
Serum Amyloid A: A Double-Edged Sword in Health and Disease


Abstract
1. Introduction
2. Expression Profile
3. Regulation of SAA Gene Expression
4. SAA Receptors and Functions
5. Serum Amyloid A-Mediated Regulation of Inflammatory Cell Migration and Recruitment
6. Pro-Inflammatory Properties of SAA
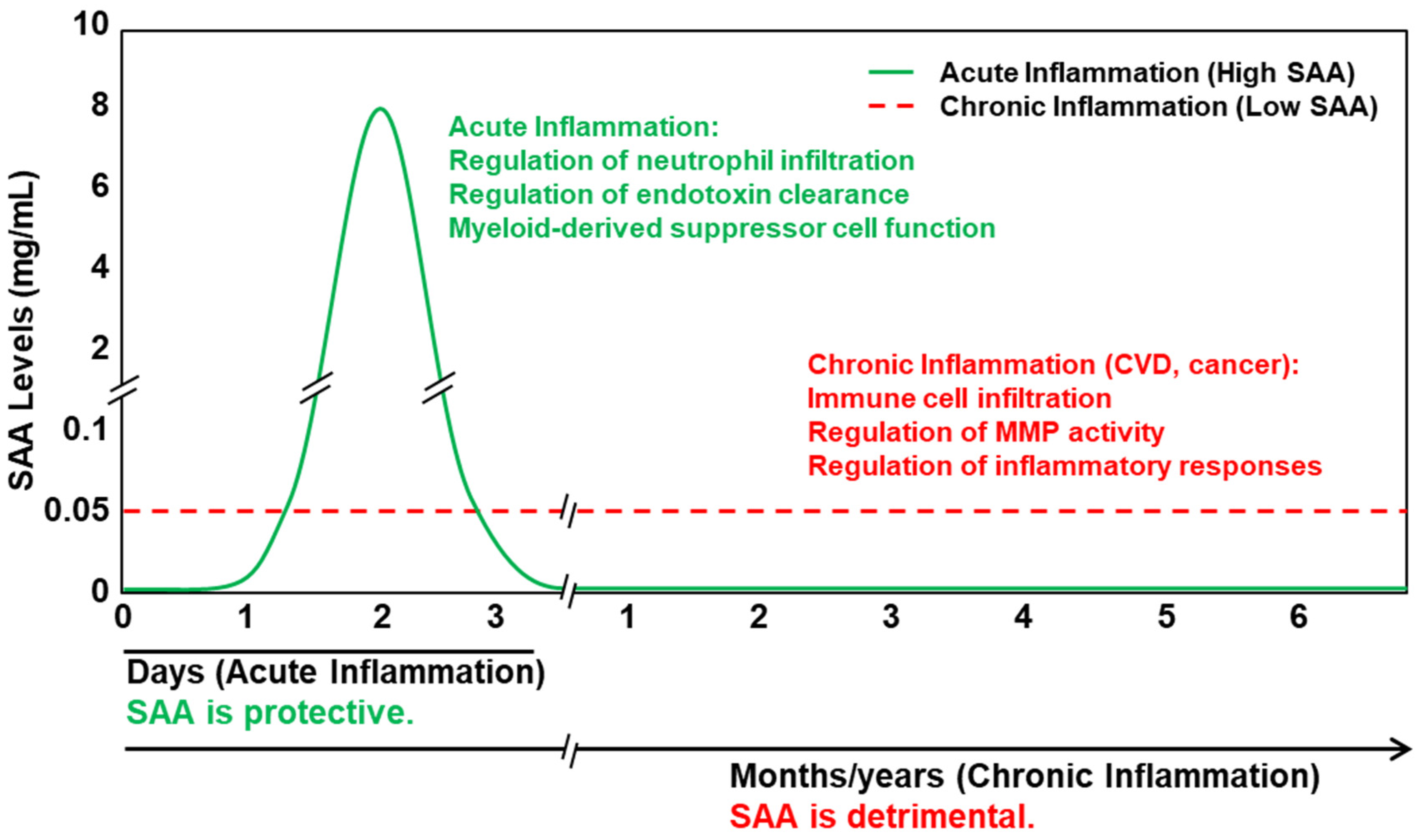
7. The Controversy: Is Serum Amyloid Truly Pro-Inflammatory?
8. Results from Animal Models of SAA
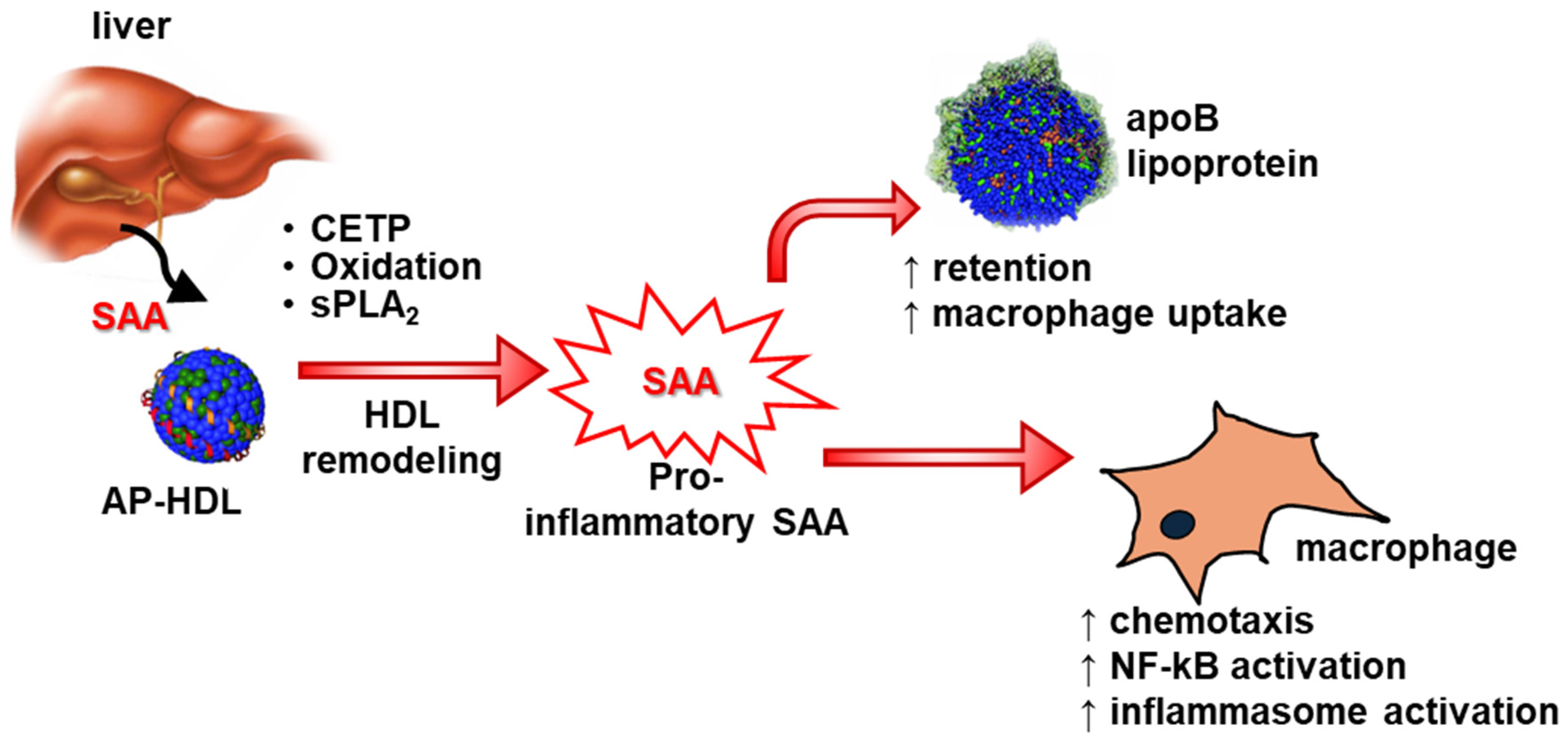
9. SAA’s Impact on HDL’s Properties
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Uhlar, C.M.; Whitehead, A.S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 1999, 265, 501–523. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Mahankali, A.P. Interactions of Serum Amyloid A Proteins with the Blood-Brain Barrier: Implications for Central Nervous System Disease. Int. J. Mol. Sci. 2024, 25, 6607. [Google Scholar] [CrossRef] [PubMed]
- Sellar, G.C.; Whitehead, A.S. Localization of four human serum amyloid A (SAA) protein superfamily genes to chromosome 11p: Characterization of a fifth SAA-related gene sequence. Genomics 1993, 16, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Kluve-Beckerman, B.; Naylor, S.L.; Marshall, A.; Gardner, J.C.; Shows, T.B.; Benson, M.D. Localization of human SAA gene(s) to chromosome 11 and detection of DNA polymorphisms. Biochem. Biophys. Res. Commun. 1986, 137, 1196–1204. [Google Scholar] [CrossRef]
- Sellar, G.C.; Jordan, S.A.; Bickmore, W.A.; Fantes, J.A.; van Heyningen, V.; Whitehead, A.S. The human serum amyloid A protein (SAA) superfamily gene cluster: Mapping to chromosome 11p15.1 by physical and genetic linkage analysis. Genomics 1994, 19, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Benditt, E.P.; Meek, R.L. Expression of the third member of the serum amyloid A gene family in mouse adipocytes. J. Exp. Med. 1989, 169, 1841–1846. [Google Scholar] [CrossRef]
- De Buck, M.; Gouwy, M.; Wang, J.M.; Snick, J.; Opdenakker, G.; Struyf, S.; Damme, J. Structure and Expression of Different Serum Amyloid A (SAA) Variants and their Concentration-Dependent Functions During Host Insults. Curr. Med. Chem. 2016, 23, 1725–1755. [Google Scholar] [CrossRef]
- Huang, J.H.; Liao, W.S. Synergistic induction of mouse serum amyloid A3 promoter by the inflammatory mediators IL-1 and IL-6. J. Interferon Cytokine Res. 1999, 19, 1403–1411. [Google Scholar] [CrossRef]
- Sack, G.H., Jr. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef]
- Uhlar, C.M.; Burgess, C.J.; Sharp, P.M.; Whitehead, A.S. Evolution of the serum amyloid A (SAA) protein superfamily. Genomics 1994, 19, 228–235. [Google Scholar] [CrossRef]
- Lowell, C.A.; Potter, D.A.; Stearman, R.S.; Morrow, J.F. Structure of the murine serum amyloid A gene family. Gene conversion. J. Biol. Chem. 1986, 261, 8442–8452. [Google Scholar] [CrossRef]
- Rennegarbe, M.; Lenter, I.; Schierhorn, A.; Sawilla, R.; Haupt, C. Influence of C-terminal truncation of murine Serum amyloid A on fibril structure. Sci. Rep. 2017, 7, 6170. [Google Scholar] [CrossRef]
- Lu, J.; Yu, Y.; Zhu, I.; Cheng, Y.; Sun, P.D. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5189–5194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Srinivasan, S.; Ye, Z.; Aguilera, J.J.; Lopez, M.M.; Colón, W. Serum amyloid A 2.2 refolds into a octameric oligomer that slowly converts to a more stable hexamer. Biochem. Biophys. Res. Commun. 2011, 407, 725–729. [Google Scholar] [CrossRef]
- Nady, A.; Reichheld, S.E.; Sharpe, S. Structural studies of a serum amyloid A octamer that is primed to scaffold lipid nanodiscs. Protein Sci. 2024, 33, e4983. [Google Scholar] [CrossRef] [PubMed]
