
A Novel KGD-Based αIIbβ3 Antagonist Prevents Arterial Thrombosis While Preserving Hemostasis and Avoiding Thrombocytopenia

 ,
,  ,
, 
 ,
, 
Abstract
1. Introduction
2. Results
2.1. Structure-Activity Relationships in Optimizing TMV-7 Derivatives for Enhanced Antiplatelet Efficacy with Reduced Bleeding Risk
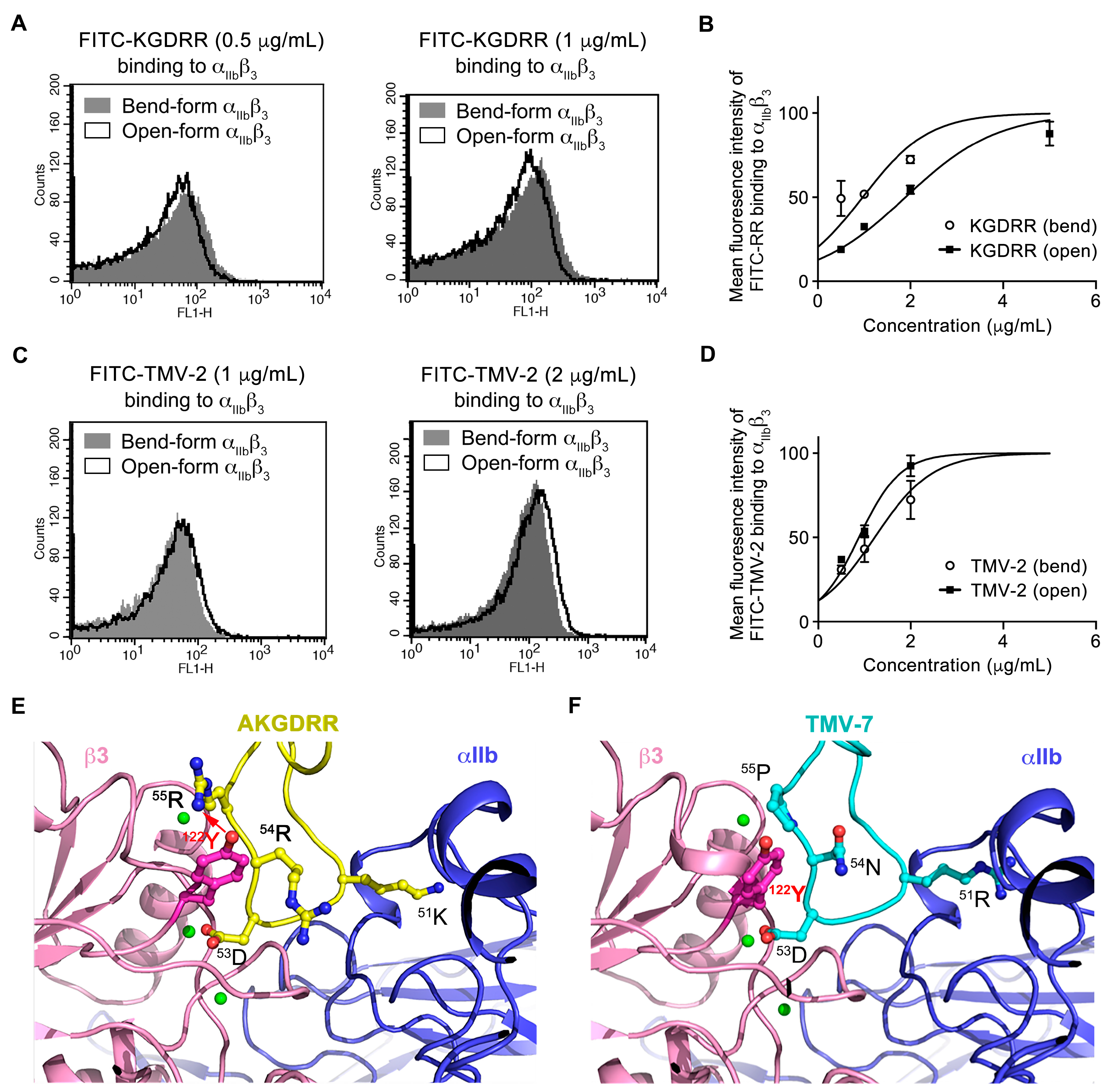
2.2. Distinct Binding Characteristics of Pure and Partial Antagonists to Integrin αIIbβ3
2.3. Structural Basis of KGDRR’s Enhanced Safety Profile and Unique Binding Mechanism to Integrin αIIbβ3
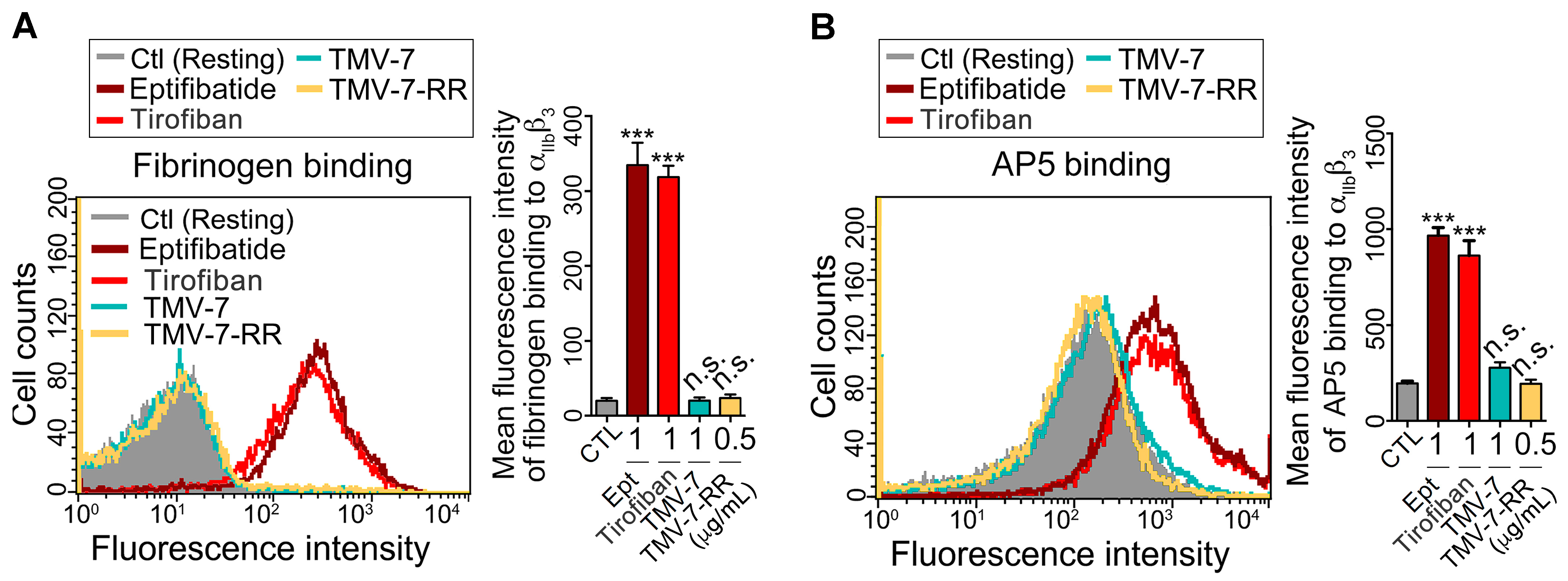
2.4. Pure Antagonists Do Not Induce Conformational Changes or Exposure of the Ligand-Induced Binding Site (LIBS) in Integrin αIIbβ3
2.5. Molecular Mechanisms of Pure Antagonists in Human Platelets
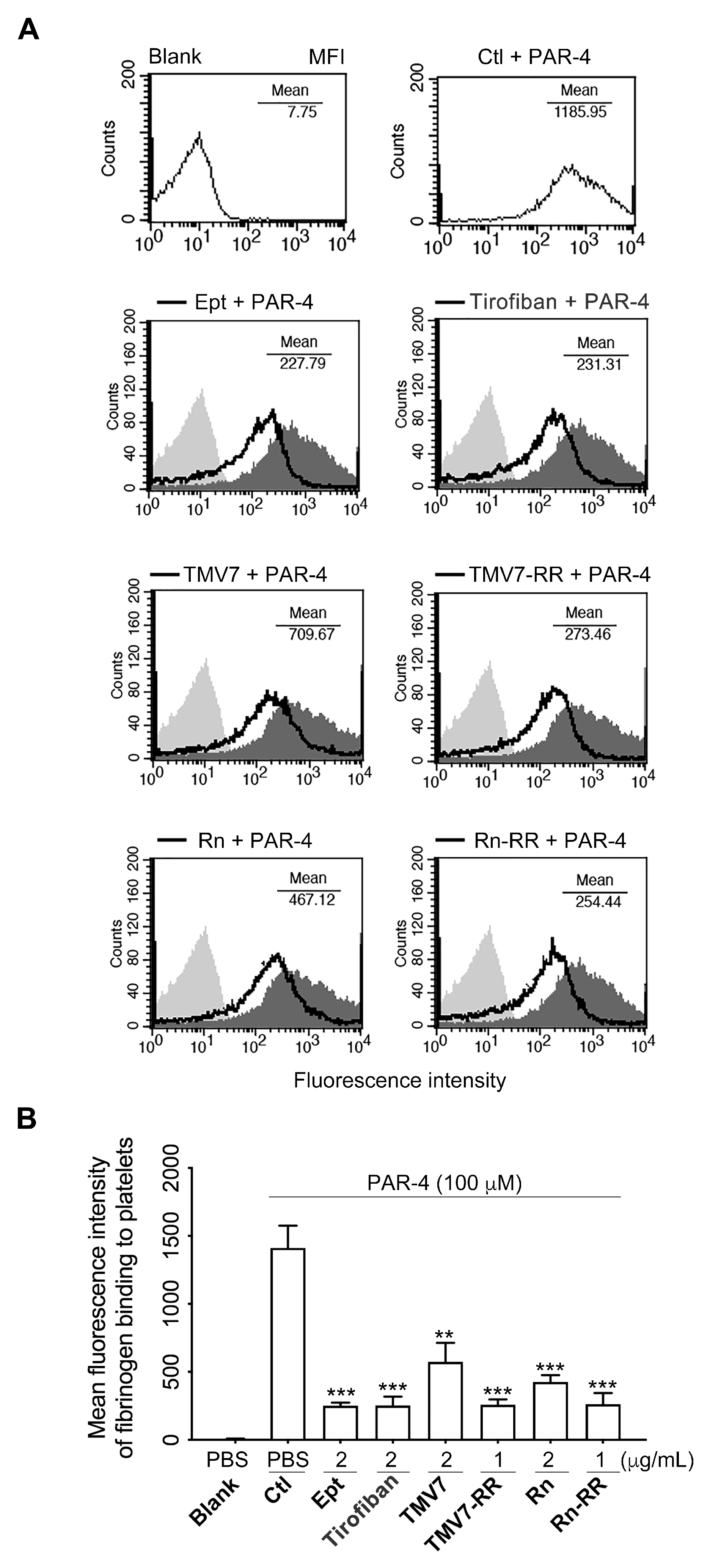
2.6. Both Partial Agonists and Pure Antagonists Inhibit Agonist-Induced Integrin αIIbβ3 Ligation and Platelet Aggregation
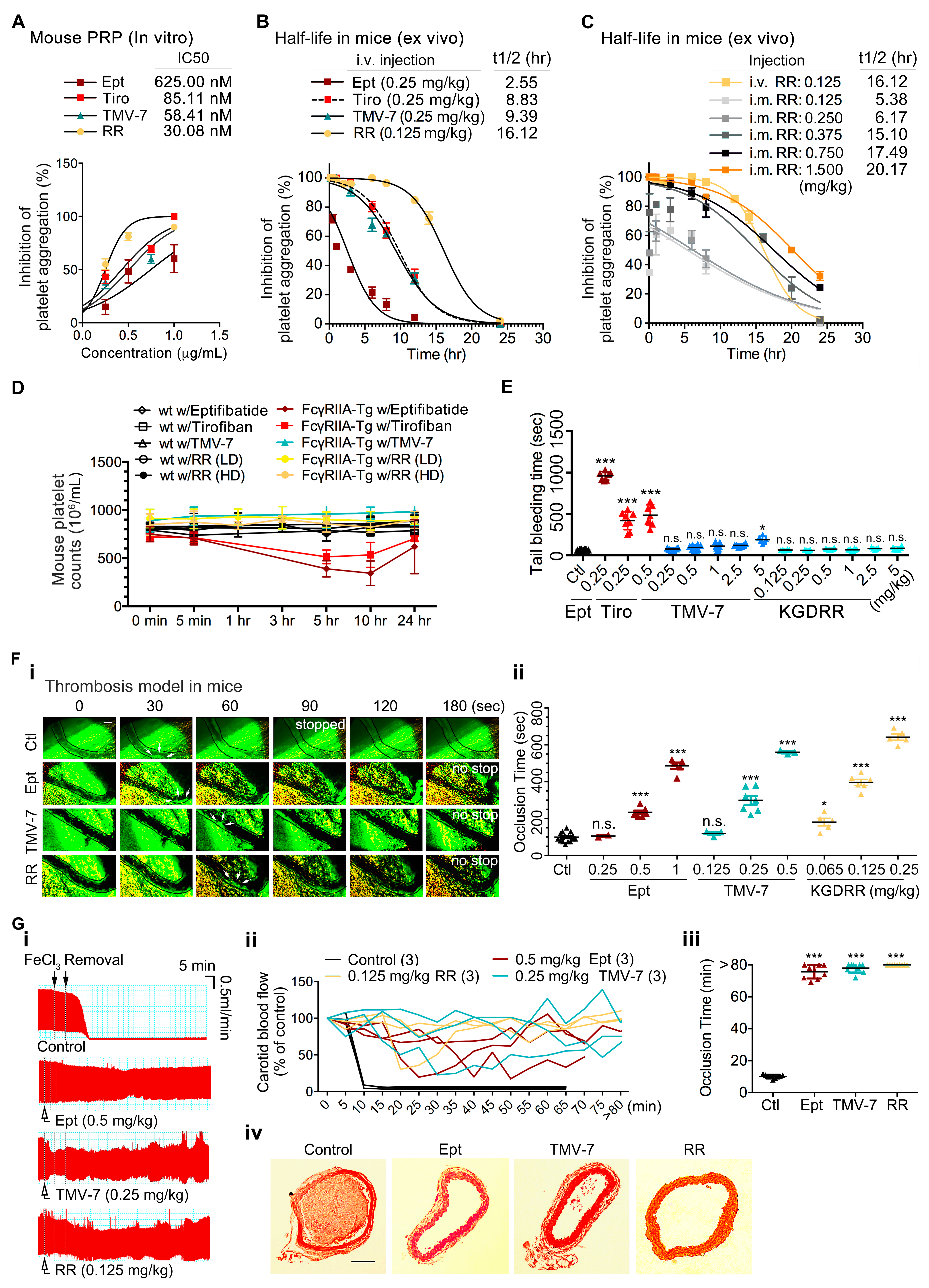
2.7. The Pure Antagonist KGDRR Exhibits a Prolonged Half-Life, Favorable Safety Profile, and Potent Antithrombotic Activity in Murine Models
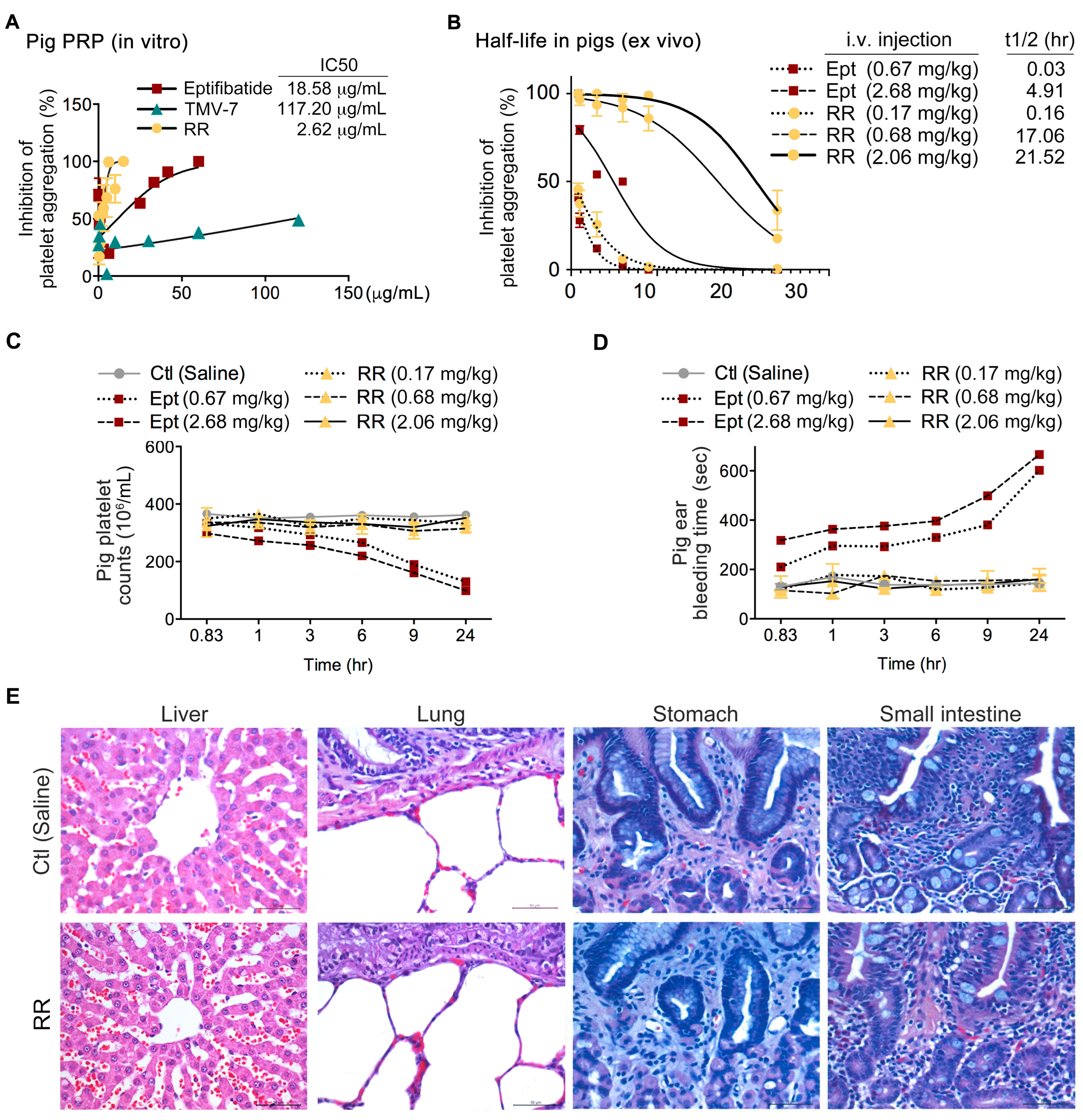
2.8. The Pure Antagonist KGDRR Exhibits Potent Antiplatelet Activity, a Prolonged Half-Life, and a Favorable Safety Profile in Swine Models
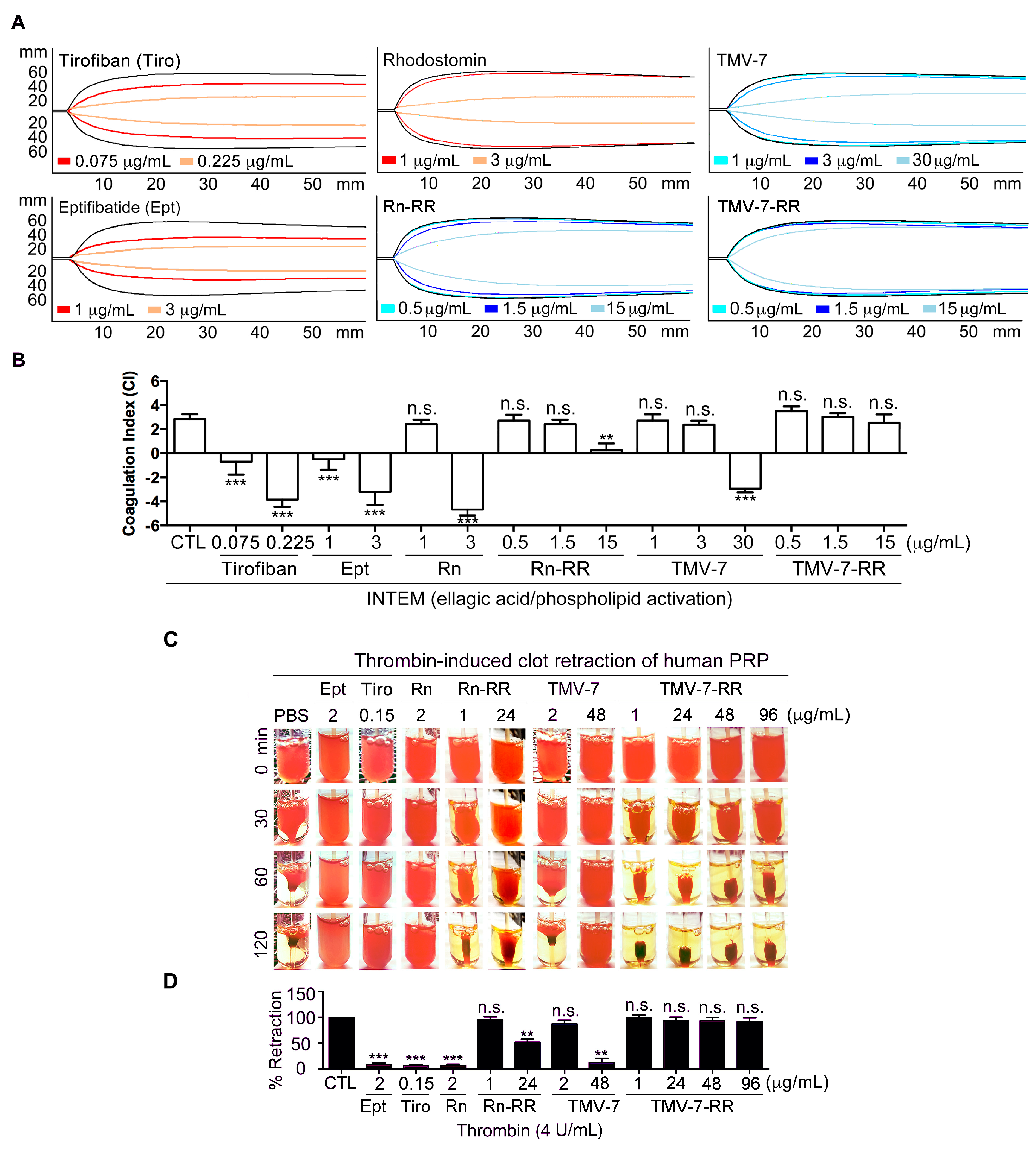
2.9. Pure Antagonists Preserve Physiological Hemostasis in Human Whole Blood
2.10. Thrombin-Induced Clot Retraction Confirms Superior Safety Profile of Pure Antagonists
3. Discussion
4. Materials and Methods
4.1. The Expression of TMV-7 and Its Mutants in P. pastoris and Purification
