
Long-Term Functional and Structural Renoprotection After Experimental Acute Kidney Injury in Subclinical Chronic Kidney Disease In Vivo

 ,
,  , and
, and
Abstract
1. Introduction
2. Results
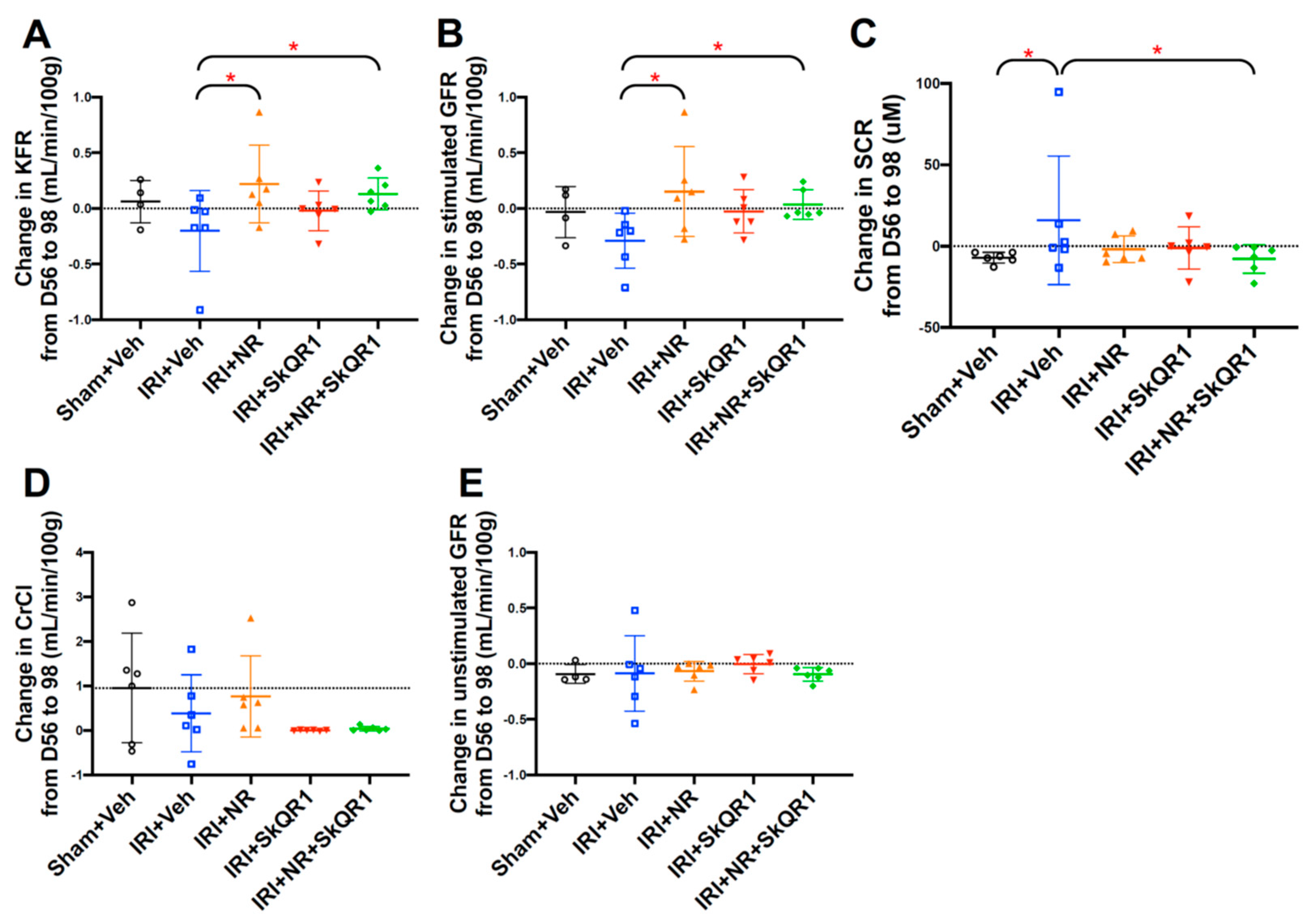
2.1. NR + SkQR1 Protected Stimulated GFR and KFR After IRI
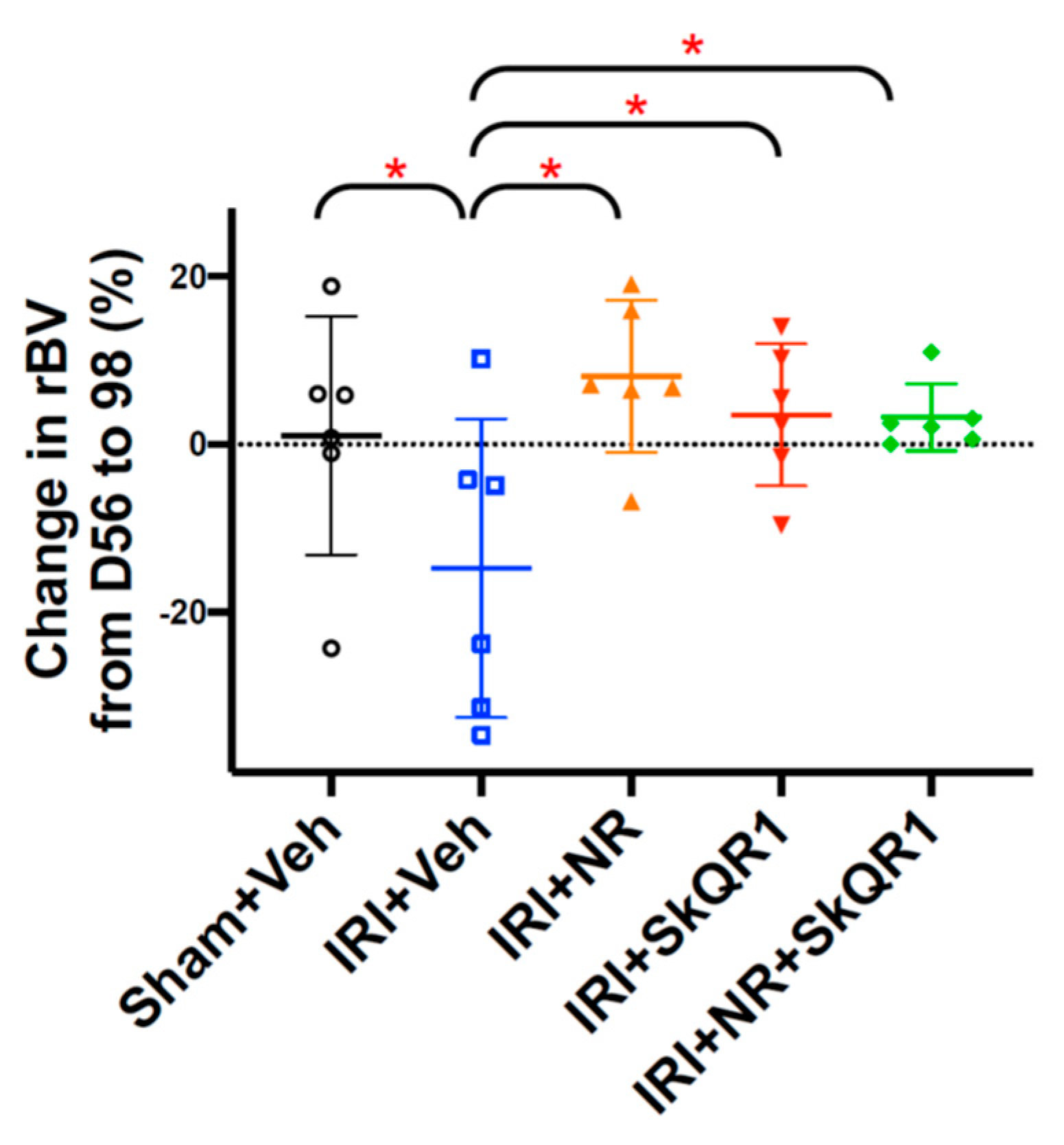
2.2. NR + SkQR1 Protected Relative Blood Volume (rBV) After IRI
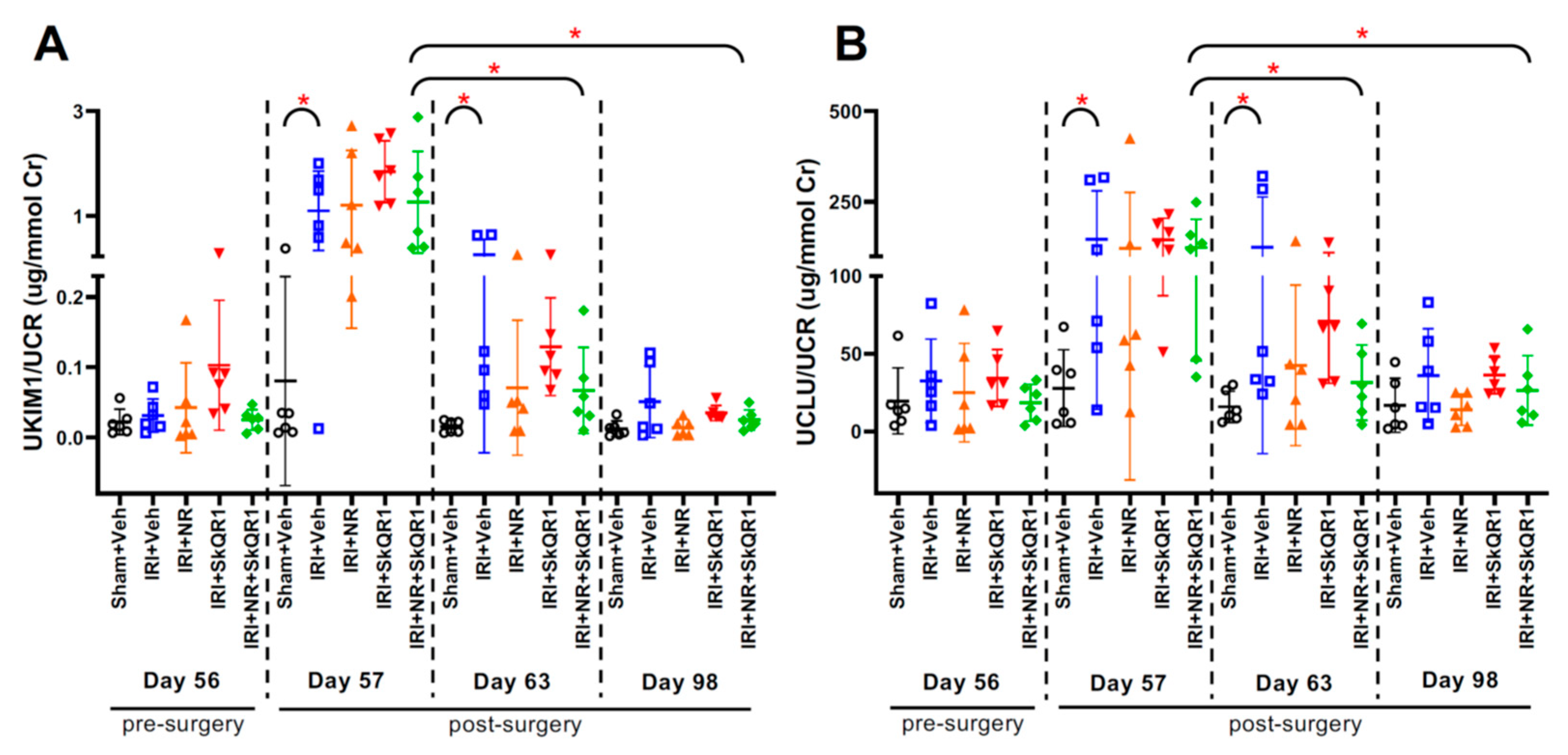
2.3. Kidney Injury After IRI Confirmed by Urine Biomarkers
2.4. NR + SkQR1 Treatment Protected Against Structural Damage
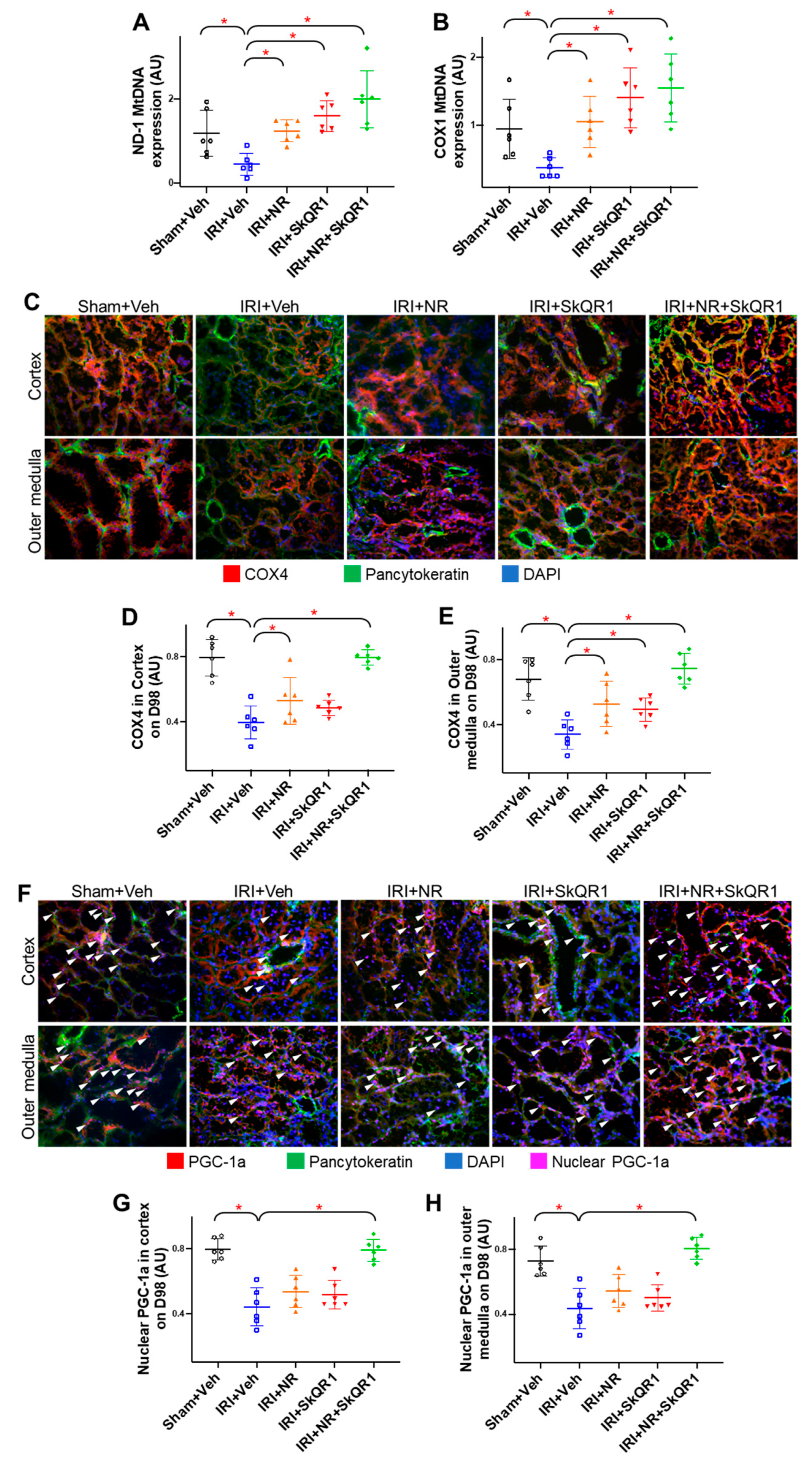
2.5. Mitochondrial Density Was Protected by NR + SkQR1
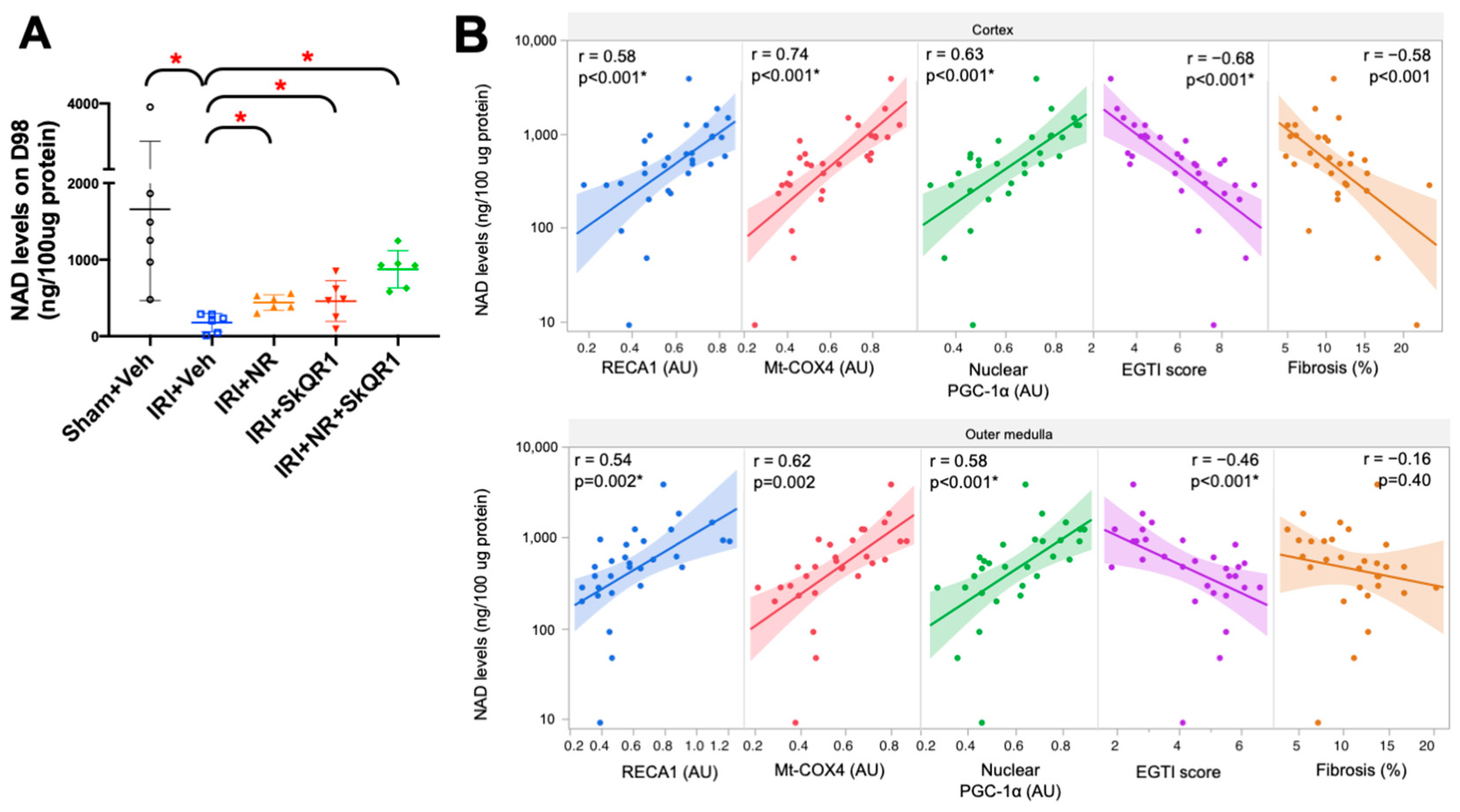
2.6. Kidney Redox State and Treatment
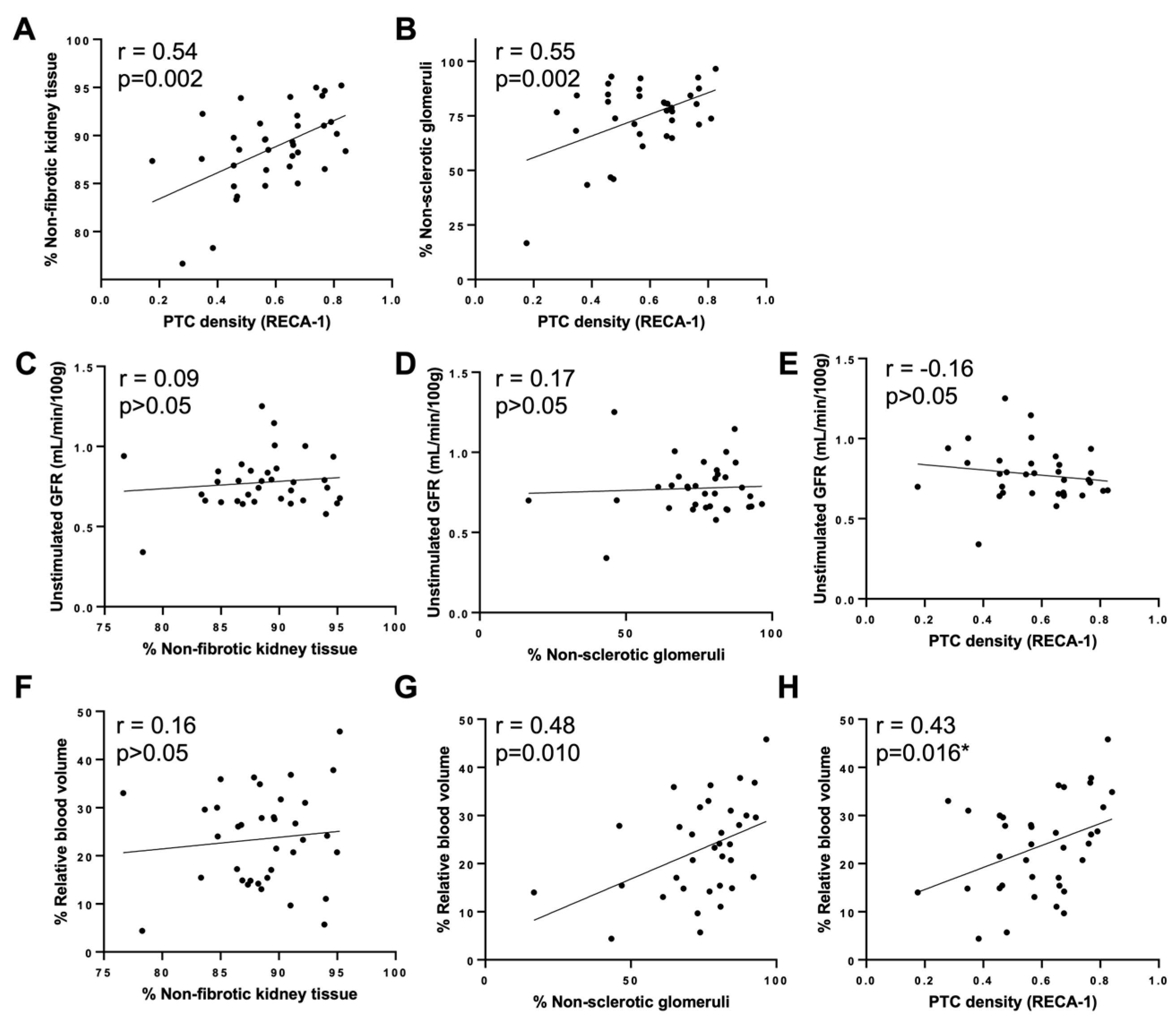
2.7. Correlation of Functional Markers with Structural Markers
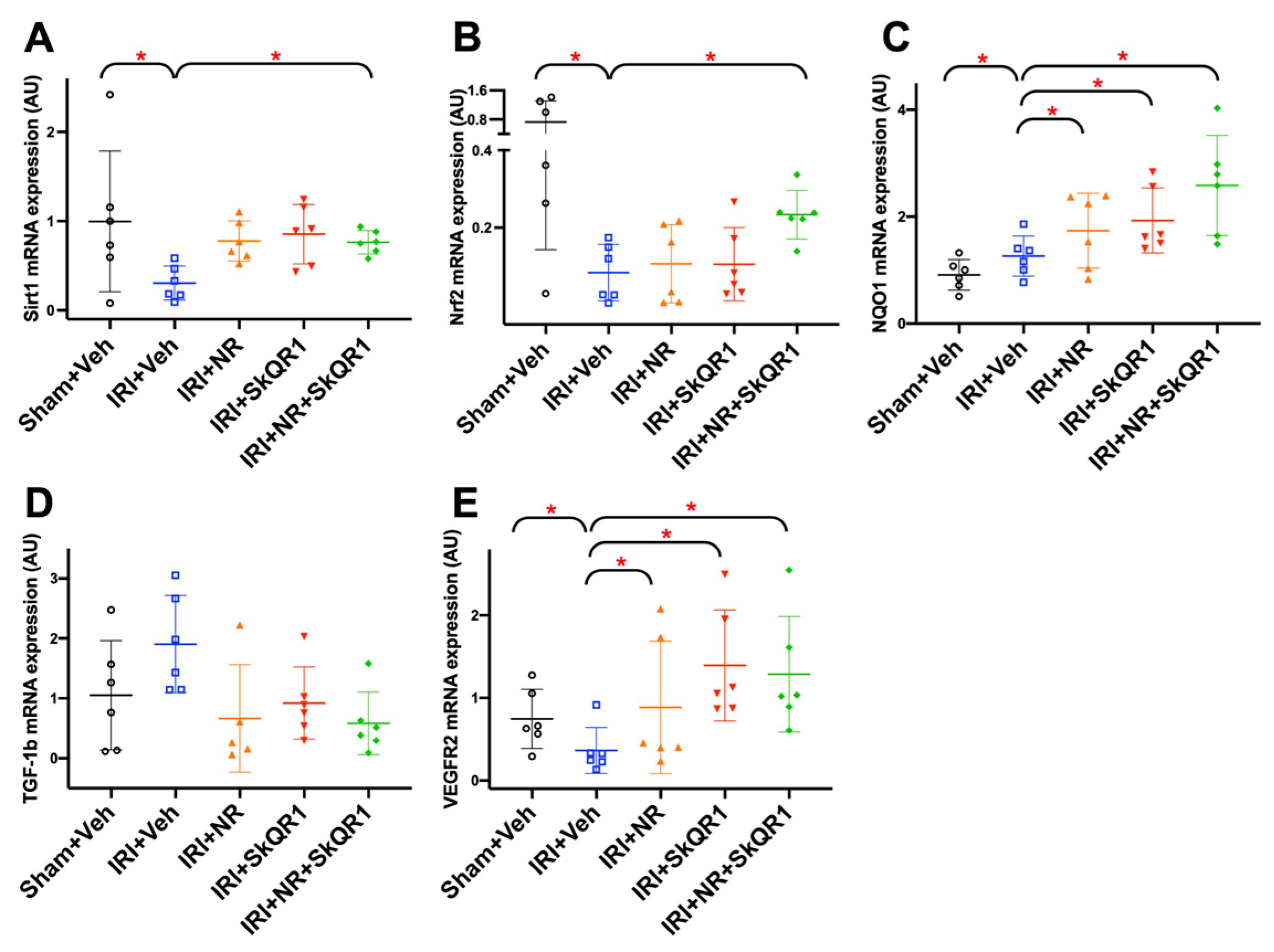
2.8. Identification of Protection Pathways Against IRI
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. sCKD Induction and IRI Protocol
4.3. Treatment Groups
4.4. GFR Measurements
4.5. Biomarker Measurements
4.6. Histopathology
4.7. Immunofluorescence
4.8. Quantitation of Protein of Interest
4.9. NAD Measurement
4.10. Vascular Ultrasound
4.11. RNA Extraction, cDNA Synthesis, and qPCRs
4.12. Mitochondrial DNA PCR
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; ADQI XIII Work Group. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Amdur, R.L.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011, 79, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Jensen, S.K.; Heide-Jørgensen, U.; Gammelager, H.; Birn, H.; Christiansen, C.F. Acute Kidney Injury Duration and 20-Year Risks of CKD and Cardiovascular Disease. Kidney Int. Rep. 2024, 9, 817–829. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- Sharma, A.; Mucino, M.J.; Ronco, C. Renal functional reserve and renal recovery after acute kidney injury. Nephron Clin. Pract. 2014, 127, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Chawla, L.S. Glomerular and Tubular Kidney Stress Test: New Tools for a Deeper Evaluation of Kidney Function. Nephron 2016, 134, 191–194. [Google Scholar] [CrossRef]