- Derebe, M.G.; Zlatkov, C.M.; Gattu, S.; Ruhn, K.A.; Vaishnava, S.; Diehl, G.E.; MacMillan, J.B.; Williams, N.S.; Hooper, L.V. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. eLife 2014, 3, e03206. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R. High-Density Lipoproteins and Serum Amyloid A (SAA). Curr. Atheroscler. Rep. 2021, 23, 7. [Google Scholar] [CrossRef]
- Urieli-Shoval, S.; Cohen, P.; Eisenberg, S.; Matzner, Y. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J. Histochem. Cytochem. 1998, 46, 1377–1384. [Google Scholar] [CrossRef]
- Meek, R.L.; Benditt, E.P. Amyloid A gene family expression in different mouse tissues. J. Exp. Med. 1986, 164, 2006–2017. [Google Scholar] [CrossRef]
- Tannock, L.R.; De Beer, M.C.; Ji, A.; Shridas, P.; Noffsinger, V.P.; Hartigh, L.D.; Chait, A.; De Beer, F.C.; Webb, N.R. Serum amyloid A3 is a high density lipoprotein-associated acute-phase protein. J. Lipid Res. 2018, 59, 339–347. [Google Scholar] [CrossRef]
- de Beer, M.C.; de Beer, F.C.; Gerardot, C.J.; Cecil, D.R.; Webb, N.R.; Goodson, M.L.; Kindy, M.S. Structure of the mouse Saa4 gene and its linkage to the serum amyloid A gene family. Genomics 1996, 34, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Poli, V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 1998, 273, 29279–29282. [Google Scholar] [CrossRef]
- Numerof, R.P.; Sipe, J.D.; Trehu, E.G.; Dinarello, C.A.; Mier, J.W. Suppression of IL-2-induced SAA gene expression in mice by the administration of an IL-1 receptor antagonist. Cytokine 1992, 4, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G.; Benigni, F.; Sironi, M.; Conni, M.; Carelli, M.; Cantoni, L.; Shapiro, L.; Dinarello, C.A.; Sipe, J.D.; Ghezzi, P. Ciliary neurotrophic factor (CNTF) induces serum amyloid A, hypoglycaemia and anorexia, and potentiates IL-1 induced corticosterone and IL-6 production in mice. Cytokine 1995, 7, 150–156. [Google Scholar] [CrossRef]
- Foyn Bruun, C.; Sletten, K.; Marhaug, G. Mouse serum amyloid A (SAA) proteins isolated by two-dimensional electrophoresis: Characterization of isotypes and the effect of separate and combined administrations of cytokines, dexamethasone and lipopolysaccharide (LPS) on serum levels and isotype distribution. Clin. Exp. Immunol. 1998, 111, 231–236. [Google Scholar]
- Lee, H.Y.; Kim, S.D.; Baek, S.H.; Choi, J.H.; Bae, Y.-S. Role of formyl peptide receptor 2 on the serum amyloid A-induced macrophage foam cell formation. Biochem. Biophys. Res. Commun. 2013, 433, 255–259. [Google Scholar] [CrossRef]
- Anthony, D.; McQualter, J.L.; Bishara, M.; Lim, E.X.; Yatmaz, S.; Seow, H.J.; Hansen, M.; Thompson, M.; Hamilton, J.A.; Irving, L.B.; et al. SAA drives proinflammatory heterotypic macrophage differentiation in the lung via CSF-1R-dependent signaling. FASEB J. 2014, 28, 3867–3877. [Google Scholar] [CrossRef]
- Yu, N.; Zhang, S.; Lu, J.; Li, Y.; Yi, X.; Tang, L.; Su, L.; Ding, Y. Serum amyloid A, an acute phase protein, stimulates proliferative and proinflammatory responses of keratinocytes. Cell Prolif. 2017, 50, e12320. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Kim, H.J.; Yun, J.; Baek, S.-H.; Kim, K.; Bae, Y.-S. A pertussis toxin sensitive G-protein-independent pathway is involved in serum amyloid A-induced formyl peptide receptor 2-mediated CCL2 production. Exp. Mol. Med. 2010, 42, 302–309. [Google Scholar] [CrossRef]
- Christenson, K.; Bjorkman, L.; Tangemo, C.; Bylund, J. Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J. Leukoc. Biol. 2008, 83, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Song, C.; Endoh, I.; Goyette, J.; Jessup, W.; Ben Freedman, S.; McNeil, H.P.; Geczy, C.L. Serum amyloid A induces monocyte tissue factor. J. Immunol. 2007, 178, 1852–1860. [Google Scholar] [CrossRef]