4.2. Definition of RR Mutant
4.3. Safety Index Calculation
4.4. Priming Assay
4.5. Induction of Ligand-Induced Binding Site (LIBS)
4.6. Rotational Thromboelastometry (ROTEM)
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CI | coagulation index |
| LIBS | ligand-induced binding site |
| mAb | monoclonal antibody |
| PCI | percutaneous coronary interventions |
| PRP | platelet-rich plasma |
| PS | platelet suspension |
| RGD | Arg-Gly-Asp |
| ROTEM | rotational-thromboelastometry |
| SAR | Structure–activity relationship |
| TMV | Trimeresurus mucrosquamatus venom |
References
- Bledzka, K.; Smyth, S.S.; Plow, E.F. Integrin αIIbβ3: From Discovery to Efficacious Therapeutic Target. Circ. Res. 2013, 112, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Swieringa, F.; Kuijpers, M.J.; Heemskerk, J.W.; van der Meijden, P.E. Targeting platelet receptor function in thrombus formation: The risk of bleeding. Blood Rev. 2014, 28, 9–21. [Google Scholar] [CrossRef]
- Calvete, J.J. The continuing saga of snake venom disintegrins. Toxicon 2013, 62, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.F.; Holt, J.C.; Lukasiewicz, H.; Niewiarowski, S. Trigramin. A low molecular weight peptide inhibiting fibrinogen interaction with platelet receptors expressed on glycoprotein IIb-IIIa complex. J. Biol. Chem. 1987, 262, 16157–16163. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.J.; Chen, Y.R.; Hsu, C.C.; Peng, H.C.; Huang, T.F. An αIIbβ3 antagonist prevents thrombosis without causing Fc receptor gamma-chain IIa-mediated thrombocytopenia. J. Thromb. Haemost. JTH 2017, 15, 2230–2244. [Google Scholar] [CrossRef]
