
Phytochemical-Induced Metabolic Modulation: Dihydroartemisinin Regulates Cellular Metabolism in Madin-Darby Bovine Kidney Cells
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
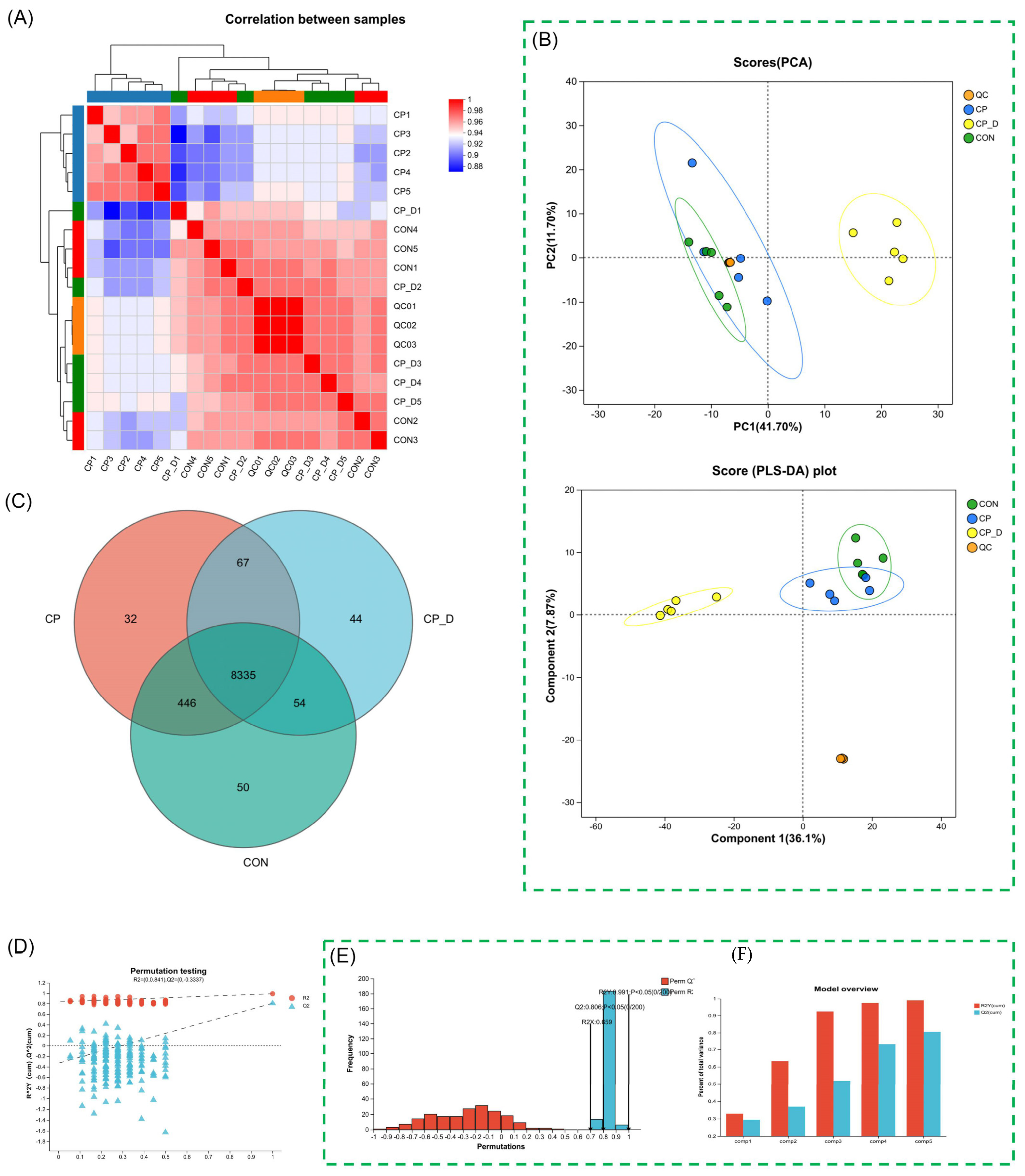
2.1. Metabolomic Analysis Reveals Significant Alterations in Metabolic Profiles Following Dihydroartemisinin (DHA) Treatment
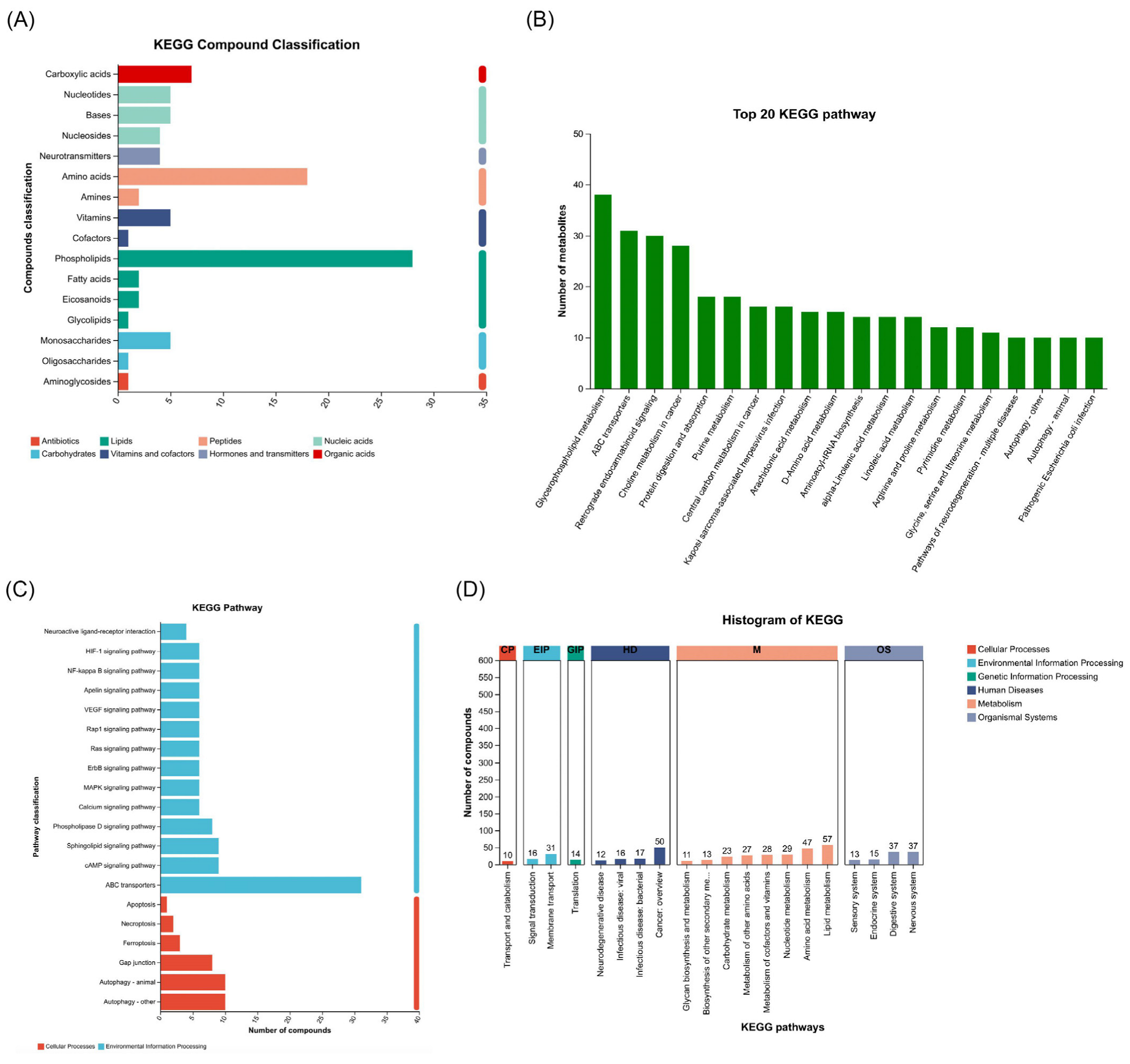
2.2. Comprehensive Metabolic Reprogramming Highlights Lipid Dysregulation, Amino Acid Alterations, and Pathway Adaptations
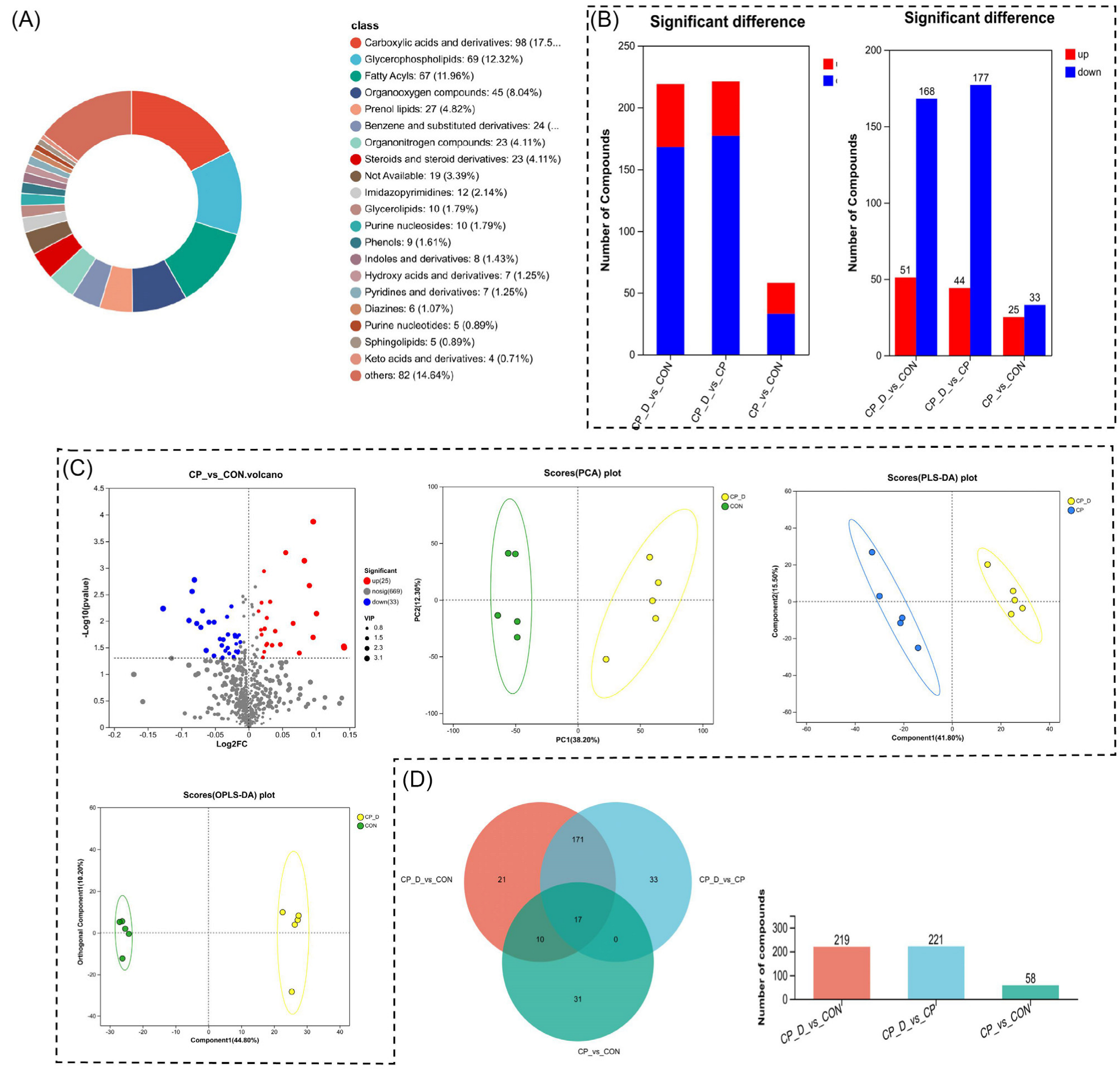
2.3. Metabolomic Profiling Reveals Perturbations in Carboxylic Acids, Glycerophospholipids, and Key Differential Metabolic Pathways
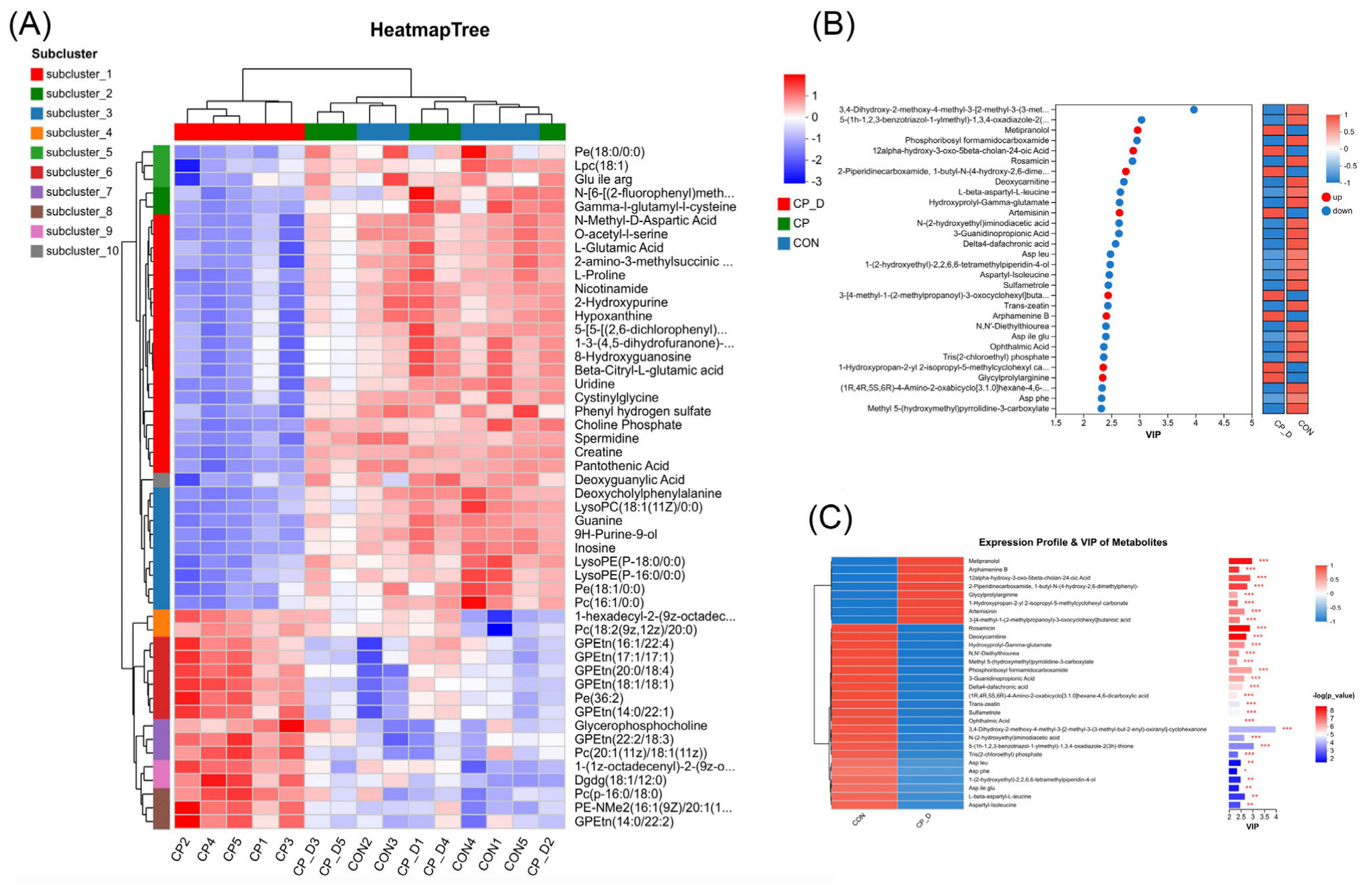
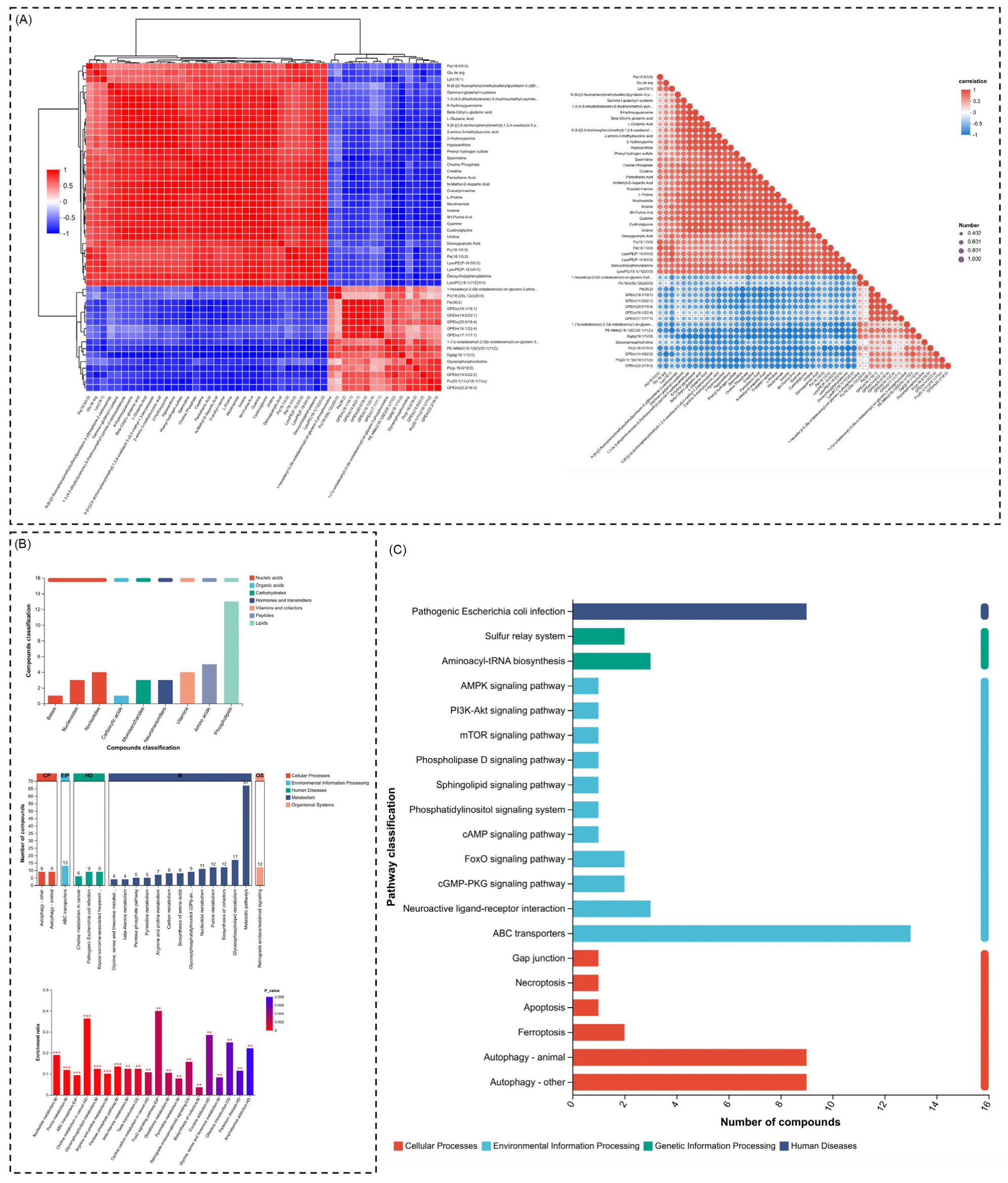
2.4. Correlation Highlights Co-Regulated Pathways, Dominance of Cellular Processes, and Enriched Apoptosis-Related Pathways
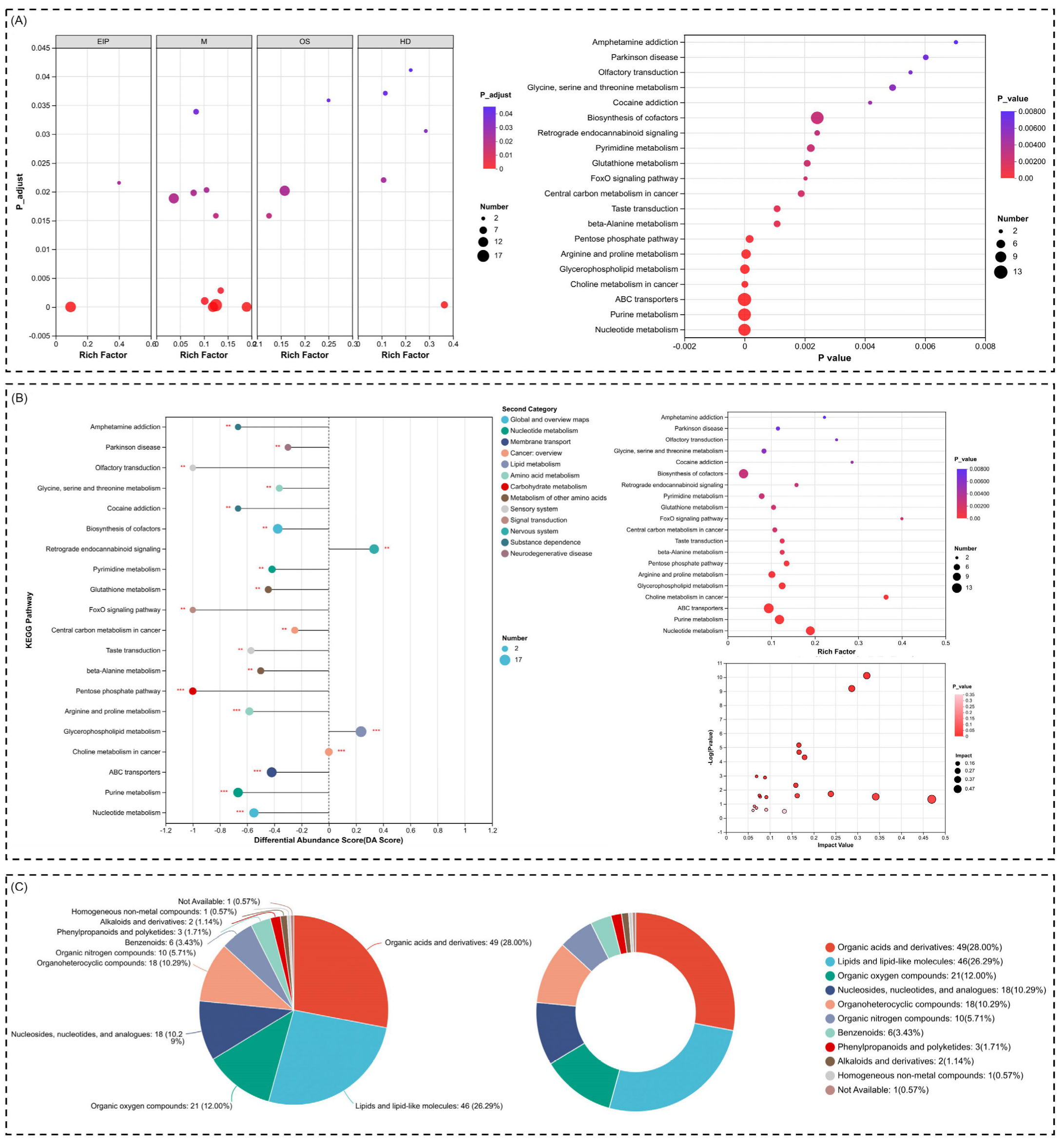
2.5. Metabolic Reprogramming and Oxidative Stress Regulation Highlighted Through Pathway Enrichment and Compound Analysis
3. Discussion
4. Materials and Methods
4.1. Sample Source and Preparation
4.2. Metabolite Extraction
4.3. LC-MS Analysis
4.4. Data Preprocessing
4.5. Statistical Analysis
4.6. Pathway Analysis
4.7. Visualization and Bioinformatics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ali, S.; Ni, X.; Khan, M.; Zhao, X.; Yang, H.; Danzeng, B.; Raja, I.H.; Quan, G. Effects of Dietary Protein Levels on Sheep Gut Metabolite