- Yan, S.D.; Zhu, H.; Zhu, A.; Golabek, A.; Du, H.; Roher, A.; Yu, J.; Soto, C.; Schmidt, A.M.; Stern, D.; et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat. Med. 2000, 6, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Katagiri, Y.; Kiire, A.; Momohara, S.; Kamatani, N. Serum amyloid A activates nuclear factor-kappaB in rheumatoid synovial fibroblasts through binding to receptor of advanced glycation end-products. J. Rheumatol. 2008, 35, 752–756. [Google Scholar]
- Cai, L.; de Beer, M.C.; de Beer, F.C.; van der Westhuyzen, D.R. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J. Biol. Chem. 2005, 280, 2954–2961. [Google Scholar] [CrossRef]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Kurlander, R.; Chen, Z.; Kimelman, M.L.; Remaley, A.T.; Csako, G.; Thomas, F.; Eggerman, T.L.; et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J. Biol. Chem. 2005, 280, 8031–8040. [Google Scholar] [CrossRef]
- Hong, C.; Shen, C.; Ding, H.; Huang, S.; Mu, Y.; Su, H.; Wei, W.; Ma, J.; Zheng, F. An involvement of SR-B1 mediated p38 MAPK signaling pathway in serum amyloid A-induced angiogenesis in rheumatoid arthritis. Mol. Immunol. 2015, 66, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Baranova, I.N.; Bocharov, A.V.; Vishnyakova, T.G.; Kurlander, R.; Chen, Z.; Fu, D.; Arias, I.M.; Csako, G.; Patterson, A.P.; Eggerman, T.L. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J. Biol. Chem. 2010, 285, 8492–8506. [Google Scholar] [CrossRef]
- Cheng, N.; He, R.; Tian, J.; Ye, P.P.; Ye, R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 2008, 181, 22–26. [Google Scholar] [CrossRef]
- Sandri, S.; Rodriguez, D.; Gomes, E.; Monteiro, H.P.; Russo, M.; Campa, A. Is serum amyloid A an endogenous TLR4 agonist? J. Leukoc. Biol. 2008, 83, 1174–1180. [Google Scholar] [CrossRef]
- Abouelasrar Salama, S.; Gouwy, M.; Van Damme, J.; Struyf, S. The turning away of serum amyloid A biological activities and receptor usage. Immunology 2021, 163, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Burgess, E.J.; Hoyt, L.R.; Randall, M.J.; Mank, M.M.; Bivona, J.J.; Eisenhauer, P.L.; Botten, J.W.; Ballif, B.A.; Lam, Y.-W.; Wargo, M.J.; et al. Bacterial Lipoproteins Constitute the TLR2-Stimulating Activity of Serum Amyloid A. J. Immunol. 2018, 201, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J. Serum amyloid A is a chemoattractant: Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Badolato, R.; Murphy, W.J.; Longo, D.L.; Anver, M.; Hale, S.; Oppenheim, J.J.; Wang, J.M. A novel biologic function of serum amyloid A. Induction of T lymphocyte migration and adhesion. J. Immunol. 1995, 155, 1184–1190. [Google Scholar] [CrossRef]
- Su, S.B.; Gong, W.; Gao, J.L.; Shen, W.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J. Exp. Med. 1999, 189, 395–402. [Google Scholar] [CrossRef]
- Sander, L.E.; Sackett, S.D.; Dierssen, U.; Beraza, N.; Linke, R.P.; Müller, M.; Blander, J.M.; Tacke, F.; Trautwein, C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 2010, 207, 1453–1464. [Google Scholar] [CrossRef]
- Anthony, D.; Seow, H.J.; Uddin, M.; Thompson, M.; Dousha, L.; Vlahos, R.; Irving, L.B.; Levy, B.D.; Anderson, G.P.; Bozinovski, S. Serum amyloid A promotes lung neutrophilia by increasing IL-17A levels in the mucosa and gammadelta T cells. Am. J. Respir. Crit. Care Med. 2013, 188, 179–186. [Google Scholar] [CrossRef]
- Ji, A.; Trumbauer, A.C.; Noffsinger, V.P.; Meredith, L.W.; Dong, B.; Wang, Q.; Guo, L.; Li, X.; De Beer, F.C.; Webb, N.R.; et al. Deficiency of Acute-Phase Serum Amyloid A Exacerbates Sepsis-Induced Mortality and Lung Injury in Mice. Int. J. Mol. Sci. 2023, 24, 17501. [Google Scholar] [CrossRef]