- Shen, B.; Zhao, X.; O’Brien, K.A.; Stojanovic-Terpo, A.; Delaney, M.K.; Kim, K.; Cho, J.; Lam, S.C.; Du, X. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature 2013, 503, 131–135. [Google Scholar] [CrossRef]
- Hsu, C.C.; Chuang, W.J.; Chung, C.H.; Chang, C.H.; Peng, H.C.; Huang, T.F. Improved antithrombotic activity and diminished bleeding side effect of a PEGylated αIIbβ3 antagonist, disintegrin. Thromb. Res. 2016, 143, 3–10. [Google Scholar] [CrossRef]
- Gao, C.; Boylan, B.; Bougie, D.; Gill, J.C.; Birenbaum, J.; Newman, D.K.; Aster, R.H.; Newman, P.J. Eptifibatide-induced thrombocytopenia and thrombosis in humans require FcgammaRIIa and the integrin β3 cytoplasmic domain. J. Clin. Investig. 2009, 119, 504–511. [Google Scholar] [CrossRef]
- Arnaout, M.A. Integrins: A Bedside to Bench to Bedside Story. Trans. Am. Clin. Climatol. Assoc. 2023, 133, 34–55. [Google Scholar]
- Mao, L.; Wang, L.; Xu, J.; Zou, J. The role of integrin family in bone metabolism and tumor bone metastasis. Cell Death Discov. 2023, 9, 119. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, J.; Springer, T.A. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013, 201, 1053–1068. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, O.M.; Hughes, C.E.; Montague, S.; Watson, S.K.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Bougie, D.W.; Rasmussen, M.; Zhu, J.; Aster, R.H. Antibodies causing thrombocytopenia in patients treated with RGD-mimetic platelet inhibitors recognize ligand-specific conformers of αIIb/β3 integrin. Blood 2012, 119, 6317–6325. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Shen, B.; Du, X. Targeting integrin and integrin signaling in treating thrombosis. Arter. Thromb. Vasc. Biol. 2015, 35, 24–29. [Google Scholar] [CrossRef]