Profiles during the Lactating Stage. Animals 2024, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Breton, J.; Panic, G.; Cogan, T.A.; Bailey, M.; Swann, J.R.; Lee, M.R.F. The Mycotoxin deoxynivalenol significantly alters the function and metabolism of bovine kidney epithelial cells in vitro. Toxins 2019, 11, 554. [Google Scholar] [CrossRef]
- Burk, O.; Arnold, K.A.; Nussler, A.K.; Schaeffeler, E.; Efimova, E.; Avery, B.A.; Avery, M.A.; Fromm, M.F.; Eichelbaum, M. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol. Pharmacol. 2005, 67, 1954–1965. [Google Scholar] [CrossRef]
- Chairat, K.; Jittamala, P.; Hanboonkunupakarn, B.; Pukrittayakamee, S.; Hanpithakpong, W.; Blessborn, D.; White, N.J.; Day, N.P.J.; Tarning, J. Enantiospecific pharmacokinetics and drug–drug interactions of primaquine and blood-stage antimalarial drugs. J. Antimicrob. Chemother. 2018, 73, 3102–3113. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Contreras, G.A.; Sordillo, L.M. Lipid mobilization and inflammatory responses during the transition period of dairy cows. Comp. Immunol. Microbiol. Infect. Dis. 2011, 34, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, Y.; Liang, J.; Li, Q.; Hu, H.; Zhou, Y.; Zhang, B. Dihydroartemisinin Potentiates VEGFR-TKIs Antitumorigenic Effect on Osteosarcoma by Regulating Loxl2/VEGFA Expression and Lipid Metabolism Pathway. J. Cancer 2023, 14, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Dumas, S.J.; Meta, E.; Borri, M.; Luo, Y.; Li, X.; Rabelink, T.J.; Carmeliet, P. Phenotypic diversity and metabolic specialization of renal endothelial cells. Nat. Rev. Nephrol. 