- Cheng, N.; Liang, Y.; Du, X.; Ye, R.D. Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep. 2018, 19, e45517. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, G.; Vong, C.T.; Ye, R.D. Serum amyloid A3 confers protection against acute lung injury in Pseudomonas aeruginosa-infected mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L314–L322. [Google Scholar] [CrossRef]
- Preciado-Patt, L.; Pras, M.; Fridkin, M. Binding of human serum amyloid A (hSAA) and its high-density lipoprotein3 complex (hSAA-HDL3) to human neutrophils. Possible implication to the function of a protein of an unknown physiological role. Int. J. Pept. Protein Res. 1996, 48, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Preciado-Patt, L.; Levartowsky, D.; Prass, M.; Hershkoviz, R.; Lider, O.; Fridkin, M. Inhibition of cell adhesion to glycoproteins of the extracellular matrix by peptides corresponding to serum amyloid A. Toward understanding the physiological role of an enigmatic protein. Eur. J. Biochem. 1994, 223, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.D.; Sun, L. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol. 2015, 98, 923–929. [Google Scholar] [CrossRef]
- Patel, H.; Fellowes, R.; Coade, S.; Woo, P. Human serum amyloid A has cytokine-like properties. Scand. J. Immunol. 1998, 48, 410–418. [Google Scholar] [CrossRef]
- Furlaneto, C.J.; Campa, A. A novel function of serum amyloid A: A potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 by human blood neutrophil. Biochem. Biophys. Res. Commun. 2000, 268, 405–408. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Sang, H.; Ye, R.D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 2003, 101, 1572–1581. [Google Scholar] [CrossRef]
- O’Hara, R.; Murphy, E.P.; Whitehead, A.S.; FitzGerald, O.; Bresnihan, B. Local expression of the serum amyloid A and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum. 2004, 50, 1788–1799. [Google Scholar] [CrossRef]
- He, R.; Shepard, L.W.; Chen, J.; Pan, Z.K.; Ye, R.D. Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. J. Immunol. 2006, 177, 4072–4079. [Google Scholar] [CrossRef]
- He, R.L.; Zhou, J.; Hanson, C.Z.; Chen, J.; Cheng, N.; Ye, R.D. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood 2009, 113, 429–437. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, H.; Zhu, Z.; Yan, Q.; Wang, L.; Liang, Q.; Ye, R.D. Ex vivo and in vitro effect of serum amyloid a in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. J. Immunol. 2015, 194, 4891–4900. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Macaubas, C.; Nadeau, K.C.; Truong, P.; Yoon, T.; Lee, T.; Park, J.L.; Mellins, E.D. Serum amyloid A overrides Treg anergy via monocyte-dependent and Treg-intrinsic, SOCS3-associated pathways. Blood 2011, 117, 3793–3798. [Google Scholar] [CrossRef] [PubMed]
- Shridas, P.; Patrick, A.C.; Tannock, L.R. Role of Serum Amyloid A in Abdominal Aortic Aneurysm and Related Cardiovascular Diseases. Biomolecules 2021, 11, 1883. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.P.; Al-Shawi, R.; Ellmerich, S.; Speck, I.; Aslam, S.; Hutchinson, W.L.; Mangione, P.P.; Disterer, P.; Gilbertson, J.A.; Hunt, T.; et al. Pathogenetic mechanisms of amyloid A amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16115–16120. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; de Beer, M.C.; Wroblewski, J.M.; Webb, N.R.; de Beer, F.C. SAA does not induce cytokine production in physiological conditions. Cytokine 2013, 61, 506–512. [Google Scholar] [CrossRef]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef]
