

Tissue-Resident Macrophages in Cardiovascular Diseases: Heterogeneity and Therapeutic Potential
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. The Role of TRMs in Cardiovascular Disease
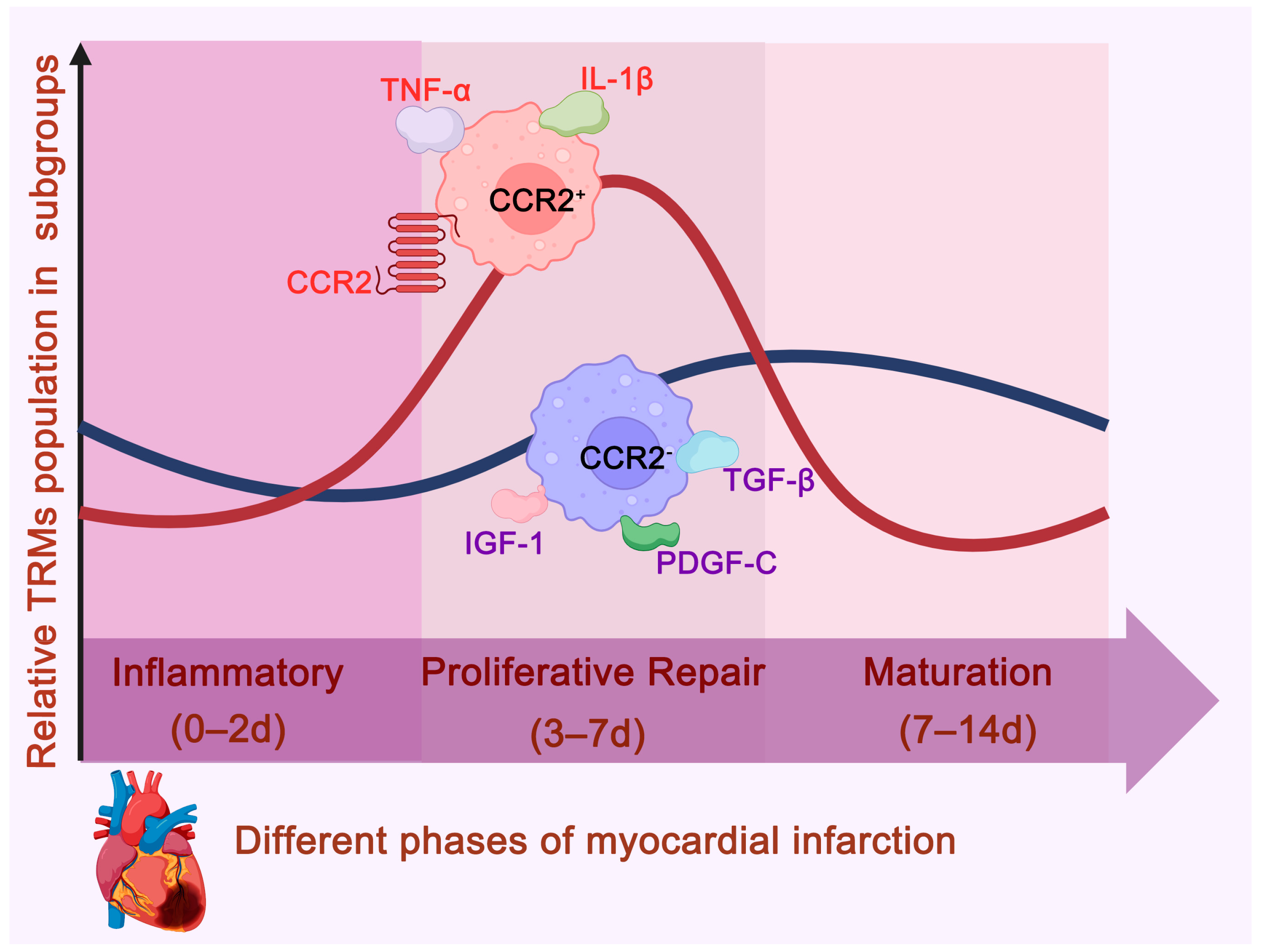