- Negri, A.; Li, J.; Naini, S.; Coller, B.S.; Filizola, M. Structure-based virtual screening of small-molecule antagonists of platelet integrin αIIbβ3 that do not prime the receptor to bind ligand. J. Comput.-Aided Mol. Des. 2012, 26, 1005–1015. [Google Scholar] [CrossRef]
- Nieswandt, B.; Moser, M.; Pleines, I.; Varga-Szabo, D.; Monkley, S.; Critchley, D.; Fassler, R. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J. Exp. Med. 2007, 204, 3113–3118. [Google Scholar] [CrossRef]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; de Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin β tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef]
- Haling, J.R.; Monkley, S.J.; Critchley, D.R.; Petrich, B.G. Talin-dependent integrin activation is required for fibrin clot retraction by platelets. Blood 2011, 117, 1719–1722. [Google Scholar] [CrossRef]
- Haas, T.; Spielmann, N.; Mauch, J.; Madjdpour, C.; Speer, O.; Schmugge, M.; Weiss, M. Comparison of thromboelastometry (ROTEM®) with standard plasmatic coagulation testing in paediatric surgery. Br. J. Anaesth. 2012, 108, 36–41. [Google Scholar] [CrossRef]
- Lance, M.D. A general review of major global coagulation assays: Thrombelastography, thrombin generation test and clot waveform analysis. Thromb. J. 2015, 13, 1. [Google Scholar] [CrossRef]
- Van Agthoven, J.F.; Xiong, J.P.; Alonso, J.L.; Rui, X.; Adair, B.D.; Goodman, S.L.; Arnaout, M.A. Structural basis for pure antagonism of integrin αVβ3 by a high-affinity form of fibronectin. Nat. Struct. Mol. Biol. 2014, 21, 383–388. [Google Scholar] [CrossRef]