- Franco, A.G.; Sandri, S.; Campa, A. High-density lipoprotein prevents SAA-induced production of TNF-alpha in THP-1 monocytic cells and peripheral blood mononuclear cells. Mem. Inst. Oswaldo Cruz 2011, 106, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Vallon, R.; Freuler, F.; Desta-Tsedu, N.; Robeva, A.; Dawson, J.; Wenner, P.; Engelhardt, P.; Boes, L.; Schnyder, J.; Tschopp, C.; et al. Serum amyloid A (apoSAA) expression is up-regulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J. Immunol. 2001, 166, 2801–2807. [Google Scholar] [CrossRef]
- Meek, R.L.; Urieli-Shoval, S.; Benditt, E.P. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: Implications for serum amyloid A function. Proc. Natl. Acad. Sci. USA 1994, 91, 3186–3190. [Google Scholar] [CrossRef]
- Wilson, P.G.; Thompson, J.C.; Webb, N.R.; de Beer, F.C.; King, V.L.; Tannock, L.R. Serum amyloid A, but not C-reactive protein, stimulates vascular proteoglycan synthesis in a pro-atherogenic manner. Am. J. Pathol. 2008, 173, 1902–1910. [Google Scholar] [CrossRef]
- Dong, Z.; Wu, T.; Qin, W.; An, C.; Wang, Z.; Zhang, M.; Zhang, Y.; Zhang, C.; An, F. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol. Med. 2011, 17, 1357–1364. [Google Scholar] [CrossRef]
- Shridas, P.; Ji, A.; Trumbauer, A.C.; Noffsinger, V.P.; Meredith, L.W.; de Beer, F.C.; Mullick, A.E.; Webb, N.R.; Karounos, D.G.; Tannock, L.R. Antisense oligonucleotide targeting hepatic Serum Amyloid A limits the progression of angiotensin II-induced abdominal aortic aneurysm formation. Atherosclerosis 2024, 391, 117492. [Google Scholar] [CrossRef]
- Webb, N.R.; De Beer, M.C.; Wroblewski, J.M.; Ji, A.; Bailey, W.; Shridas, P.; Charnigo, R.J.; Noffsinger, V.P.; Witta, J.; Howatt, D.A.; et al. Deficiency of Endogenous Acute-Phase Serum Amyloid A Protects apoE-/- Mice from Angiotensin II-Induced Abdominal Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1156–1165. [Google Scholar] [CrossRef]
- Lee, J.W.; Stone, M.L.; Porrett, P.M.; Thomas, S.K.; Komar, C.A.; Li, J.H.; Delman, D.; Graham, K.; Gladney, W.L.; Hua, X.; et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019, 567, 249–252. [Google Scholar] [CrossRef]
- Stone, M.L.; Lee, J.; Lee, J.W.; Coho, H.; Tariveranmoshabad, M.; Wattenberg, M.W.; Choi, H.; Herrera, V.M.; Xue, Y.; Choi-Bose, S.; et al. Hepatocytes coordinate immune evasion in cancer via release of serum amyloid A proteins. Nat. Immunol. 2024, 25, 755–763. [Google Scholar] [CrossRef]
- Niederau, C.; Backmerhoff, F.; Schumacher, B. Inflammatory mediators and acute phase proteins in patients with Crohn’s disease and ulcerative colitis. Hepatogastroenterology 1997, 44, 90–107. [Google Scholar]
- Marzi, C.; Huth, C.; Herder, C.; Baument, J.; Thorand, B.; Rathmann, W.; Meisinger, C.; Wichmann, H.-E.; Roden, M.; Peters, A.; et al. Acute-phase serum amyloid A protein and its implication in the development of type 2 diabetes in the KORA S4/F4 study. Diabetes Care 2013, 36, 1321–1326. [Google Scholar] [CrossRef]
- Brunger, A.F.; Nienhuis, H.L.A.; Bijzet, J.; Hazenberg, B.P.C. Causes of AA amyloidosis: A systematic review. Amyloid 2020, 27, 1–12. [Google Scholar] [CrossRef]
- Lachmann, H.J.; Goodman, H.J.; Gilbertson, J.A.; Gallimore, J.R.; Sabin, C.A.; Gillmore, J.D.; Hawkins, P.N. Natural history and outcome in systemic AA amyloidosis. N. Engl. J. Med. 2007, 356, 2361–2371. [Google Scholar] [CrossRef]
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef]
- Westermark, G.T.; Fandrich, M.; Westermark, P. AA amyloidosis: Pathogenesis and targeted therapy. Annu. Rev. Pathol. 2015, 10, 321–344. [Google Scholar] [CrossRef]