2.1. Myocardial Infarction
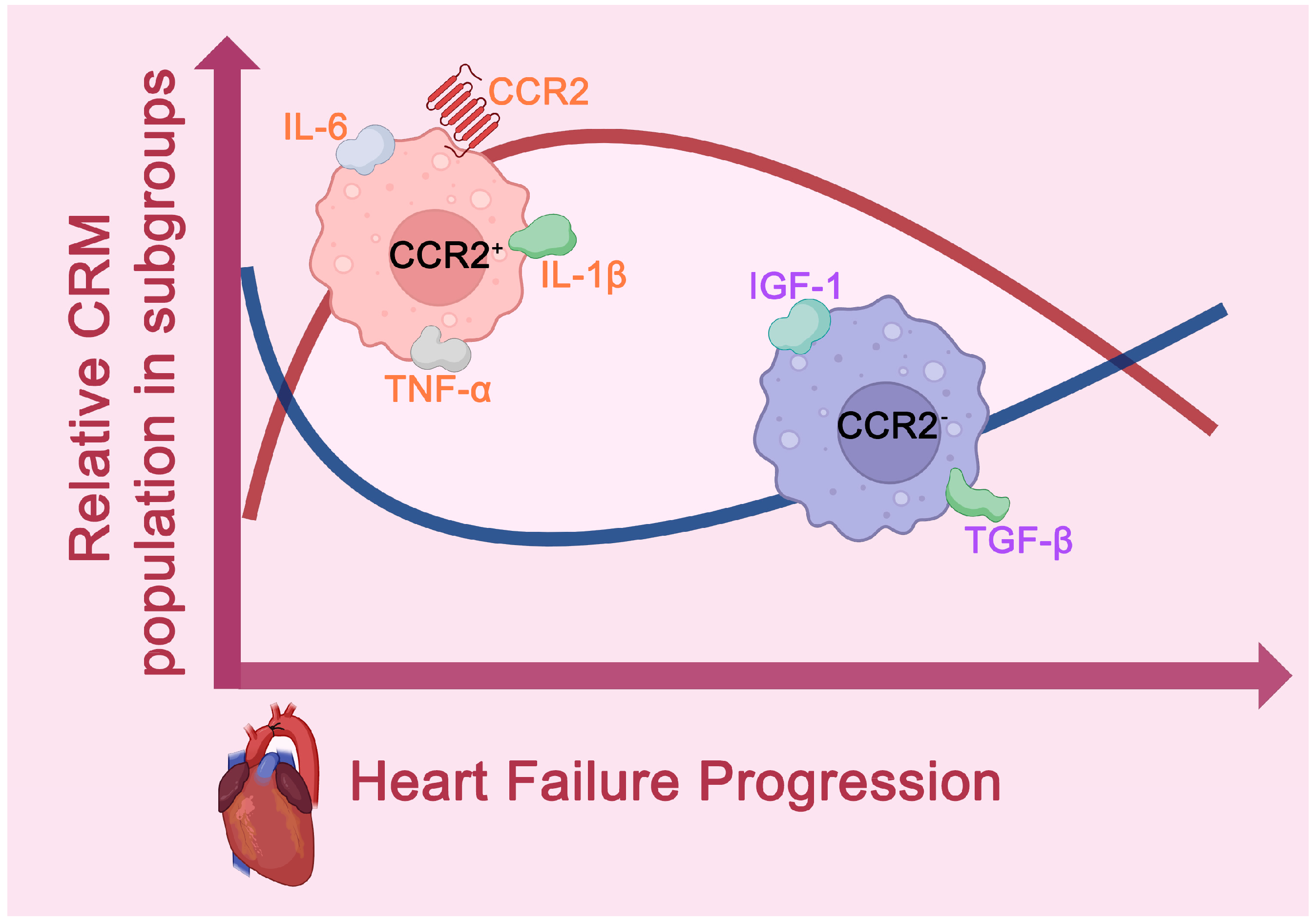
2.2. Heart Failure