- Xiao, T.; Takagi, J.; Coller, B.S.; Wang, J.H.; Springer, T.A. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 2004, 432, 59–67. [Google Scholar] [CrossRef]
- Adair, B.D.; Alonso, J.L.; van Agthoven, J.; Hayes, V.; Ahn, H.S.; Yu, I.S.; Lin, S.W.; Xiong, J.P.; Poncz, M.; Arnaout, M.A. Structure-guided design of pure orthosteric inhibitors of αIIbβ3 that prevent thrombosis but preserve hemostasis. Nat. Commun. 2020, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.T.; Chou, L.J.; Chen, Y.C.; Chen, C.Y.; Pari, K.; Jen, C.J.; Lo, S.J.; Huang, S.L.; Lee, C.Y.; Chang, T.W.; et al. Expression in Pichia pastoris and characterization by circular dichroism and NMR of rhodostomin. Proteins 2001, 43, 499–508. [Google Scholar] [CrossRef]
- Shiu, J.H.; Chen, C.Y.; Chen, Y.C.; Chang, Y.T.; Chang, Y.S.; Huang, C.H.; Chuang, W.J. Effect of P to A mutation of the N-terminal residue adjacent to the Rgd motif on rhodostomin: Importance of dynamics in integrin recognition. PLoS ONE 2012, 7, e28833. [Google Scholar] [CrossRef] [PubMed]
- Du, X.P.; Plow, E.F.; Frelinger, A.L., 3rd; O’Toole, T.E.; Loftus, J.C.; Ginsberg, M.H. Ligands “activate” integrin αIIbβ3 (platelet GPIIb-IIIa). Cell 1991, 65, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.F.; Chang, C.H.; Ho, P.L.; Chung, C.H. FcgammaRII mediates platelet aggregation caused by disintegrins and GPIIb/IIIa monoclonal antibody, AP2. Exp. Hematol. 2008, 36, 1704–1713. [Google Scholar] [CrossRef]
- Huang, T.F.; Wang, W.J.; Teng, C.M.; Liu, C.S.; Ouyang, C. Purification and characterization of an antiplatelet peptide, arietin, from Bitis arietans venom. Biochim. Biophys. Acta 1991, 1074, 136–143. [Google Scholar] [CrossRef]