- Kluve-Beckerman, B.; Hardwick, J.; Du, L.; Benson, M.D.; Monia, B.P.; Watt, A.; Crooke, R.M.; Mullick, A. Antisense oligonucleotide suppression of serum amyloid A reduces amyloid deposition in mice with AA amyloidosis. Amyloid 2011, 18, 136–146. [Google Scholar] [CrossRef]
- Johnson, B.D.; Kip, K.E.; Marroquin, O.C.; Ridker, P.M.; Kelsey, S.F.; Shaw, L.J.; Pepine, C.J.; Sharaf, B.; Merz, C.N.B.; Sopko, G.; et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: The National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation 2004, 109, 726–732. [Google Scholar] [CrossRef]
- Kosuge, M.; Ebina, T.; Ishikawa, T.; Hibi, K.; Tsukahara, K.; Okuda, J.; Iwahashi, N.; Ozaki, H.; Yano, H.; Kusama, I.; et al. Serum amyloid A is a better predictor of clinical outcomes than C-reactive protein in non-ST-segment elevation acute coronary syndromes. Circ. J. 2007, 71, 186–190. [Google Scholar] [CrossRef]
- Ogasawara, K.; Mashiba, S.; Wada, Y.; Sahara, M.; Uchida, K.; Aizawa, T.; Kodama, T. A serum amyloid A and LDL complex as a new prognostic marker in stable coronary artery disease. Atherosclerosis 2004, 174, 349–356. [Google Scholar] [CrossRef]
- Song, C.; Shen, Y.; Yamen, E.; Hsu, K.; Yan, W.; Witting, P.K.; Geczy, C.L.; Ben Freedman, S. Serum amyloid A may potentiate prothrombotic and proinflammatory events in acute coronary syndromes. Atherosclerosis 2009, 202, 596–604. [Google Scholar] [CrossRef]
- O’Brien, K.D.; McDonald, T.O.; Kunjathoor, V.; Eng, K.L.; Knopp, E.A.; Lewis, K.; Lopez, R.; Kirk, E.A.; Chait, A.; Wight, T.N.; et al. Serum amyloid A and lipoprotein retention in murine models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 785–790. [Google Scholar] [CrossRef]
- Thompson, J.C.; Jayne, C.; Thompson, J.; Wilson, P.G.; Yoder, M.H.; Webb, N.; Tannock, L.R. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J. Lipid Res. 2015, 56, 286–293. [Google Scholar] [CrossRef]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Ji, A.; Shridas, P.; Thompson, J.C.; van der Westhuyzen, D.R.; et al. Deficiency of endogenous acute phase serum amyloid A does not affect atherosclerotic lesions in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 255–261. [Google Scholar] [CrossRef]
- Thompson, J.C.; Wilson, P.G.; Shridas, P.; Ji, A.; de Beer, M.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum amyloid A3 is pro-atherogenic. Atherosclerosis 2018, 268, 32–35. [Google Scholar] [CrossRef]
- Cai, X.; Ahmad, G.; Hossain, F.; Liu, Y.; Wang, X.; Dennis, J.; Freedman, B.; Witting, P.K. High-Density Lipoprotein (HDL) Inhibits Serum Amyloid A (SAA)-Induced Vascular and Renal Dysfunctions in Apolipoprotein E-Deficient Mice. Int. J. Mol. Sci. 2020, 21, 1316. [Google Scholar] [CrossRef]
- Shridas, P.; Ji, A.; Trumbauer, A.C.; Noffsinger, V.P.; Leung, S.W.; Dugan, A.J.; Thatcher, S.E.; Cassis, L.A.; de Beer, F.C.; Webb, N.R.; et al. Adipocyte-Derived Serum Amyloid A Promotes Angiotensin II-Induced Abdominal Aortic Aneurysms in Obese C57BL/6J Mice. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 632–643. [Google Scholar] [CrossRef]
- Wilson, P.G.; Thompson, J.C.; Shridas, P.; McNamara, P.J.; de Beer, M.C.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum Amyloid A Is an Exchangeable Apolipoprotein. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1890–1900. [Google Scholar] [CrossRef]
- Ji, A.; Trumbauer, A.C.; Noffsinger, V.P.; de Beer, F.C.; Webb, N.R.; Tannock, L.R.; Shridas, P. Serum amyloid A augments the atherogenic effects of cholesteryl ester transfer protein. J. Lipid Res. 2023, 64, 100365. [Google Scholar] [CrossRef]