- Succar, L.; Pianta, T.J.; Davidson, T.; Pickering, J.W.; Endre, Z.H. Subclinical chronic kidney disease modifies the diagnosis of experimental acute kidney injury. Kidney Int. 2017, 92, 680–692. [Google Scholar] [CrossRef]
- Sarafidis, P.; Ferro, C.J.; Ortiz, A. Hypernatremia and subclinical chronic kidney disease. Eur. Heart J. 2022, 43, 4436–4437. [Google Scholar] [CrossRef]
- Taylor, K.M.; Au, A.Y.M.; Herath, S.; Succar, L.; Wong, J.; Erlich, J.H.; Endre, Z.H. Kidney functional reserve and damage biomarkers in subclinical chronic kidney disease and acute kidney injury. Kidney functional reserve and damage biomarkers in subclinical chronic kidney disease and acute kidney injury. Am. J. Physiol. Renal Physiol. 2023, 325, F888–F898. [Google Scholar] [CrossRef]
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 9, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Kramer, R.S.; Coca, S.G.; Parikh, C.R. Duration of acute kidney injury impacts long-term survival after cardiac surgery. Ann. Thorac. Surg. 2010, 90, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Lagny, M.G.; Jouret, F.; Koch, J.N.; Blaffart, F.; Donneau, A.F.; Albert, A.; Roediger, L.; Krzesinski, J.M.; Defraigne, J.O. Incidence and outcomes of acute kidney injury after cardiac surgery using either criteria of the RIFLE classification. BMC Nephrol. 2015, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Loubani, M.; Grant, S.W.; Goodwin, A.T.; Trivedi, U.; Kendall, S.; Jenkins, D.P. Cardiac surgery in older patients: Hospital outcomes during a 15-year period from a complete national series. Interact. CardioVascular Thorac. Surg. 2022, 34, 532–539. [Google Scholar] [CrossRef]
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular hyperfiltration: Definitions, mechanisms and clinical implications. Nat. Rev. Nephrol. 2012, 8, 293–300. [Google Scholar] [CrossRef]
