
An MHC-Related Gene’s Signature Predicts Prognosis and Immune Microenvironment Infiltration in Glioblastoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
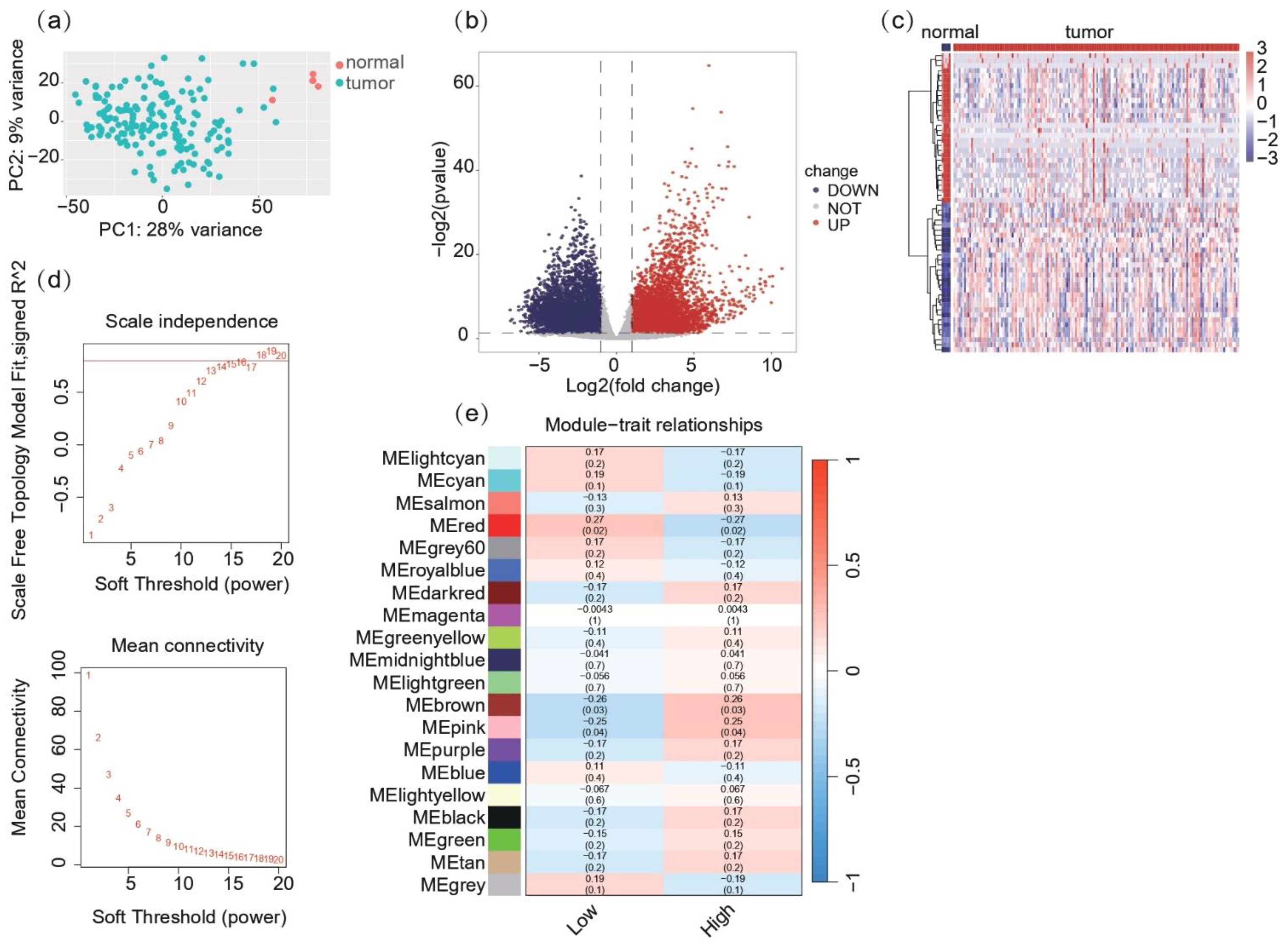
2.1. Co-Expression Network and Module Identification Reveal MHC-Related Gene Clusters in Glioblastoma
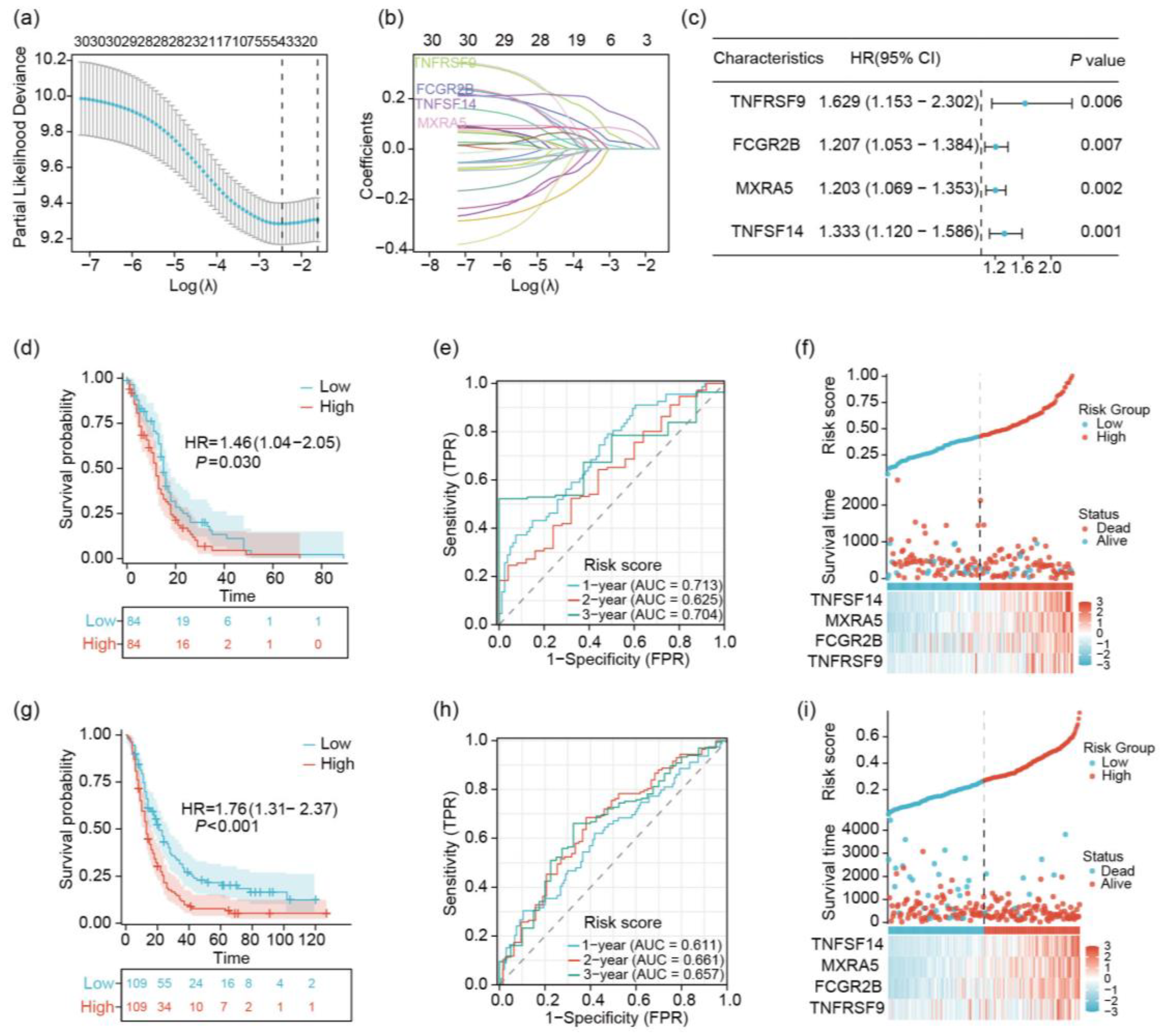
2.2. Identification of 4 Prognostic Key Genes Using LASSO Regression Analysis
2.3. Analysis of TNFSF14, MXRA5, FCGR2B, and TNFRSF9 in GBM
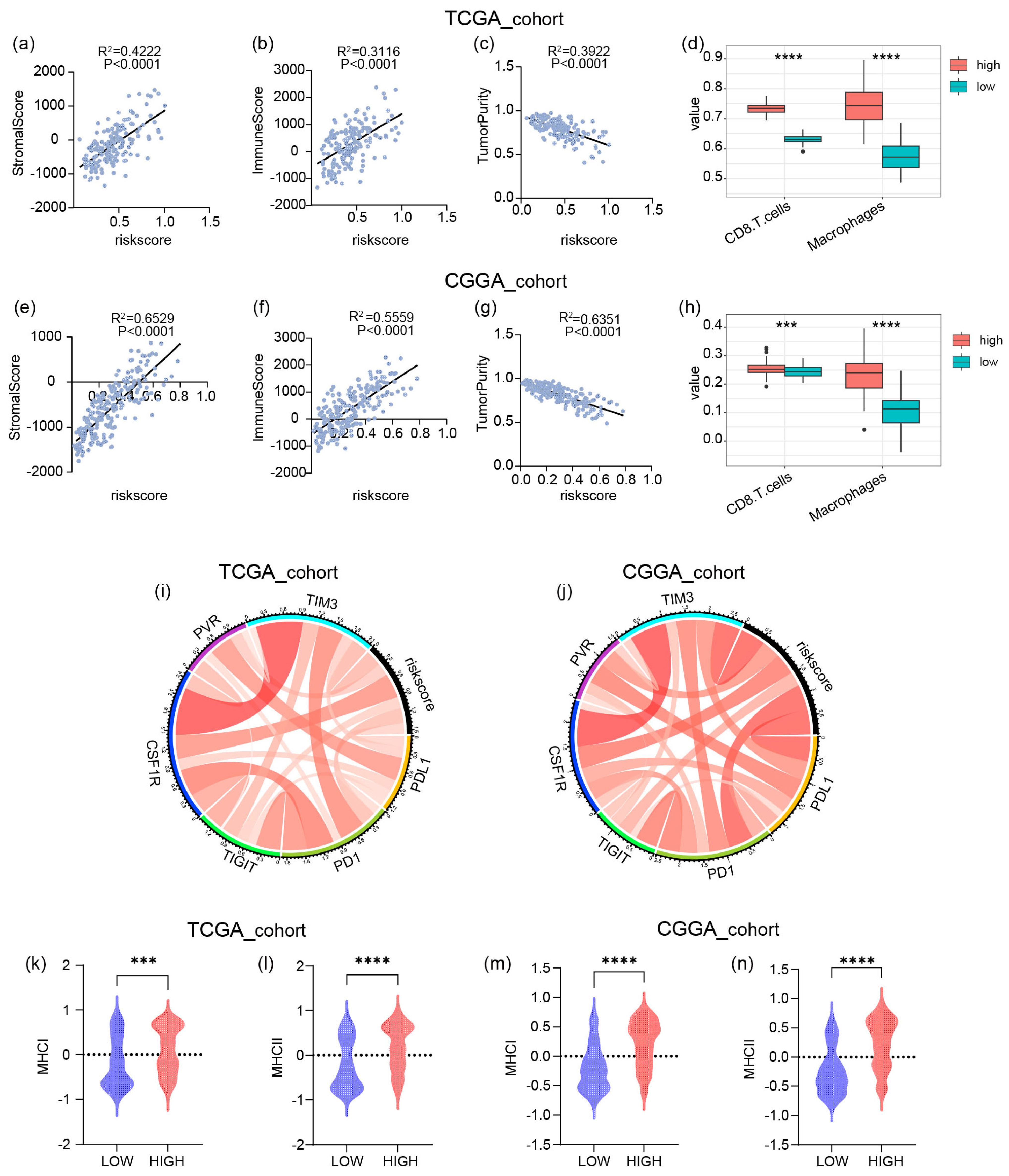
2.4. The Risk Signature Is Strongly Associated with Immune Functions in GBM
2.5. The High Risk Score Predicts the Enrichment of Macrophages and CD8+ T Cells in GBM
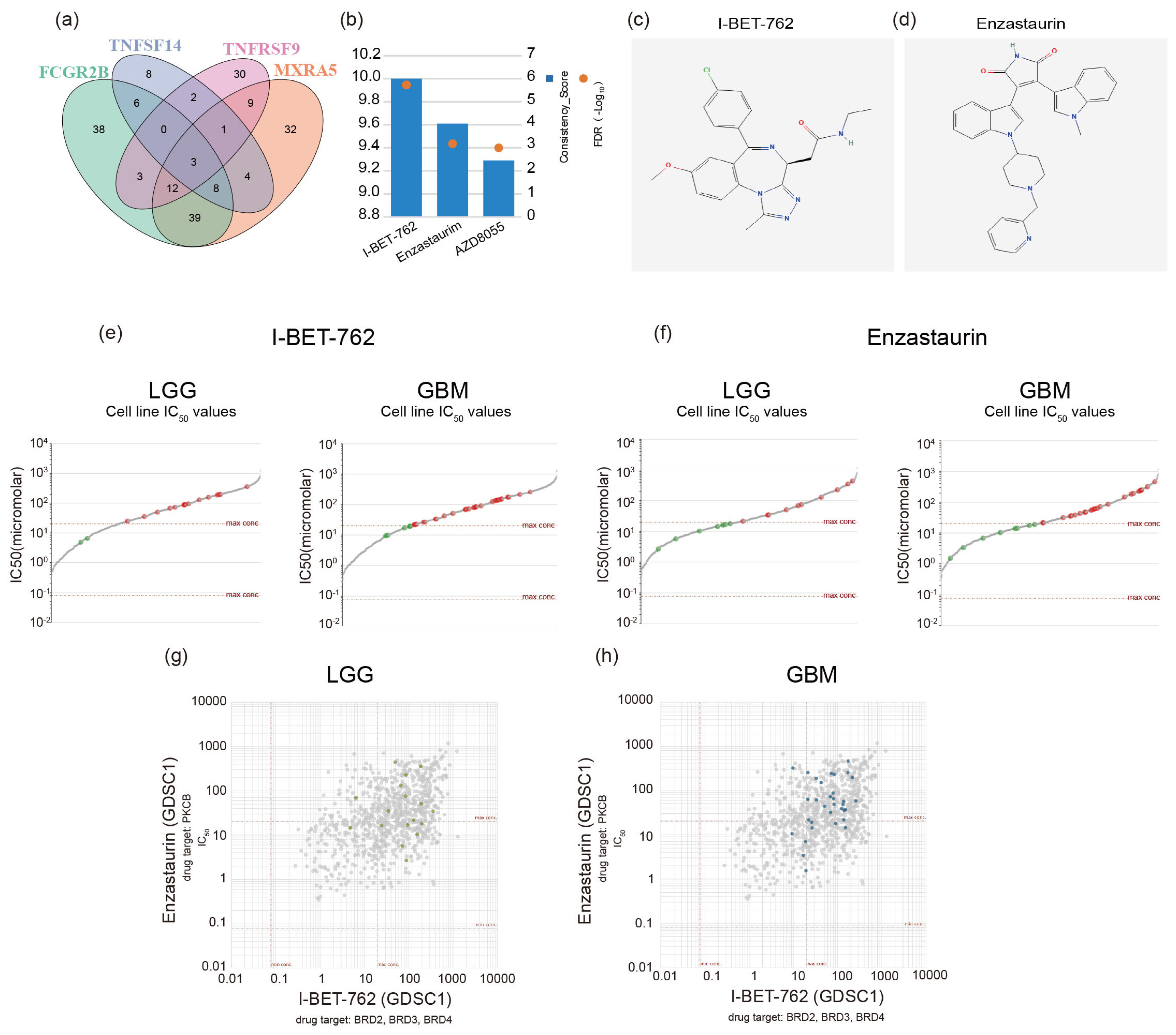
2.6. Screening GBM Therapeutic Drugs via Critical Genes
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Preprocessing
4.2. Differential Gene Expression (DEG) Analysis
4.3. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.4. LASSO Regression Analysis
4.5. Risk Score Calculation and Patient Stratification
4.6. Survival Analysis
4.7. Time-Dependent Receiver Operating Characteristic (ROC) Curve Analysis
4.8. Risk Score Distribution and Gene Expression Analysis
4.9. Validation in Independent Cohort
4.10. Gene Ontology and KEGG Pathway Enrichment Analyses
4.11. Gene Set Enrichment Analysis (GSEA)
4.12. Screening of Small Molecule Drugs and Sensitivity Calculation
4.13. Gene–Gene Interaction Network Analysis
4.14. Immune Cell Infiltration Analysis
4.14.1. ESTIMATE Analysis
4.14.2. ssGSEA Analysis
4.15. Gene Expression and Survival Analysis
Differential Expression Analysis
4.16. Immunohistochemical Analysis
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TCGA | The Cancer Genome Atlas |
| CGGA | Chinese Glioma Genome Atlas |