- Hu, W.; Abe-Dohmae, S.; Tsujita, M.; Iwamoto, N.; Ogikubo, O.; Otsuka, T.; Kumon, Y.; Yokoyama, S. Biogenesis of HDL by SAA is dependent on ABCA1 in the liver in vivo. J. Lipid Res. 2008, 49, 386–393. [Google Scholar] [CrossRef]
- Chien, J.Y.; Jerng, J.S.; Yu, C.J.; Yang, P.C. Low serum level of high-density lipoprotein cholesterol is a poor prognostic factor for severe sepsis. Crit. Care Med. 2005, 33, 1688–1693. [Google Scholar] [CrossRef]
- Linke, R.P.; Meinel, A.; Chalcroft, J.P.; Urieli-Shoval, S. Serum amyloid A (SAA) treatment enhances the recovery of aggravated polymicrobial sepsis in mice, whereas blocking SAA’s invariant peptide results in early death. Amyloid 2017, 24, 149–150. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, J.; Wu, L.; Fan, Y.; Sun, L.; Qian, F.; Chen, D.; Ye, R.D. Elevated Expression of Serum Amyloid A 3 Protects Colon Epithelium Against Acute Injury Through TLR2-Dependent Induction of Neutrophil IL-22 Expression in a Mouse Model of Colitis. Front. Immunol. 2018, 9, 1503. [Google Scholar] [CrossRef]
- Hu, Z.; Bang, Y.J.; Ruhn, K.A.; Hooper, L.V. Molecular basis for retinol binding by serum amyloid A during infection. Proc. Natl. Acad. Sci. USA 2019, 116, 19077–19082. [Google Scholar] [CrossRef]
- Bang, Y.J.; Hu, Z.; Li, Y.; Gattu, S.; Ruhn, K.A.; Raj, P.; Herz, J.; Hooper, L.V. Serum amyloid A delivers retinol to intestinal myeloid cells to promote adaptive immunity. Science 2021, 373, eabf9232. [Google Scholar] [CrossRef]
- Saco, Y.; Bassols, A. Acute phase proteins in cattle and swine: A review. Vet. Clin. Pathol. 2023, 52 (Suppl. S1), 50–63. [Google Scholar] [CrossRef]
- Skovgaard, K.; Mortensen, S.; Boye, M.; Poulsen, K.T.; Campbell, F.M.; Eckersall, P.D.; Heegaard, P.M. Rapid and widely disseminated acute phase protein response after experimental bacterial infection of pigs. Vet. Res. 2009, 40, 23. [Google Scholar] [CrossRef] [PubMed]
- Hulten, C.; Johansson, E.; Fossum, C.; Wallgren, P. Interleukin 6, serum amyloid A and haptoglobin as markers of treatment efficacy in pigs experimentally infected with Actinobacillus pleuropneumoniae. Vet. Microbiol. 2003, 95, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Soler, L.; Gutierrez, A.; Escribano, D.; Fuentes, M.; Cerón, J. Response of salivary haptoglobin and serum amyloid A to social isolation and short road transport stress in pigs. Res. Vet. Sci. 2013, 95, 298–302. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Wagner, A.C.; Navab, M.; Anantharamaiah, G.M.; Hama, S.; Reddy, S.T.; Fogelman, A.M. Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J. Lipid Res. 2007, 48, 2344–2353. [Google Scholar] [CrossRef]
- Ma, J.; Liu, X.; Qiao, L.; Meng, L.; Xu, X.; Xue, F.; Cheng, C.; Han, Z.; Lu, Y.; Zhang, W.; et al. Association Between Stent Implantation and Progression of Nontarget Lesions in a Rabbit Model of Atherosclerosis. Circ. Cardiovasc. Interv. 2021, 14, e010764. [Google Scholar] [CrossRef]
- Han, C.Y.; Kang, I.; Omer, M.; Wang, S.; Wietecha, T.; Wight, T.N.; Chait, A. Serum amyloid A-containing HDL binds adipocyte-derived versican and macrophage-derived biglycan, reducing its antiinflammatory properties. JCI Insight 2020, 5, e142635. [Google Scholar] [CrossRef]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef]
- Zewinger, S.; Drechsler, C.; Kleber, M.E.; Dressel, A.; Riffel, J.; Triem, S.; Lehmann, M.; Kopecky, C.; Säemann, M.D.; Lepper, P.M.; et al. Serum amyloid A: High-density lipoproteins interaction and cardiovascular risk. Eur. Heart J. 2015, 36, 3007–3016. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Kopecky, C.; Kubicek, M.; Haidinger, M.; Döller, D.; Katholnig, K.; Suarna, C.; Eller, P.; Tölle, M.; Gerner, C.; et al. Serum amyloid A in uremic HDL promotes inflammation. J. Am. Soc. Nephrol. 2012, 23, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Ashby, D.; Gamble, J.; Vadas, M.; Fidge, N.; Siggins, S.; Rye, K.A.; Barter, P.J. Lack of effect of serum amyloid A (SAA) on the ability of high-density lipoproteins to inhibit endothelial cell adhesion molecule expression. Atherosclerosis 2001, 154, 113–121. [Google Scholar] [CrossRef]