- Tucker, K.L.; Sage, T.; Gibbins, J.M. Clot retraction. Methods Mol. Biol. 2012, 788, 101–107. [Google Scholar] [CrossRef]
- Hsu, C.C.; Wu, W.B.; Huang, T.F. A snake venom metalloproteinase, kistomin, cleaves platelet glycoprotein VI and impairs platelet functions. J. Thromb. Haemost. 2008, 6, 1578–1585. [Google Scholar] [CrossRef]
- Chang, M.C.; Huang, T.F. In-Vivo Effect of a Thrombin-Like Enzyme on Platelet Plug Formation Induced in Mesenteric Microvessels of Mice. Thromb. Res. 1994, 73, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Hong, S.Y.; Kim, Y.D.; Nam, H.S.; Kang, S.; Yang, S.H.; Heo, J.H. Thrombolytic Effects of the Snake Venom Disintegrin Saxatilin Determined by Novel Assessment Methods: A FeCl3-Induced Thrombosis Model in Mice. PloS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, S.E.; Taylor, S.M.; Malladi, P.; Yuhan, H.; Cassel, D.L.; Chien, P.; Schwartz, E.; Schreiber, A.D.; Surrey, S.; Reilly, M.P. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: A transgenic mouse model. J. Immunol. 1999, 162, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Lin, H.K.; Peng, H.C.; Huang, T.F. Antithrombotic effect of crotalin, a platelet membrane glycoprotein Ib antagonist from venom of Crotalus atrox. Blood 1998, 91, 1582–1589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence of Antithrombotic Agents | IC50 | Safety | Tail-Bleeding Time (sec) | |||
|---|---|---|---|---|---|---|
| (41Linker45—50RGD Loop55—68C-Terminal Domain) | (nM) | Index | Dosage: Twice IC50 | Dosage: 10 × Times IC50 | Dosage: 20 × Times IC50 | |