| WGCNA | Weighted gene co-expression network analysis |
| LASSO | The least absolute shrinkage and selection operator |
| MHC | Major histocompatibility complex |
References
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Szklener, K.; Mazurek, M.; Wieteska, M.; Wacławska, M.; Bilski, M.; Mańdziuk, S. New Directions in the Therapy of Glioblastoma. Cancers 2022, 14, 5377. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, L.; Chen, H.; Zhao, J.; Li, K.; Sun, J.; Zhou, M. Pan-cancer landscape of T-cell exhaustion heterogeneity within the tumor microenvironment revealed a progressive roadmap of hierarchical dysfunction associated with prognosis and therapeutic efficacy. EBioMedicine 2022, 83, 104207. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Dersh, D.; Hollý, J.; Yewdell, J.W. Author Correction: A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2020, 20, 644. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef]
- Wu, X.; Jiang, R.; Yang, X.; Guo, H.; Yang, R. Targeting MHC-I molecules for cancer: Function, mechanism, and therapeutic prospects. Mol. Cancer 2023, 22, 194. [Google Scholar] [CrossRef]
- Abd Hamid, M.; Wang, R.Z.; Yao, X.; Fan, P.; Li, X.; Chang, X.M.; Feng, Y.; Jones, S.; Maldonado-Perez, D.; Waugh, C.; et al. Enriched HLA-E and CD94/NKG2A Interaction Limits Antitumor CD8(+) Tumor-Infiltrating T Lymphocyte Responses. Cancer Immunol. Res. 2019, 7, 1293–1306. [Google Scholar] [CrossRef]
- Liu, X.; Song, J.; Zhang, H.; Liu, X.; Zuo, F.; Zhao, Y.; Zhao, Y.; Yin, X.; Guo, X.; Wu, X.; et al. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell 2023, 41, 272–287.e9. [Google Scholar] [CrossRef] [PubMed]