- Husain-Syed, F.; Ferrari, F.; Sharma, A.; Danesi, T.H.; Bezerra, P.; Lopez-Giacoman, S.; Samoni, S.; de Cal, M.; Corradi, V.; Virzì, G.M.; et al. Preoperative Renal Functional Reserve Predicts Risk of Acute Kidney Injury After Cardiac Operation. Ann. Thorac. Surg. 2018, 105, 1094–1101. [Google Scholar] [CrossRef]
- Landoni, G.; Monaco, F.; Ti, L.K.; Baiardo Redaelli, M.; Bradic, N.; Comis, M.; Kotani, Y.; Brambillasca, C.; Garofalo, E.; Scandroglio, A.M.; et al. A Randomized Trial of Intravenous Amino Acids for Kidney Protection. N. Engl. J. Med. 2024, 391, 687–698. [Google Scholar] [CrossRef]
- Ostermann, M.; Shaw, A.D. Amino Acid Infusion to Protect Kidney Function after Cardiac Surgery. N. Engl. J. Med. 2024, 391, 759–760. [Google Scholar] [CrossRef]
- Pruna, A.; Losiggio, R.; Landoni, G.; Kotani, Y.; Redaelli, M.B.; Veneziano, M.; Lee, T.C.; Zangrillo, A.; Gaudino, M.F.L.; Bellomo, R.; et al. Amino Acid Infusion for Perioperative Functional Renal Protection: A Meta-analysis. J. Cardiothorac. Vasc. Anesth. 2024, 38, 3076–3085. [Google Scholar] [CrossRef]
- Woods, L.L. Mechanisms of renal hemodynamic regulation in response to protein feeding. Kidney Int. 1993, 44, 659–675. [Google Scholar] [CrossRef]
- Schrijvers, B.F.; Rasch, R.; Tilton, R.G.; Flyvbjerg, A. High protein-induced glomerular hypertrophy is vascular endothelial growth factor-dependent. Kidney Int. 2002, 61, 1600–1604. [Google Scholar] [CrossRef]
- Husain-Syed, F.; Emlet, D.R.; Wilhelm, J.; Danesi, T.H.; Ferrari, F.; Bezerra, P.; Lopez-Giacoman, S.; Villa, G.; Tello, K.; Birk, H.W.; et al. Effects of preoperative high-oral protein loading on short- and long-term renal outcomes following cardiac surgery: A cohort study. J. Transl. Med. 2022, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bledsoe, G.; Hagiwara, M.; Yang, Z.R.; Shen, B.; Chao, L.; Chao, J. Blockade of endogenous tissue kallikrein aggravates renal injury by enhancing oxidative stress and inhibiting matrix degradation. Am. J. Physiol. Ren. Physiol. 