding, J.W.; Wang, X.A.; Zhang, Z.Q.; Wan, J.Q. Effect of Bmal1 Overexpression on the Severity and Stability of Atherosclerotic Plaque Lesions. Int. J. Cardiovasc. Dis. 2022, 49, 367–370. [Google Scholar] [CrossRef]
- Kroetsch, J.T.; Lidington, D.; Alibhai, F.J.; Reitz, C.J.; Zhang, H.; Dinh, D.D.; Hanchard, J.; Khatua, T.N.; Heximer, S.P.; Martino, T.A.; et al. Disrupting circadian control of peripheral myogenic reactivity mitigates cardiac injury following myocardial infarction. Cardiovasc. Res. 2023, 119, 1403–1415. [Google Scholar] [CrossRef]
- Storch, K.F.; Lipan, O.; Leykin, I.; Viswanathan, N.; Davis, F.C.; Wong, W.H.; Weitz, C.J. Extensive and divergent circadian gene expression in liver and heart. Nature 2002, 417, 78–83. [Google Scholar] [CrossRef]
- Durgan, D.J.; Hotze, M.A.; Tomlin, T.M.; Egbejimi, O.; Graveleau, C.; Abel, E.D.; Shaw, C.A.; Bray, M.S.; Hardin, P.E.; Young, M.E. The intrinsic circadian clock within the cardiomyocyte. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1530–H1541. [Google Scholar] [CrossRef]
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef]
- Gallego, M.; Virshup, D.M. Post-translational modifications regulate the ticking of the circadian clock. Nat. Rev. Mol. Cell Biol. 2007, 8, 139–148. [Google Scholar] [CrossRef]
- Yoo, S.H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.K.; Kornblum, I.; Kumar, V.; Koike, N.; Xu, M.; et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 2013, 152, 1091–1105. [Google Scholar] [CrossRef]
- Koh, K.; Zheng, X.; Sehgal, A. JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science 2006, 312, 1809–1812. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sabater-Lleal, M.; Asselbergs, F.W.; Tregouet, D.; Shin, S.Y.; Ding, J.; Baumert, J.; Oudot-Mellakh, T.; Folkersen, L.; Johnson, A.D.; et al. Genome-wide association study for circulating levels of PAI-1 provides novel insights into its regulation. Blood 2012, 120, 4873–4881. [Google Scholar] [CrossRef] [PubMed]
- Škrlec, I.; Milić, J.; Steiner, R. The influence of the circadian genes CLOCK and ARNTL on myocardial infarction. Eur. J. Hum. Genet. 2020, 28, 847. [Google Scholar] [CrossRef]
- Škrlec, I.; Milić, J.; Heffer, M.; Wagner, J.; Peterlin, B. Circadian clock genes and circadian phenotypes in patients with myocardial infarction. Adv. Med. Sci. 2019, 64, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Leibetseder, V.; Humpeler, S.; Svoboda, M.; Schmid, D.; Thalhammer, T.; Zuckermann, A.; Marktl, W.; Ekmekcioglu, C. Clock genes display rhythmic expression in human hearts. Chronobiol. Int. 2009, 26, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Chavva, H.; Brazeau, D.A.; Denvir, J.; Primerano, D.A.; Fan, J.; Seeley, S.L.; Rorabaugh, B.R. Methamphetamine-induced changes in myocardial gene transcription are sex-dependent. BMC Genom. 2021, 22, 259. [Google Scholar] [CrossRef]
- Škrlec, I.; Talapko, J.; Juzbašić, M.; Steiner, R. Sex differences in circadian clock genes and myocardial infarction susceptibility. J. Cardiovasc. Dev. Dis. 2021, 8, 53. [Google Scholar] [CrossRef]
- Curtis, A.M.; Cheng, Y.; Kapoor, S.; Reilly, D.; Price, T.S.; Fitzgerald, G.A. Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc. Natl. Acad. Sci. USA 2007, 104, 3450–3455. [Google Scholar] [CrossRef]