- Eugène, J.; Jouand, N.; Ducoin, K.; Dansette, D.; Oger, R.; Deleine, C.; Leveque, E.; Meurette, G.; Podevin, J.; Matysiak, T.; et al. The inhibitory receptor CD94/NKG2A on CD8(+) tumor-infiltrating lymphocytes in colorectal cancer: A promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod. Pathol. 2020, 33, 468–482. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Q.; Li, L.; Chen, K.; Yang, J.; Dixit, D.; Gimple, R.C.; Ci, S.; Lu, C.; Hu, L.; et al. β2-Microglobulin Maintains Glioblastoma Stem Cells and Induces M2-like Polarization of Tumor-Associated Macrophages. Cancer Res. 2022, 82, 3321–3334. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.S.; Rancan, C.; et al. Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e13. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.J.; Pereira, E.R.; Andersson, P.; Amoozgar, Z.; Van Wijnbergen, J.W.; O’Melia, M.J.; Zhou, H.; Chatterjee, S.; Ho, W.W.; Posada, J.M.; et al. Cancer cell plasticity and MHC-II-mediated immune tolerance promote breast cancer metastasis to lymph nodes. J. Exp. Med. 2023, 220, e20221847. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Mu, Q.; Bao, Z.; Chen, Y.; Liu, Y.; Chen, J.; Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678.e18. [Google Scholar] [CrossRef]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef]
- Lu, Q.; Kou, D.; Lou, S.; Ashrafizadeh, M.; Aref, A.R.; Canadas, I.; Tian, Y.; Niu, X.; Wang, Y.; Torabian, P.; et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J. Hematol. Oncol. 2024, 17, 16. [Google Scholar] [CrossRef]
- Yang, K.; Halima, A.; Chan, T.A. Antigen presentation in cancer-mechanisms and clinical implications for immunotherapy. Nat. Rev. Clin. Oncol. 2023, 20, 604–623. [Google Scholar] [CrossRef]