2010, 298, F1033–F1040. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.; Zondler, L.; Ludwig, N.; Kardell, M.; Lüneburg, C.; Henke, K.; Mersmann, S.; Margraf, A.; Spieker, T.; Tekath, T.; et al. Glutamine prevents acute kidney injury by modulating oxidative stress and apoptosis in tubular epithelial cells. JCI Insight 2022, 7, e163161. [Google Scholar] [CrossRef]
- Lee, K.; Thompson, E.A.; Gharaie, S.; Patel, C.H.; Kurzhagen, J.T.; Pierorazio, P.M.; Arend, L.J.; Thomas, A.G.; Noel, S.; Slusher, B.S.; et al. T cell metabolic reprogramming in acute kidney injury and protection by glutamine blockade. JCI Insight 2023, 8, e160345. [Google Scholar] [CrossRef]
- Bakeeva, L.E.; Barskov, I.V.; Egorov, M.V.; Isaev, N.K.; Kapelko, V.I.; Kazachenko, A.V.; Kirpatovsky, V.I.; Kozlovsky, S.V.; Lakomkin, V.L.; Levina, S.B.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and Age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke). Biochemistry 2016, 73, 1288–1299. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1beta and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sha, Y.; Wei, K.; Wu, C.; Ding, D.; Yang, Y.; Zhu, C.; Zhang, Y.; Ding, G.; Zhang, A.; et al. Rotenone ameliorates chronic renal injury caused by acute ischemia/reperfusion. Oncotarget 2018, 9, 24199–24208. [Google Scholar] [CrossRef]
- Ding, M.; Tolbert, E.; Birkenbach, M.; Gohh, R.; Akhlaghi, F.; Ghonem, N.S. Treprostinil reduces mitochondrial injury during rat renal ischemia-reperfusion injury. Biomed. Pharmacother. 2021, 141, 111912. [Google Scholar] [CrossRef]
- Coca, S.G. Acute kidney injury in elderly persons. Am. J. Kidney Dis. 2010, 56, 122–131. [Google Scholar] [CrossRef]
- Kane-Gill, S.L.; Sileanu, F.E.; Murugan, R.; Trietley, G.S.; Handler, S.M.; Kellum, J.A. Risk factors for acute kidney injury in older adults with critical illness: A retrospective cohort study. Am. J. Kidney Dis. 2015, 65, 860–869. [Google Scholar] [CrossRef]