- Li, X.L.; Ding, Y.K.; Huang, X.Y.; Wei, L.; Zhu, D.Y.; Li, Q.P. Study on BMAL1 Protein Expression in Hypertrophic Myocardium of Hypertensive Rats. Chin. Pharmacol. Bull. 2005, 21, 811–815. [Google Scholar] [CrossRef]
- Gottlieb, L.A.; Larsen, K.; Halade, G.V.; Young, M.E.; Thomsen, M.B. Prolonged QT intervals in mice with cardiomyocyte-specific deficiency of the molecular clock. Acta Physiol. 2021, 233, e13707. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chuang, J.H.; Wang, H.T.; Chen, H.C.; Liu, W.H.; Yang, M.Y. Altered Expression of Circadian Clock Genes in Patients with Atrial Fibrillation Is Associated with Atrial High-Rate Episodes and Left Atrial Remodeling. Diagnostics 2021, 11, 90. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, D.; Zhang, N.; Liu, T.; Zhang, X.; Zou, J. PO-01-244 BMAL1 REGULATES THE CIRCADIAN RHYTHM OF RYR2 AND CONTRIBUTES TO CIRCADIAN VENTRICULAR ARRHYTHMIA IN CHRONIC HEART FAILURE AFTER MYOCARDIAL INFARCTION. Heart Rhythm 2023, 20, S168. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, P.; Cheng, Z.; Hao, W.; Wang, R.; Fang, Q.; Cao, J.M. Altered circadian rhythm of cardiac β3-adrenoceptor activity following myocardial infarction in the rat. Basic Res. Cardiol. 2011, 106, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ingle, K.A.; Kain, V.; Goel, M.; Prabhu, S.D.; Young, M.E.; Halade, G.V. Cardiomyocyte-specific Bmal1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1827–H1836. [Google Scholar] [CrossRef]
- Essop, M.F. Cardiac metabolic adaptations in response to chronic hypoxia. J. Physiol. 2007, 584, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Sonkar, R.; Berry, R.; Latimer, M.N.; Prabhu, S.D.; Young, M.E.; Frank, S.J. Augmented Cardiac Growth Hormone Signaling Contributes to Cardiomyopathy Following Genetic Disruption of the Cardiomyocyte Circadian Clock. Front. Pharmacol. 2022, 13, 836725. [Google Scholar] [CrossRef]
- Hang, P.Z.; Liu, J.; Wang, J.P.; Li, F.F.; Li, P.F.; Kong, Q.N.; Shi, J.; Ji, H.Y.; Du, Z.M.; Zhao, J. 7,8-Dihydroxyflavone alleviates cardiac fibrosis by restoring circadian signals via downregulating Bmal1/Akt pathway. Eur. J. Pharmacol. 2023, 938, 175420. [Google Scholar] [CrossRef]
- Farías, J.G.; Molina, V.M.; Carrasco, R.A.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant Therapeutic Strategies for Cardiovascular Conditions Associated with Oxidative Stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Zhang, D.; Jin, T.; Cai, D.J.; Wu, Q.; Lu, Y.; Liu, J.; Klaassen, C.D. Diurnal variation of hepatic antioxidant gene expression in mice. PLoS ONE 2012, 7, e44237. [Google Scholar] [CrossRef]
- Hemmeryckx, B.; Hohensinner, P.; Swinnen, M.; Heggermont, W.; Wojta, J.; Lijnen, H.R. Antioxidant Treatment Improves Cardiac Dysfunction in a Murine Model of Premature Aging. J. Cardiovasc. Pharmacol. 2016, 68, 374–382. [Google Scholar] [CrossRef]
- Lin, C.; Xu, L.; Tang, X.; Li, X.; Lu, C.; Cheng, Q.; Jiang, J.; Shen, Y.; Yan, D.; Qian, R.; et al. Clock Gene Bmal1 Disruption in Vascular Smooth Muscle Cells Worsens Carotid Atherosclerotic Lesions. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 565–579. [Google Scholar] [CrossRef]
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, E15–E29. [Google Scholar] [CrossRef]