| Eptifibatide | 625.00 | 5.00 | 538.60 *** | 1341.20 *** | ||
| Tirofiban | 85.11 | 4.00 | 480.67 *** | 1443.60 *** | ||
| Abciximab | 105.02 | 4.25 | 373.38 *** | 580.80 *** | ||
| Non-RGD small molecule RUC-2 | 62.50 | 25.50 | 66.33 n.s. | |||
| TMV-2: KKKGT-ARGDWN-PRNGLYG | 41.65 | 6.25 | 353.70 ** | 1152.60 *** | ||
| TMV-7: KKKRT-ARGDNP-PRNGLYG | 58.41 | 23.40 | 76.50 n.s. | 121.00 n.s. | 188.00 * | |
| TMV-7 mutant | KKKRT-ARGDWN-PRNGLYG | 48.52 | 4.00 | 834.00 *** | 725.80 *** | |
| KKKRT-ARGDFP-PRNGLYG | 68.75 | 7.00 | 417.50 *** | 599.10 ** | ||
| KKKRT-ARGDWR-PRNRLYG | 32.27 | 277.89 | 66.00 n.s. | 72.50 n.s. | ||
| KKKRT-ARGDFR-PRNRLYG | 48.39 | 230.89 | 68.20 n.s. | 85.00 n.s. | ||
| KKKRT-ARGDNR-PRNRLYG | 44.05 | 268.82 | 70.20 n.s. | 73.20 n.s. | ||
| KKKRT-ARGDAR-PRNRLYG | 34.42 | >3640.33 | 68.20 n.s. | 86.00 n.s. | ||
| KKKRT-ARGDRR-PRNRLYG | 31.41 | >3709.20 | 71.40 n.s. | 73.00 n.s. | 81.60 n.s. | |
| KKKRT-AKGDRR-PRNRLYG | 30.08 | >3365.87 | 63.00 n.s. | 61.50 n.s. | 82.17 n.s. | |
| Rn: | SRAGK-PRGDMP-PRYHRR | 55.31 | 3.20 | 890.34 *** | 1800.00 *** | |
| Rn mutant: | SRAGK-ARGDRR-PRYHRR | 39.07 | >3403.68 | 60.50 n.s. | 63.50 n.s. | 78.35 n.s. |
| Parameters | Integrin αIIbβ3 | Integrin αIIbβ3 |
|---|---|---|
| KGDRR | TMV-7 | |
| Electrostatic energy (kcal/mol) | −826.1 ± 76.7 | −774.1 ± 42.8 |
| Van der Waals energy (kcal/mol) | −22.6 ± 9.7 | −20.1 ± 4.7 |
| Restraints violation energy (kcal/mol) | 2.2 ± 0.5 | 0.7 ± 0.1 |
| Cluster size | 200 | 143 |
| Buried Surface Area (Å2) | 1887.7 ± 70.0 | 1775.6 ± 43.7 |
| RMSD (Å) | 0.7 ± 0.5 | 0.8 ± 0.5 |
| Disintegrin | KGDRR (nM) | Rn-RR (nM) | ||