- Imam, Z.; Odish, F.; Gill, I.; O’Connor, D.; Armstrong, J.; Vanood, A.; Ibironke, O.; Hanna, A.; Ranski, A.; Halalau, A. Older age and comorbidity are independent mortality predictors in a large cohort of 1305 COVID-19 patients in Michigan, United States. J. Intern. Med. 2020, 288, 469–476. [Google Scholar] [CrossRef]
- Bábíčková, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Schuh, C.D. Mitochondria as therapeutic targets in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2016, 25, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transpl. 2016, 31, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Silachev, D.N.; Chupyrkina, A.A.; Danshina, M.I.; Jankauskas, S.S.; Morosanova, M.A.; Stelmashook, E.V.; Vasileva, A.K.; Goryacheva, E.S.; Pirogov, Y.A.; et al. New-generation Skulachev ions exhibiting nephroprotective and neuroprotective properties. Biochemistry 2010, 75, 145–150. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Andrianova, N.V.; Alieva, I.B.; Prusov, A.N.; Matsievsky, D.D.; Zorova, L.D.; Pevzner, I.B.; Savchenko, E.S.; Pirogov, Y.A.; Silachev, D.N.; et al. Dysfunction of Kidney Endothelium after Ischemia/Reperfusion and Its Prevention by Mitochondria-Targeted Antioxidant. Biochemistry 2016, 81, 1538–1548. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef]
- He, S.; Gao, Q.; Wu, X.; Shi, J.; Zhang, Y.; Yang, J.; Li, X.; Du, S.; Zhang, Y.; Yu, J. NAD+ ameliorates endotoxin-induced acute kidney injury in a sirtuin1–dependent manner via GSK-3β/Nrf2 signalling pathway. J. Cell. Mol. Med. 2022, 26, 1979–1993. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Chupyrkina, A.A.; Jankauskas, S.S.; Pevzner, I.B.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim. Biophys. Acta 2011, 1812, 77–86. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Morevati, M.; Egstrand, S.; Nordholm, A.; Mace, M.L.; Andersen, C.B.; Salmani, R.; Olgaard, K.; Lewin, E. Effect of NAD+ boosting on kidney ischemia-reperfusion injury. PLoS ONE 2021, 16, e0252554. [Google Scholar] [CrossRef] [PubMed]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD(+) in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef]