2021, 17, 441–464. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Schroder, A.; Reyes-Reyes, E.M.; Franco, R. mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Curr. Opin. Toxicol. 2018, 8, 102–110. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Godde, C.M.; Mason-D’Croz, D.; Mayberry, D.E.; Thornton, P.K.; Herrero, M. Impacts of climate change on the livestock food supply chain; a review of the evidence. Glob. Food Secur. 2021, 28, 100488. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Virgilio, L.; Silva-Lucero, M.D.C.; Flores-Morelos, D.S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.M.; Zacapala-Gómez, A.E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef]
- Hanboonkunupakarn, B.; Ashley, E.A.; Jittamala, P.; Tarning, J.; Pukrittayakamee, S.; Hanpithakpong, W.; Chotsiri, P.; Wattanakul, T.; Panapipat, S.; Lee, S.J.; et al. Open-label crossover study of primaquine and dihydroartemisinin- Piperaquine pharmacokinetics in healthy adult thai subjects. Antimicrob. Agents Chemother. 2014, 58, 7340–7346. [Google Scholar] [CrossRef]
- Hanboonkunupakarn, B.; Tarning, J.; Pukrittayakamee, S.; Chotivanich, K. Artemisinin resistance and malaria elimination: Where are we now? Front. Pharmacol. 2022, 13, 876282. [Google Scholar] [CrossRef]