|---|---|---|---|---|
| Inducer | PRP | PRP | PS | PS |
| ADP (20 μM) | 21.46 | N/A | 35.60 | N/A |
| Thrombin (0.1 U) | N/A | 19.81 | N/A | 32.90 |
| Collagen (10 μg/mL) | 30.08 | 24.87 | 58.41 | 41.90 |
| U46619 (1 μM) | 30.62 | 19.28 | 52.30 | 31.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, Y.-J.; Chung, C.-H.; Chen, C.-C.; Liu, J.-C.; Chiou, K.-R.; Sheu, J.-R.; Chuang, W.-J.; Huang, T.-F. A Novel KGD-Based αIIbβ3 Antagonist Prevents Arterial Thrombosis While Preserving Hemostasis and Avoiding Thrombocytopenia. Int. J. Mol. Sci. 2025, 26, 4530. https://doi.org/10.3390/ijms26104530
Kuo Y-J, Chung C-H, Chen C-C, Liu J-C, Chiou K-R, Sheu J-R, Chuang W-J, Huang T-F. A Novel KGD-Based αIIbβ3 Antagonist Prevents Arterial Thrombosis While Preserving Hemostasis and Avoiding Thrombocytopenia. International Journal of Molecular Sciences. 2025; 26(10):4530. https://doi.org/10.3390/ijms26104530
Chicago/Turabian StyleKuo, Yu-Ju, Ching-Hu Chung, Chun-Chao Chen, Ju-Chi Liu, Kuan-Rau Chiou, Joen-Rong Sheu, Woei-Jer Chuang, and Tur-Fu Huang. 2025. "A Novel KGD-Based αIIbβ3 Antagonist Prevents Arterial Thrombosis While Preserving Hemostasis and Avoiding Thrombocytopenia" International Journal of Molecular Sciences 26, no. 10: 4530. https://doi.org/10.3390/ijms26104530
APA StyleKuo, Y.-J., Chung, C.-H., Chen, C.-C., Liu, J.-C., Chiou, K.-R., Sheu, J.-R., Chuang, W.-J., & Huang, T.-F. (2025). A Novel KGD-Based αIIbβ3 Antagonist Prevents Arterial Thrombosis While Preserving Hemostasis and Avoiding Thrombocytopenia. International Journal of Molecular Sciences, 26(10), 4530. https://doi.org/10.3390/ijms26104530