- Mitchell, T.; Rotaru, D.; Saba, H.; Smith, R.A.; Murphy, M.P.; MacMillan-Crow, L.A. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. J. Pharmacol. Exp. Ther. 2011, 336, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef]
- Szeto, H.H. Pharmacologic Approaches to Improve Mitochondrial Function in AKI and CKD. J. Am. Soc. Nephrol. 2017, 28, 2856–2865. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation with a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horváth, B.; Zsengellér, Z.; Zielonka, J.; Tanchian, G.; Holovac, E.; Kechrid, M.; Patel, V.; Stillman, I.E.; Parikh, S.M.; et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 2012, 52, 497–506. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.; Logan, A.; Gruszczyk, A.V.; Beach, T.E.; James, A.M.; Dare, A.J.; Barlow, A.; Martin, J.; Georgakopoulos, N.; Gane, A.M.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates ischaemia-reperfusion injury in kidney transplantation models. Br. J. Surg. 2021, 108, 1072–1081. [Google Scholar] [CrossRef]
- Kirkman, D.L.; Stock, J.M.; Shenouda, N.; Bohmke, N.J.; Kim, Y.; Kidd, J.; Townsend, R.R.; Edwards, D.G. Effects of a mitochondrial-targeted ubiquinol on vascular function and exercise capacity in chronic kidney disease: A randomized controlled pilot study. Am. J. Physiol. Renal Physiol. 2023, 325, F448–F456. [Google Scholar] [CrossRef] [PubMed]
- Jufar, A.H.; Lankadeva, Y.R.; May, C.N.; Cochrane, A.D.; Bellomo, R.; Evans, R.G. Renal functional reserve: From physiological phenomenon to clinical biomarker and beyond. American Journal of Physiology-Regulatory. Integr. Comp. Physiol. 2020, 319, R690–R702. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Z.; Zhang, H.; Chen, H.; Hao, J.; Liu, H.; Li, X. Mitochondrial metabolism and targeted treatment strategies in ischemic-induced acute kidney injury. Cell Death Discov. 