- Hanboonkunupakarn, B.; White, N.J. Advances and roadblocks in the treatment of malaria. Br. J. Clin. Pharmacol. 2022, 88, 374–382. [Google Scholar] [CrossRef]
- Hristov, A.N.; Bannink, A.; Crompton, L.A.; Huhtanen, P.; Kreuzer, M.; McGee, M.; Nozière, P.; Reynolds, C.K.; Bayat, A.R.; Yáñez-Ruiz, D.R.; et al. Invited review: Nitrogen in ruminant nutrition: A review of measurement techniques. J. Dairy Sci. 2019, 102, 5811–5852. [Google Scholar] [CrossRef]
- Josi, C.; Bürki, S.; Stojiljkovic, A.; Wellnitz, O.; Stoffel, M.H.; Pilo, P. Bovine epithelial in vitro infection models for Mycoplasma bovis. Front. Cell. Infect. Microbiol. 2018, 8, 329. [Google Scholar] [CrossRef] [PubMed]
- Koistinen, V.M.; Haldar, S.; Tuomainen, M.; Lehtonen, M.; Klåvus, A.; Draper, J.; Lloyd, A.; Beckmann, M.; Bal, W.; Ross, A.B.; et al. Metabolic changes in response to varying whole-grain wheat and rye intake. Npj Sci. Food 2024, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Hristov, A.N.; Heyler, K.S.; Cassidy, T.W.; Lapierre, H.; Varga, G.A.; Parys, C. Effects of metabolizable protein supply and amino acid supplementation on nitrogen utilization, milk production, and ammonia emissions from manure in dairy cows. J. Dairy Sci. 2012, 95, 5253–5268. [Google Scholar] [CrossRef]
- Li, G.; Yu, X.; Portela Fontoura, A.B.; Javaid, A.; de la Maza-Escolà, V.S.; Salandy, N.S.; Fubini, S.L.; Grilli, E.; McFadden, J.W.; Duan, J.E. Transcriptomic regulations of heat stress response in the liver of lactating dairy cows. BMC Genom. 2023, 24, 410. [Google Scholar] [CrossRef]