- Martinez-Usatorre, A.; De Palma, M. A LIGHTning Strike to the Metastatic Niche. Cell Rep. 2020, 30, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Skeate, J.G.; Otsmaa, M.E.; Prins, R.; Fernandez, D.J.; Da Silva, D.M.; Kast, W.M. TNFSF14: LIGHTing the Way for Effective Cancer Immunotherapy. Front. Immunol. 2020, 11, 922. [Google Scholar] [CrossRef]
- Yang, Y.; Lv, W.; Xu, S.; Shi, F.; Shan, A.; Wang, J. Molecular and Clinical Characterization of LIGHT/TNFSF14 Expression at Transcriptional Level via 998 Samples with Brain Glioma. Front. Mol. Biosci. 2021, 8, 567327. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Li, M.; Liu, J.; Yang, Y.; Li, G. Identification of immunologic subtype and prognosis of GBM based on TNFSF14 and immune checkpoint gene expression profiling. Aging 2020, 12, 7112–7128. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Z.; Zhang, J.H.; Li, J.B.; Yuan, F.; Tong, L.Q.; Wang, X.Y.; Chen, L.L.; Fan, X.G.; Zhang, Y.M.; Ren, X.; et al. MXRA5 Is a Novel Immune-Related Biomarker That Predicts Poor Prognosis in Glioma. Dis. Markers 2021, 2021, 6680883. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Li, G.; Li, K.; Xu, Q.; Pan, Z.; Ding, F.; Vedell, P.; Liu, P.; Cui, P.; Hua, X.; et al. Exome sequencing identifies MXRA5 as a novel cancer gene frequently mutated in non-small cell lung carcinoma from Chinese patients. Carcinogenesis 2012, 33, 1797–1805. [Google Scholar] [CrossRef]
- Wang, G.H.; Yao, L.; Xu, H.W.; Tang, W.T.; Fu, J.H.; Hu, X.F.; Cui, L.; Xu, X.M. Identification of MXRA5 as a novel biomarker in colorectal cancer. Oncol. Lett. 2013, 5, 544–548. [Google Scholar] [CrossRef]
- Liu, L.; Pang, H.; He, Q.; Pan, B.; Sun, X.; Shan, J.; Wu, L.; Wu, K.; Yao, X.; Guo, Y. A novel strategy to identify candidate diagnostic and prognostic biomarkers for gastric cancer. Cancer Cell Int. 2021, 21, 335. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Smith, K.G.; Clatworthy, M.R. FcgammaRIIB in autoimmunity and infection: Evolutionary and therapeutic implications. Nat. Rev. Immunol. 2010, 10, 674. [Google Scholar] [CrossRef]