2024, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Diwan, V.; Small, D.; Kauter, K.; Gobe, G.C.; Brown, L. Gender differences in adenine-induced chronic kidney disease and cardiovascular complications in rats. Am. J. Physiol. Renal Physiol. 2014, 307, F1169–F1178. [Google Scholar] [CrossRef]
- Schock-Kusch, D.; Sadick, M.; Henninger, N.; Kraenzlin, B.; Claus, G.; Kloetzer, H.M.; Weiss, C.; Pill, J.; Gretz, N. Transcutaneous measurement of glomerular filtration rate using FITC-sinistrin in rats. Nephrol. Dial. Transpl. 2009, 24, 2997–3001. [Google Scholar] [CrossRef]
- Paller, M.S.; Hostetter, T.H. Dietary protein increases plasma renin and reduces pressor reactivity to angiotensin II. Am. J. Physiol. 1986, 251, F34–F39. [Google Scholar] [CrossRef]
- Dahab, G.M.; Kheriza, M.M.; El-Beltagi, H.M.; Fouda, A.M.; El-Din, O.A. Digital quantification of fibrosis in liver biopsy sections: Description of a new method by Photoshop software. J. Gastroenterol. Hepatol. 2003, 19, 78–85. [Google Scholar] [CrossRef]
- Model, M.A. Intensity calibration and shading correction for fluorescence microscopes. Curr. Protoc. Cytom. 2006, 37, 10.14.1–10.14.7. [Google Scholar] [CrossRef] [PubMed]
- Ehling, J.; Theek, B.; Gremse, F.; Baetke, S.; Möckel, D.; Maynard, J.; Ricketts, S.A.; Grüll, H.; Neeman, M.; Knuechel, R.; et al. Micro-CT imaging of tumor angiogenesis: Quantitative measures describing micromorphology and vascularization. Am. J. Pathol. 2014, 184, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.B.; Paschal, B.M. Fluorescence-based quantification of nucleocytoplasmic transport. Methods 2019, 157, 106–114. [Google Scholar] [CrossRef]
- Herath, S.; Dai, H.; Erlich, J.; Au, A.Y.; Taylor, K.; Succar, L.; Endre, Z.H. Selection and validation of reference genes for normalisation of gene expression in ischaemic and toxicological studies in kidney disease. PLoS ONE 2020, 15, e0233109. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dual Treatment (NR + SkQR1; n = 6) vs. Vehicle Group (n = 6) | Univariable Analysis | AUC (95% CI) |