- Pearlin, B.V.; Muthuvel, S.; Govidasamy, P.; Villavan, M.; Alagawany, M.; Ragab Farag, M.; Dhama, K.; Gopi, M. Role of acidifiers in livestock nutrition and health: A review. J. Anim. Physiol. Anim. Nutr. 2020, 104, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Piedade, R.; Traub, S.; Bitter, A.; Nüssler, A.K.; Gil, J.P.; Schwab, M.; Burk, O. Carboxymefloquine, the major metabolite of the antimalarial drug mefloquine, induces drug-metabolizing enzyme and transporter expression by activation of pregnane X receptor. Antimicrob. Agents Chemother. 2015, 59, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of pi3k-akt-mtor pathway as a pro-survival signaling and resistance-mediating mechanism to therapy of prostate cancer. Int. J. Mol. Sci. 2021, 22, 1088. [Google Scholar] [CrossRef]
- Romao, J.M.; Jin, W.; He, M.; McAllister, T.; Guan, L.L. Elucidation of molecular mechanisms of physiological variations between bovine subcutaneous and visceral fat depots under different nutritional regimes. PLoS ONE 2013, 8, e83211. [Google Scholar] [CrossRef]
- Ruan, Z.; Li, J.; Liu, F.; Cao, J.; Chen, S.; Chen, J.; Huang, K.; Wang, Y.; Li, H.; Wang, Y.; et al. Study design, general characteristics of participants, and preliminary findings from the metabolome, microbiome, and dietary salt intervention study (MetaSalt). Chronic Dis. Transl. Med. 2021, 7, 227–234. [Google Scholar] [CrossRef]