- Yi, N.; Xiao, W.; Tian, Y.; Yuan, L.L. BMAL1 Alleviates H2O2-Induced Myocardial Cell Injury: A Study on the Mechanism. Tianjin Med. J. 2024, 52, 119–123. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zou, L.H.; Liu, X.H.; Yuan, L.L.; Jiang, Y. NRF2 Alleviates Doxorubicin-Induced Oxidative Stress and Lysosomal Dysfunction in Myocardial H9C2 Cells. Chin. J. Pathophysiol. 2019, 35, 1359–1364. [Google Scholar]
- Shi, S. Role of BMAL1 in Regulating the Nrf2/HO-1 Signaling Pathway in Increased Vulnerability to Myocardial Ischemia-Reperfusion in Diabetic Rats. Ph.D. Thesis, Wuhan University, Wuhan, China, 2019. [Google Scholar]
- Turek, F.W.; Joshu, C.; Kohsaka, A.; Lin, E.; Ivanova, G.; McDearmon, E.; Laposky, A.; Losee-Olson, S.; Easton, A.; Jensen, D.R.; et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef]
- Jeong, K.; He, B.; Nohara, K.; Park, N.; Shin, Y.; Kim, S.; Shimomura, K.; Koike, N.; Yoo, S.H.; Chen, Z. Dual attenuation of proteasomal and autophagic BMAL1 degradation in Clock Δ19/+ mice contributes to improved glucose homeostasis. Sci. Rep. 2015, 5, 12801. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Lavie, J.; Bénard, G. Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta 2015, 1853, 2812–2821. [Google Scholar] [CrossRef]
- Verma, D.D.; Levchenko, T.S.; Bernstein, E.A.; Torchilin, V.P. ATP-loaded liposomes effectively protect mechanical functions of the myocardium from global ischemia in an isolated rat heart model. J. Control Release Off. J. Control Release Soc. 2005, 108, 460–471. [Google Scholar] [CrossRef]
- Rabinovich-Nikitin, I.; Rasouli, M.; Reitz, C.J.; Posen, I.; Margulets, V.; Dhingra, R.; Khatua, T.N.; Thliveris, J.A.; Martino, T.A.; Kirshenbaum, L.A. Mitochondrial autophagy and cell survival is regulated by the circadian Clock gene in cardiac myocytes during ischemic stress. Autophagy 2021, 17, 3794–3812. [Google Scholar] [CrossRef]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef]
- Kohsaka, A.; Das, P.; Hashimoto, I.; Nakao, T.; Deguchi, Y.; Gouraud, S.S.; Waki, H.; Muragaki, Y.; Maeda, M. The circadian clock maintains cardiac function by regulating mitochondrial metabolism in mice. PLoS ONE 2014, 9, e112811. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Li, W.; Huang, X.; Zhao, S.; Chen, W.; Xia, Y.; Yu, W.; Rao, T.; Ning, J.; Zhou, X.; et al. BMAL1 regulates mitochondrial homeostasis in renal ischaemia-reperfusion injury by mediating the SIRT1/PGC-1α axis. J. Cell. Mol. Med. 2022, 26, 1994–2009. [Google Scholar] [CrossRef] [PubMed]
- Ran, H.X. Regulatory Factors Associated with the Browning of White Adipocytes. J. Med. Mol. Biol. 2014, 11, 53–57. [Google Scholar] [CrossRef]
- Bonney, S.; Kominsky, D.; Brodsky, K.; Eltzschig, H.; Walker, L.; Eckle, T. Cardiac Per2 functions as novel link between fatty acid metabolism and myocardial inflammation during ischemia and reperfusion injury of the heart. PLoS ONE 2013, 8, e71493. [Google Scholar] [CrossRef] [PubMed]
- Bartman, C.M.; Oyama, Y.; Brodsky, K.; Khailova, L.; Walker, L.; Koeppen, M.; Eckle, T. Intense light-elicited upregulation of miR-21 facilitates glycolysis and cardioprotection through Per2-dependent mechanisms. PLoS ONE 2017, 12, e0176243. [Google Scholar] [CrossRef]
- Young, M.E.; Brewer, R.A.; Peliciari-Garcia, R.A.; Collins, H.E.; He, L.; Birky, T.L.; Peden, B.W.; Thompson, E.G.; Ammons, B.J.; Bray, M.S.; et al. Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J. Biol. Rhythm. 2014, 29, 257–276. [Google Scholar] [CrossRef]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Tien, C.L.; Jain, M.K.; Zhang, L. Kruppel-Like Factor 15 Regulates the Circadian Susceptibility to Ischemia Reperfusion Injury in the Heart. Circulation 2020, 141, 1427–1429. [Google Scholar] [CrossRef]