|---|---|---|
| Histological or mitochondrial biomarkers (nominal logistic regression model) | EGTI score (cortex) | 1.00 (1.00–1.00) * |
| Fibrosis (cortex) | 1.00 (1.00–1.00) * | |
| Mt-COX1 DNA | 1.00 (1.00–1.00) * | |
| All Treated (NR, SkQR1, and NR + SkQR1; n = 18) vs.Vehicle Group (n = 6) | Multivariable Analysis | AUC (95% CI) |
| Non-invasive biomarkers (LASSO regression model) | UKIM1/UCR, UCLU/UCR, KFR, and rBV | 1.00 (0.98–1.00) * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herath, S.; Au, A.Y.M.; Taylor, K.M.; Kapoor-Kaushik, N.; Endre, Z.H.; Erlich, J.H. Long-Term Functional and Structural Renoprotection After Experimental Acute Kidney Injury in Subclinical Chronic Kidney Disease In Vivo. Int. J. Mol. Sci. 2025, 26, 4616. https://doi.org/10.3390/ijms26104616
Herath S, Au AYM, Taylor KM, Kapoor-Kaushik N, Endre ZH, Erlich JH. Long-Term Functional and Structural Renoprotection After Experimental Acute Kidney Injury in Subclinical Chronic Kidney Disease In Vivo. International Journal of Molecular Sciences. 2025; 26(10):4616. https://doi.org/10.3390/ijms26104616
Chicago/Turabian StyleHerath, Sanjeeva, Amy Y. M. Au, Kylie M. Taylor, Natasha Kapoor-Kaushik, Zoltán H. Endre, and Jonathan H. Erlich. 2025. "Long-Term Functional and Structural Renoprotection After Experimental Acute Kidney Injury in Subclinical Chronic Kidney Disease In Vivo" International Journal of Molecular Sciences 26, no. 10: 4616. https://doi.org/10.3390/ijms26104616
APA StyleHerath, S., Au, A. Y. M., Taylor, K. M., Kapoor-Kaushik, N., Endre, Z. H., & Erlich, J. H. (2025). Long-Term Functional and Structural Renoprotection After Experimental Acute Kidney Injury in Subclinical Chronic Kidney Disease In Vivo. International Journal of Molecular Sciences, 26(10), 4616. https://doi.org/10.3390/ijms26104616