- Sangalli, J.R.; Nociti, R.P.; del Collado, M.; Sampaio, R.V.; da Silveira, J.C.; Perecin, F.; Smith, L.C.; Ross, P.J.; Meirelles, F.V. Characterization of histone lysine β-hydroxybutyrylation in bovine tissues, cells, and cumulus–oocyte complexes. Mol. Reprod. Dev. 2022, 89, 375–398. [Google Scholar] [CrossRef]
- Shastak, Y.; Pelletier, W. From Metabolism to Vitality: Uncovering Riboflavin’s Importance in Poultry Nutrition. Animals 2023, 13, 3554. [Google Scholar] [CrossRef]
- Shi, S.; Luo, H.; Ji, Y.; Ouyang, H.; Wang, Z.; Wang, X.; Hu, R.; Wang, L.; Wang, Y.; Xia, J.; et al. Repurposing dihydroartemisinin to combat oral squamous cell carcinoma, associated with mitochondrial dysfunction and oxidative stress. Med. Cell. Longev. 2023, 2023, 9595201. [Google Scholar] [CrossRef] [PubMed]
- Skibiel, A.L. Hepatic mitochondrial bioenergetics and metabolism across lactation and in response to heat stress in dairy cows. JDS Commun. 2024, 5, 247–252. [Google Scholar] [CrossRef]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef]
- Te Pas, M.F.W.; Veldkamp, T.; de Haas, Y.; Bannink, A.; Ellen, E.D. Adaptation of livestock to new diets using feed components without competition with human edible protein sources—A review of the possibilities and recommendations. Animals 2021, 11, 2293. [Google Scholar] [CrossRef] [PubMed]