- Dierickx, P.; Zhu, K.; Carpenter, B.J.; Jiang, C.; Vermunt, M.W.; Xiao, Y.; Luongo, T.S.; Yamamoto, T.; Martí-Pàmies, Í.; Mia, S.; et al. Circadian REV-ERBs repress E4bp4 to activate NAMPT-dependent NAD(+) biosynthesis and sustain cardiac function. Nat. Cardiovasc. Res. 2022, 1, 45–58. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K. Innate immune cells in ischaemic heart disease: Does myocardial infarction beget myocardial infarction? Eur. Heart J. 2016, 37, 868–872. [Google Scholar] [CrossRef]
- Zhong, Y.; Yu, X.; Li, X.; Zhou, H.; Wang, Y. Augmented early aged neutrophil infiltration contributes to late remodeling post myocardial infarction. Microvasc. Res. 2022, 139, 104268. [Google Scholar] [CrossRef] [PubMed]
- Spengler, M.L.; Kuropatwinski, K.K.; Comas, M.; Gasparian, A.V.; Fedtsova, N.; Gleiberman, A.S.; Gitlin, I.I.; Artemicheva, N.M.; Deluca, K.A.; Gudkov, A.V.; et al. Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E2457–E2465. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Fentress, S.J.; Qiu, Y.; Yun, K.; Cox, J.S.; Chawla, A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science 2013, 341, 1483–1488. [Google Scholar] [CrossRef]
- Sato, F.; Kohsaka, A.; Takahashi, K.; Otao, S.; Kitada, Y.; Iwasaki, Y.; Muragaki, Y. Smad3 and Bmal1 regulate p21 and S100A4 expression in myocardial stromal fibroblasts via TNF-α. Histochem. Cell Biol. 2017, 148, 617–624. [Google Scholar] [CrossRef]
- Halade, G.V.; Mat, Y.; Gowda, S.G.B.; Jain, S.; Hui, S.P.; Yadav, H.; Kain, V. Sleep deprivation in obesogenic setting alters lipidome and microbiome toward suboptimal inflammation in acute heart failure. FASEB J. 2023, 37, e22899. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, I.W.; Turrentine, M.; Wang, M. Postischemic application of estrogen ameliorates myocardial damage in an in vivo mouse model. J. Surg. Res. 2018, 231, 366–372. [Google Scholar] [CrossRef]
- Hao, K.L.; Zhai, Q.C.; Gu, Y.; Chen, Y.Q.; Wang, Y.N.; Liu, R.; Yan, S.P.; Wang, Y.; Shi, Y.F.; Lei, W.; et al. Disturbance of suprachiasmatic nucleus function improves cardiac repair after myocardial infarction by IGF2-mediated macrophage transition. Acta Pharmacol. Sin. 2023, 44, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.; Thorburn, A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54. [Google Scholar] [CrossRef]
- Pfeifer, U.; Strauss, P. Autophagic vacuoles in heart muscle and liver. A comparative morphometric study including circadian variations in meal-fed rats. J. Mol. Cell. Cardiol. 1981, 13, 37–49. [Google Scholar] [CrossRef]
- Xiong, X.; Tao, R.; DePinho, R.A.; Dong, X.C. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Biol. Chem. 2012, 287, 39107–39114. [Google Scholar] [CrossRef]
- Qiu, Z.; Ming, H.; Lei, S.; Zhou, B.; Zhao, B.; Yu, Y.; Xue, R.; Xia, Z. Roles of HDAC3-orchestrated circadian clock gene oscillations in diabetic rats following myocardial ischaemia/reperfusion injury. Cell Death Dis. 2021, 12, 43. [Google Scholar] [CrossRef]