- Sato, M.; Ohkawa, R.; Yoshimoto, A.; Yano, K.; Ichimura, N.; Nishimori, M.; Okubo, S.; Yatomi, Y.; Tozuka, M. Effects of serum amyloid A on the structure and antioxidant ability of high-density lipoprotein. Biosci. Rep. 2016, 36, e00369. [Google Scholar] [CrossRef]
- Jayaraman, S.; Haupt, C.; Gursky, O. Paradoxical effects of SAA on lipoprotein oxidation suggest a new antioxidant function for SAA. J. Lipid Res. 2016, 57, 2138–2149. [Google Scholar] [CrossRef]
- Barlage, S.; Gnewuch, C.; Liebisch, G.; Wolf, Z.; Audebert, F.-X.; Glück, T.; Fröhlich, D.; Krämer, B.K.; Rothe, G.; Schmitz, G. Changes in HDL-associated apolipoproteins relate to mortality in human sepsis and correlate to monocyte and platelet activation. Intensive Care Med. 2009, 35, 1877–1885. [Google Scholar] [CrossRef]
- Khaliq, W.; Grossmann, P.; Neugebauer, S.; Kleyman, A.; Domizi, R.; Calcinaro, S.; Brealey, D.; Gräler, M.; Kiehntopf, M.; Schäuble, S.; et al. Lipid metabolic signatures deviate in sepsis survivors compared to non-survivors. Comput. Struct. Biotechnol. J. 2020, 18, 3678–3691. [Google Scholar] [CrossRef]
- Lekkou, A.; Mouzaki, A.; Siagris, D.; Ravani, I.; Gogos, C.A. Serum lipid profile, cytokine production, and clinical outcome in patients with severe sepsis. J. Crit. Care 2014, 29, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; De Tymowski, C.; Stern, J.; Bouzid, D.; Zappella, N.; Snauwaert, A.; Robert, T.; Lortat-Jacob, B.; Tran-Dinh, A.; Augustin, P.; et al. Relationship between liver dysfunction, lipoprotein concentration and mortality during sepsis. PLoS ONE 2022, 17, e0272352. [Google Scholar] [CrossRef]
- Trinder, M.; Walley, K.R.; Boyd, J.H.; Brunham, L.R. Causal Inference for Genetically Determined Levels of High-Density Lipoprotein Cholesterol and Risk of Infectious Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Morin, E.E.; Guo, L.; Schwendeman, A.; Li, X.-A. HDL in sepsis—Risk factor and therapeutic approach. Front. Pharmacol. 2015, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; de Beer, M.C.; Noffsinger, V.; Tannock, L.R.; Ramaiah, C.; Webb, N.R.; van der Westhuyzen, D.R.; de Beer, F.C. HDL remodeling during the acute phase response. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 261–267. [Google Scholar] [CrossRef]
- Kisilevsky, R.; Manley, P.N. Acute-phase serum amyloid A: Perspectives on its physiological and pathological roles. Amyloid 2012, 19, 5–14. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, A.; Meredith, L.W.; Shridas, P. Serum Amyloid A: A Double-Edged Sword in Health and Disease. Int. J. Mol. Sci. 2025, 26, 4528. https://doi.org/10.3390/ijms26104528
Ji A, Meredith LW, Shridas P. Serum Amyloid A: A Double-Edged Sword in Health and Disease. International Journal of Molecular Sciences. 2025; 26(10):4528. https://doi.org/10.3390/ijms26104528
Chicago/Turabian StyleJi, Ailing, Luke W. Meredith, and Preetha Shridas. 2025. "Serum Amyloid A: A Double-Edged Sword in Health and Disease" International Journal of Molecular Sciences 26, no. 10: 4528. https://doi.org/10.3390/ijms26104528
APA StyleJi, A., Meredith, L. W., & Shridas, P. (2025). Serum Amyloid A: A Double-Edged Sword in Health and Disease. International Journal of Molecular Sciences, 26(10), 4528. https://doi.org/10.3390/ijms26104528