- Teja-Isavadharm, P.; Peggins, J.O.; Brewer, T.G.; White, N.J.; Webster, H.K.; Kyle, D.E. Plasmodium falciparum-Based Bioassay for Measurement of Artemisinin Derivatives in Plasma or Serum. Antimicrob. Agents Chemother. 2004, 48, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Ulfig, A.; Jakob, U. Cellular oxidants and the proteostasis network: Balance between activation and destruction. Trends Biochem. Sci. 2024, 49, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, H.Y.; Wang, Y.M.; Yang, Z.Q.; Liu, J.X.; Wu, Y.M.; Yan, T.; Ye, H.W. Effects of dietary supplementation of methionine and lysine on milk production and nitrogen utilization in dairy cows. J. Dairy Sci. 2010, 93, 3661–3670. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Shi, Z.; Shen, L.; Zhang, J.; Zhang, J. Bovine milk exosomes attenuate the alteration of purine metabolism and energy status in IEC-6 cells induced by hydrogen peroxide. Food Chem. 2021, 350, 129142. [Google Scholar] [CrossRef]
- Xu, D.L.; Fan, K.; Zhang, H.; Tang, L.X.; Wang, Y.; Xiang, Z.; Shi, A.M.; Qu, Y.C.; Su, C.J.; Pan, J. Anti-proliferation and apoptosis-inducing effects of dihydroartemisinin on SH-SY5Y cells and metabolomic analysis. Transl. Pediatrics 2022, 11, 1346–1361. [Google Scholar] [CrossRef]
- Xu, T.; Liu, R.; Zhu, H.; Zhou, Y.; Pei, T.; Yang, Z. The Inhibition of LPS-Induced Oxidative Stress and Inflammatory Responses Is Associated with the Protective Effect of (-)-Epigallocatechin-3-Gallate on Bovine Hepatocytes and Murine Liver. Antioxidants 2022, 11, 914. [Google Scholar] [CrossRef]