- Hu, J.; Xue, Y.; Tang, K.; Fan, J.; Du, J.; Li, W.; Chen, S.; Liu, C.; Ji, W.; Liang, J.; et al. The protective effects of hydrogen sulfide on the myocardial ischemia via regulating Bmal1. Biomed. Pharmacother. 2019, 120, 109540. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, F.; Matsunaga, N.; Okazaki, H.; Azuma, H.; Hamamura, K.; Tsuruta, A.; Tsurudome, Y.; Ogino, T.; Hara, Y.; Suzuki, T.; et al. Circadian Clock in a Mouse Colon Tumor Regulates Intracellular Iron Levels to Promote Tumor Progression. J. Biol. Chem. 2016, 291, 7017–7028. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Ke, Q.; Liu, Z.; Ren, J.; Zhang, W.; Hu, J.; Wang, Z.; Chen, H.; Xia, K.; Lai, X.; et al. BMAL1 moonlighting as a gatekeeper for LINE1 repression and cellular senescence in primates. Nucleic Acids Res. 2022, 50, 3323–3347. [Google Scholar] [CrossRef]
- Beam, W.R.; Weiner, D.E.; Martin, R.J. Timing of prednisone and alterations of airways inflammation in nocturnal asthma. Am. Rev. Respir. Dis. 1992, 146, 1524–1530. [Google Scholar] [CrossRef]
- Ruben, M.D.; Smith, D.F.; FitzGerald, G.A.; Hogenesch, J.B. Dosing time matters. Science 2019, 365, 547–549. [Google Scholar] [CrossRef]
- Castiello, D.S.; Buongiorno, F.; Manzi, L.; Narciso, V.; Forzano, I.; Florimonte, D.; Sperandeo, L.; Canonico, M.E.; Avvedimento, M.; Paolillo, R.; et al. Procedural and Antithrombotic Therapy Optimization in Patients with Atrial Fibrillation Undergoing Percutaneous Coronary Intervention: A Narrative Review. J. Cardiovasc. Dev. Dis. 2025, 12, 142. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Model | Species | Target/Pathway | Molecular Mechanism | Observed Effect on IHD | Reference(s) | |
|---|---|---|---|---|---|---|
| Oxidative Stress | Cardiac function decline model | Mouse (BMAL1 KO) | ROS clearance | BMAL1 deficiency leads to ROS accumulation; antioxidant therapy partially rescues cardiac function. | Affect cardiac function and myocardial senescence. | [49] |
| Atherosclerosis model | Mouse (BMAL1 KO) | ROS/NF-κB | BMAL1 downregulation enhances TLR-NF-κB signaling, increasing ROS and monocyte recruitment. | Accelerated atherosclerosis, elevated IHD risk. | [50,51] | |
| H9C2 cardiomyocyte injury model | H9C2 rat cardiomyocytes | NRF2/HO-1 | BMAL1 overexpression activates NRF2/HO-1, reducing ROS and apoptosis. | Attenuated oxidative damage. | [52] | |
| Diabetic rat I/R model | Rat | NRF2/HO-1 | BMAL1 activates NRF2/HO-1 to reduce oxidative stress and lysosomal dysfunction. | Reduced susceptibility to ischemia–reperfusion injury. | [55] | |
| Energy Metabolism | Heart disease model | Mouse | SIRT1/PGC1α, CLOCK/BMAL1-ROS/mitochondrial fission | BMAL1 damages mitochondrial function through the SIRT1-PGC1α axis, while CLOCK/BMAL1 interactions reduce ROS, inhibit division, enhance electron transfer, and alleviate oxidative stress. | BMAL1 deficiency causes mitochondrial dysfunction and impaired ATP synthesis. Insufficient myocardial energy supply. | [60,61,62,63] |
| Ischemia and reperfusion model | Mouse (PER2 KO) | PER2/Fatty acid metabolism | BMAL1 regulates PER2 via the CLOCK/BMAL1 heterodimer binding to E-Box, activating PER2 transcription. | PER2 deletion impairs glycolysis, depletes glycogen stores, and increases infarct size. | [65,66] | |
| Cardiomyocyte-specific BMAL1 knockout model | Mouse (BMAL1 KO) | PIK3R1-p85α (PI3K/AKT/GSK3β) | BMAL1 regulates the circadian expression of PIK3R1, a key insulin signaling component. | Impaired insulin signaling cascade, leading to metabolic and contractile dysfunction. | [67] | |