- Roghanian, A.; Cragg, M.S.; Frendéus, B. Resistance is futile: Targeting the inhibitory FcγRIIB (CD32B) to maximize immunotherapy. Oncoimmunology 2016, 5, e1069939. [Google Scholar] [CrossRef]
- Melero, I.; Hirschhorn-Cymerman, D.; Morales-Kastresana, A.; Sanmamed, M.F.; Wolchok, J.D. Agonist antibodies to TNFR molecules that costimulate T and NK cells. Clin. Cancer Res. 2013, 19, 1913. [Google Scholar] [CrossRef] [PubMed]
- Shuford, W.W.; Klussman, K.; Tritchler, D.D.; Loo, D.T.; Chalupny, J.; Siadak, A.W.; Brown, T.J.; Emswiler, J.; Raecho, H.; Larsen, C.P.; et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med. 1997, 186, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.C.; Carrié, N.; Cuisinier, M.; Ghazali, S.; Voisin, A.; Axisa, P.P.; Tosolini, M.; Mazzotti, C.; Golec, D.P.; Maheo, S.; et al. TCR-independent CD137 (4-1BB) signaling promotes CD8(+)-exhausted T cell proliferation and terminal differentiation. Immunity 2023, 56, 1631–1648.e10. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Guo, S.; Khan, F.; Dunterman, M.; Ali, H.; Liu, Y.; Huang, Y.; Chen, P. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep. Med. 2023, 4, 101238. [Google Scholar] [CrossRef]
- Montoya, M.; Collins, S.A.; Chuntova, P.; Patel, T.S.; Nejo, T.; Yamamichi, A.; Kasahara, N.; Okada, H. IRF8-driven reprogramming of the immune microenvironment enhances anti-tumor adaptive immunity and reduces immunosuppression in murine glioblastoma. bioRxiv 2024. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Z.; Chai, Z.; Zhan, Y.; Zhang, Y.; Liu, Z.; Liu, Y.; Li, Z.; Lin, M.; Zhang, Z.; et al. Targeting MS4A4A on tumour-associated macrophages restores CD8+ T-cell-mediated antitumour immunity. Gut 2023, 72, 2307–2320. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, P.; Zhu, G.; Wang, B.; Cai, J.; Song, L.; Wan, J.; Yang, Y.; Du, J.; Cai, Y.; et al. Targeting GSDME-mediated macrophage polarization for enhanced antitumor immunity in hepatocellular carcinoma. Cell. Mol. Immunol. 2024, 21, 1505–1521. [Google Scholar] [CrossRef]
- Gressier, E.; Schulte-Schrepping, J.; Petrov, L.; Brumhard, S.; Stubbemann, P.; Hiller, A.; Obermayer, B.; Spitzer, J.; Kostevc, T.; Whitney, P.G.; et al. CD4(+) T cell calibration of antigen-presenting cells optimizes antiviral CD8(+) T cell immunity. Nat. Immunol. 2023, 24, 979–990. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Schmalen, A.; Haines, S.; Wolff, M.; Ma, H.; Zhang, H.; Stoleriu, M.G.; Nowak, J.; Nakayama, M.; et al. Antiviral CD8(+) T-cell immune responses are impaired by cigarette smoke and in COPD. Eur. Respir. J. 2023, 62, 2201374. [Google Scholar] [CrossRef]
- Kruse, B.; Buzzai, A.C.; Shridhar, N.; Braun, A.D.; Gellert, S.; Knauth, K.; Pozniak, J.; Peters, J.; Dittmann, P.; Mengoni, M.; et al. CD4(+) T cell-induced inflammatory cell death controls immune-evasive tumours. Nature 2023, 618, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Liby, K.T. The BRD4 Inhibitor I-BET-762 Reduces HO-1 Expression in Macrophages and the Pancreas of Mice. Int. J. Mol. Sci. 2024, 25, 9985. [Google Scholar] [CrossRef]
- Leal, A.S.; Williams, C.R.; Royce, D.B.; Pioli, P.A.; Sporn, M.B.; Liby, K.T. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017, 394, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Funck-Brentano, E.; Vizlin-Hodzic, D.; Nilsson, J.A.; Nilsson, L.M. BET bromodomain inhibitor HMBA synergizes with MEK inhibition in treatment of malignant glioma. Epigenetics 2021, 16, 54–63. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Xu, Y.; Zhou, Y.; Fan, J.; Cheng, C.; Meng, R.; Gamazon, E.R.; Zhou, D. Integrating population-level and cell-based signatures for drug repositioning. bioRxiv 2023. [Google Scholar] [CrossRef]
- Odia, Y.; Iwamoto, F.M.; Moustakas, A.; Fraum, T.J.; Salgado, C.A.; Li, A.; Kreisl, T.N.; Sul, J.; Butman, J.A.; Fine, H.A. A phase II trial of enzastaurin (LY317615) in combination with bevacizumab in adults with recurrent malignant gliomas. J. Neurooncol. 2016, 127, 127–135. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, C.; Xun, M.; Yu, F.; Li, H.; Liu, Y.; Zhang, W.; Yan, J. An MHC-Related Gene’s Signature Predicts Prognosis and Immune Microenvironment Infiltration in Glioblastoma. Int. J. Mol. Sci. 2025, 26, 4609. https://doi.org/10.3390/ijms26104609
Yu C, Xun M, Yu F, Li H, Liu Y, Zhang W, Yan J. An MHC-Related Gene’s Signature Predicts Prognosis and Immune Microenvironment Infiltration in Glioblastoma. International Journal of Molecular Sciences. 2025; 26(10):4609. https://doi.org/10.3390/ijms26104609
Chicago/Turabian StyleYu, Caiyuan, Mingjuan Xun, Fei Yu, Hengyu Li, Ying Liu, Wei Zhang, and Jun Yan. 2025. "An MHC-Related Gene’s Signature Predicts Prognosis and Immune Microenvironment Infiltration in Glioblastoma" International Journal of Molecular Sciences 26, no. 10: 4609. https://doi.org/10.3390/ijms26104609
APA StyleYu, C., Xun, M., Yu, F., Li, H., Liu, Y., Zhang, W., & Yan, J. (2025). An MHC-Related Gene’s Signature Predicts Prognosis and Immune Microenvironment Infiltration in Glioblastoma. International Journal of Molecular Sciences, 26(10), 4609. https://doi.org/10.3390/ijms26104609