- Zhang, B.; Halouska, S.; Schiaffo, C.E.; Sadykov, M.R.; Somerville, G.A.; Powers, R. NMR analysis of a stress response metabolic signaling network. J. Proteome Res. 2011, 10, 3743–3754. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Sun, J.; Zhang, W.; Guo, Z.; Ma, Q. Arachidonic acid metabolism in health and disease. MedComm 2023, 4, e363. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Lei, K.; Zhang, M.; Yin, J.; Hu, C. Phytochemical-Induced Metabolic Modulation: Dihydroartemisinin Regulates Cellular Metabolism in Madin-Darby Bovine Kidney Cells. Int. J. Mol. Sci. 2025, 26, 4531. https://doi.org/10.3390/ijms26104531
Gao J, Lei K, Zhang M, Yin J, Hu C. Phytochemical-Induced Metabolic Modulation: Dihydroartemisinin Regulates Cellular Metabolism in Madin-Darby Bovine Kidney Cells. International Journal of Molecular Sciences. 2025; 26(10):4531. https://doi.org/10.3390/ijms26104531
Chicago/Turabian StyleGao, Jindong, Kuang Lei, Mengdi Zhang, Jinhua Yin, and Changmin Hu. 2025. "Phytochemical-Induced Metabolic Modulation: Dihydroartemisinin Regulates Cellular Metabolism in Madin-Darby Bovine Kidney Cells" International Journal of Molecular Sciences 26, no. 10: 4531. https://doi.org/10.3390/ijms26104531
APA StyleGao, J., Lei, K., Zhang, M., Yin, J., & Hu, C. (2025). Phytochemical-Induced Metabolic Modulation: Dihydroartemisinin Regulates Cellular Metabolism in Madin-Darby Bovine Kidney Cells. International Journal of Molecular Sciences, 26(10), 4531. https://doi.org/10.3390/ijms26104531