| Ischemia and reperfusion model | Mouse | NAMPT/NAD | BMAL1/CLOCK-KLF15 axis regulates circadian NAMPT expression; KLF15 KO disrupts NAD rhythm. | NAD deficiency causes energy stagnation and abnormal cardiac metabolism. | [68,69,70] | |
| Immune-Inflammatory Response | MI | Mouse | BMAL1/Neutrophil aging | BMAL1 regulates circadian oscillations of neutrophils; loss of BMAL1 disrupts neutrophil aging and infiltration. | Reduced angiogenesis, increased fibrosis, and impaired cardiac remodeling post-MI. | [43,72] |
| Atherosclerosis model | Mouse, human aortic endothelial cells | TLRs-NF-κB/DNMT-1 | TLRs-NF-κB axis inhibits BMAL1 via DNMT-1-mediated promoter methylation; BMAL1 loss enhances NF-κB signaling. | Elevated neutrophil chemotaxis, pro-inflammatory gene expression, and cardiac fibrosis. | [51] | |
| Cardiomyocyte-specific BMAL1 knockout model | Mouse (BMAL1 KO) | TNF-α/SMAD3 | BMAL1 deletion elevates TNF-α expression in cardiac fibroblasts and macrophages. | Enhanced myocardial fibrosis and inflammatory signaling. | [75] | |
| High-fat diet + circadian disruption + MI | Mouse | BMAL1/Clock/Pro-inflammatory cytokines | BMAL1/CLOCK downregulation increases CCL2, IL-1β, and IL-6 at MI sites. | Exacerbated cardiac inflammation and adverse remodeling under metabolic stress. | [76] | |
| SCN-ablated MI model | Mouse | BMAL1/Igf2/Macrophage phenotype | SCN ablation upregulates BMAL1, promoting Igf2 expression and anti-inflammatory macrophage polarization. | Improved post-MI inflammation resolution and cardiac repair. | [78] | |
| Apoptosis and Autophagy | Diabetic rats with I/R injury | Rat | HDAC3/REV-ERBα/BMAL1 pathway | HDAC3 upregulates BMAL1 to restore mitochondrial autophagy via REV-ERBα/BMAL1 signaling. | Alleviates myocardial infarction injury. | [82] |
| Myocardial ischemia model | H9C2 rat cardiomyocytes | PI3K/AKT signaling and oxidative stress | BMAL1 maintains the circadian rhythm of ischemic cardiomyocytes by regulating PI3K/AKT and oxidative stress. | Reduces apoptosis, protects cardiomyocyte survival. | [83] | |
| Immunodeficient mice inoculated with human tumor cells | Mouse | EGLN2/PHD1 | Reduced BMAL1 promotes ferroptosis via EGLN2/PHD1-mediated oxidative damage. | Aggravates ferroptosis-related cell death. | [85,86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Zhao, J.; Chen, Z.; Duan, L.; Yang, H.; Cai, D. BMAL1 in Ischemic Heart Disease: A Narrative Review from Molecular Clock to Myocardial Pathology. Int. J. Mol. Sci. 2025, 26, 4626. https://doi.org/10.3390/ijms26104626
Yang J, Zhao J, Chen Z, Duan L, Yang H, Cai D. BMAL1 in Ischemic Heart Disease: A Narrative Review from Molecular Clock to Myocardial Pathology. International Journal of Molecular Sciences. 2025; 26(10):4626. https://doi.org/10.3390/ijms26104626
Chicago/Turabian StyleYang, Jingyi, Junxin Zhao, Zhuoyang Chen, Lincheng Duan, Hong Yang, and Dingjun Cai. 2025. "BMAL1 in Ischemic Heart Disease: A Narrative Review from Molecular Clock to Myocardial Pathology" International Journal of Molecular Sciences 26, no. 10: 4626. https://doi.org/10.3390/ijms26104626
APA StyleYang, J., Zhao, J., Chen, Z., Duan, L., Yang, H., & Cai, D. (2025). BMAL1 in Ischemic Heart Disease: A Narrative Review from Molecular Clock to Myocardial Pathology. International Journal of Molecular Sciences, 26(10), 4626. https://doi.org/10.3390/ijms26104626