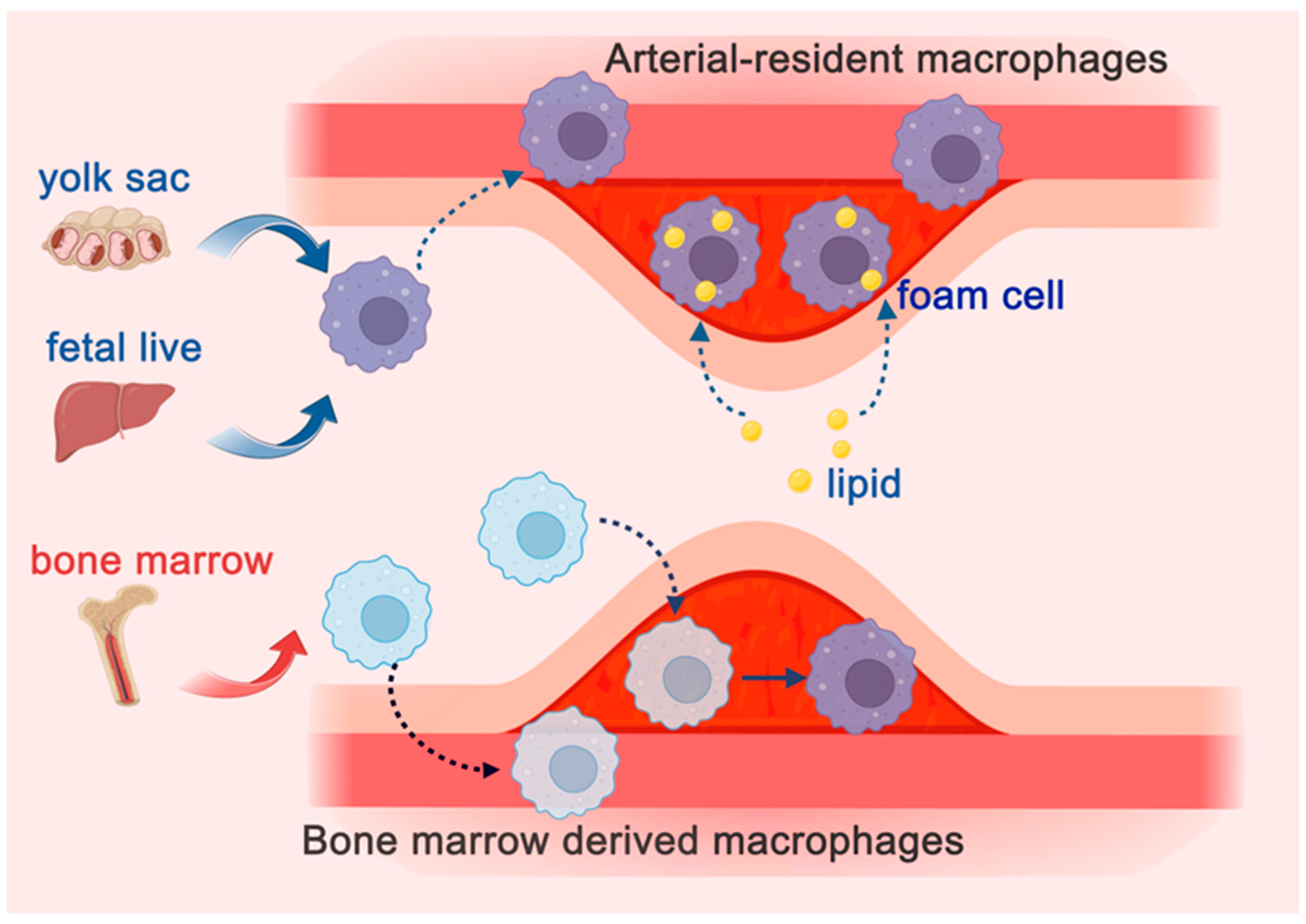
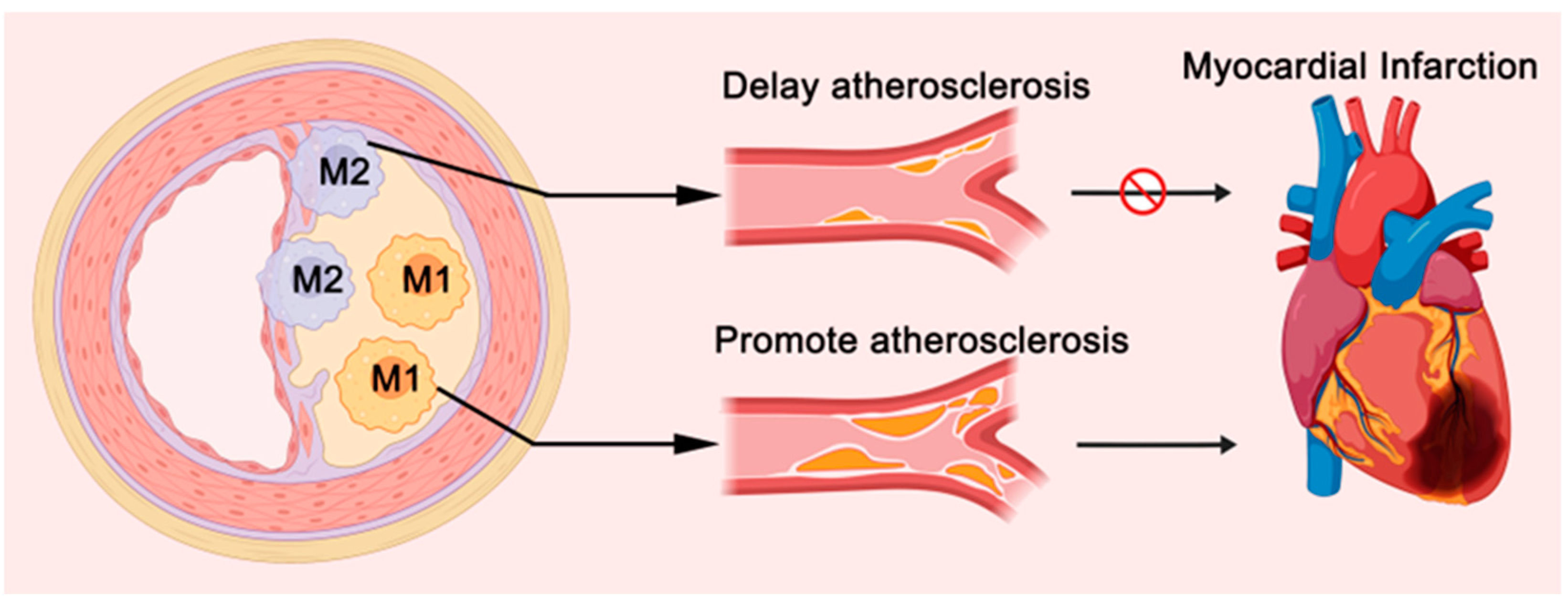
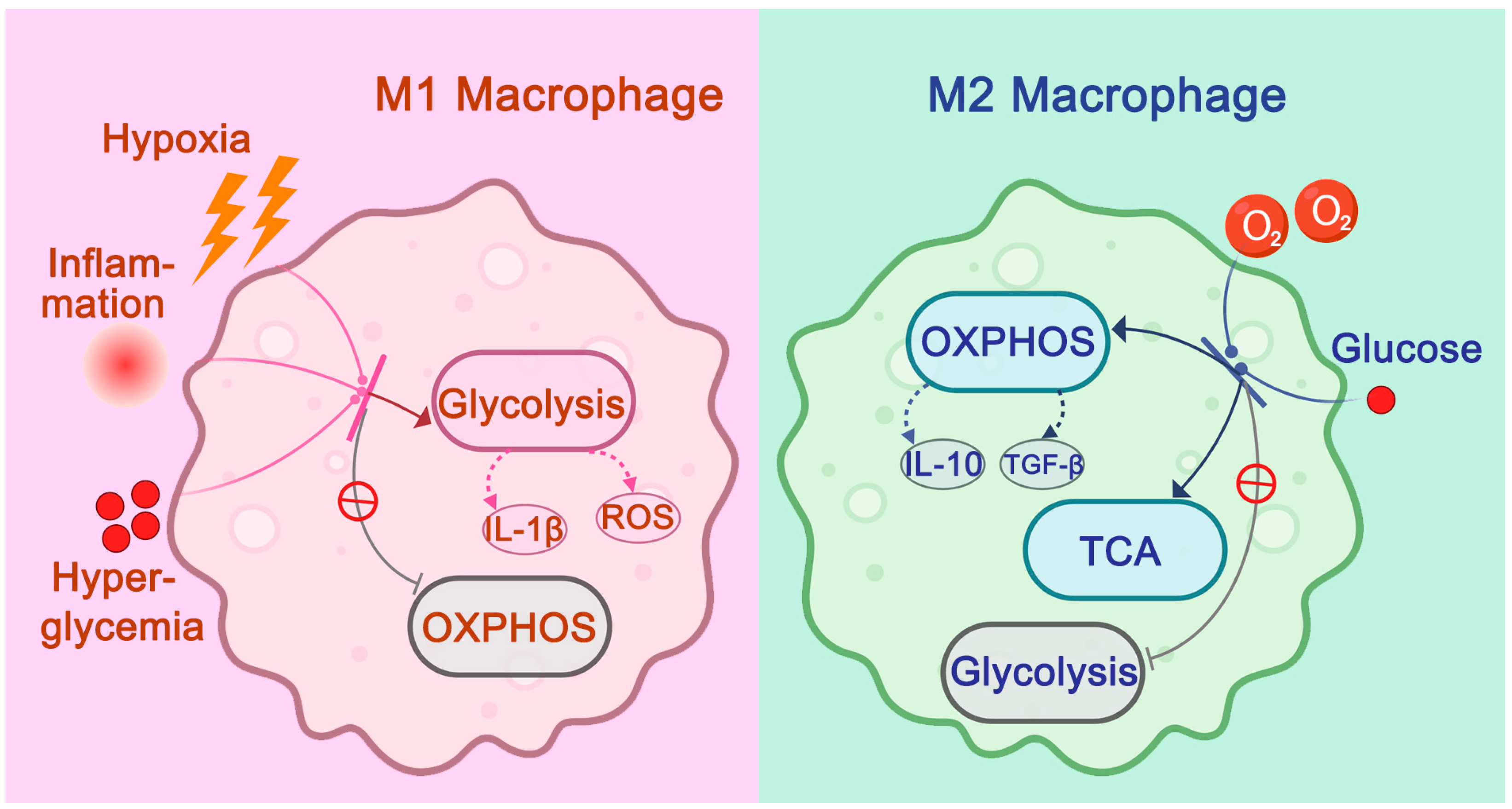
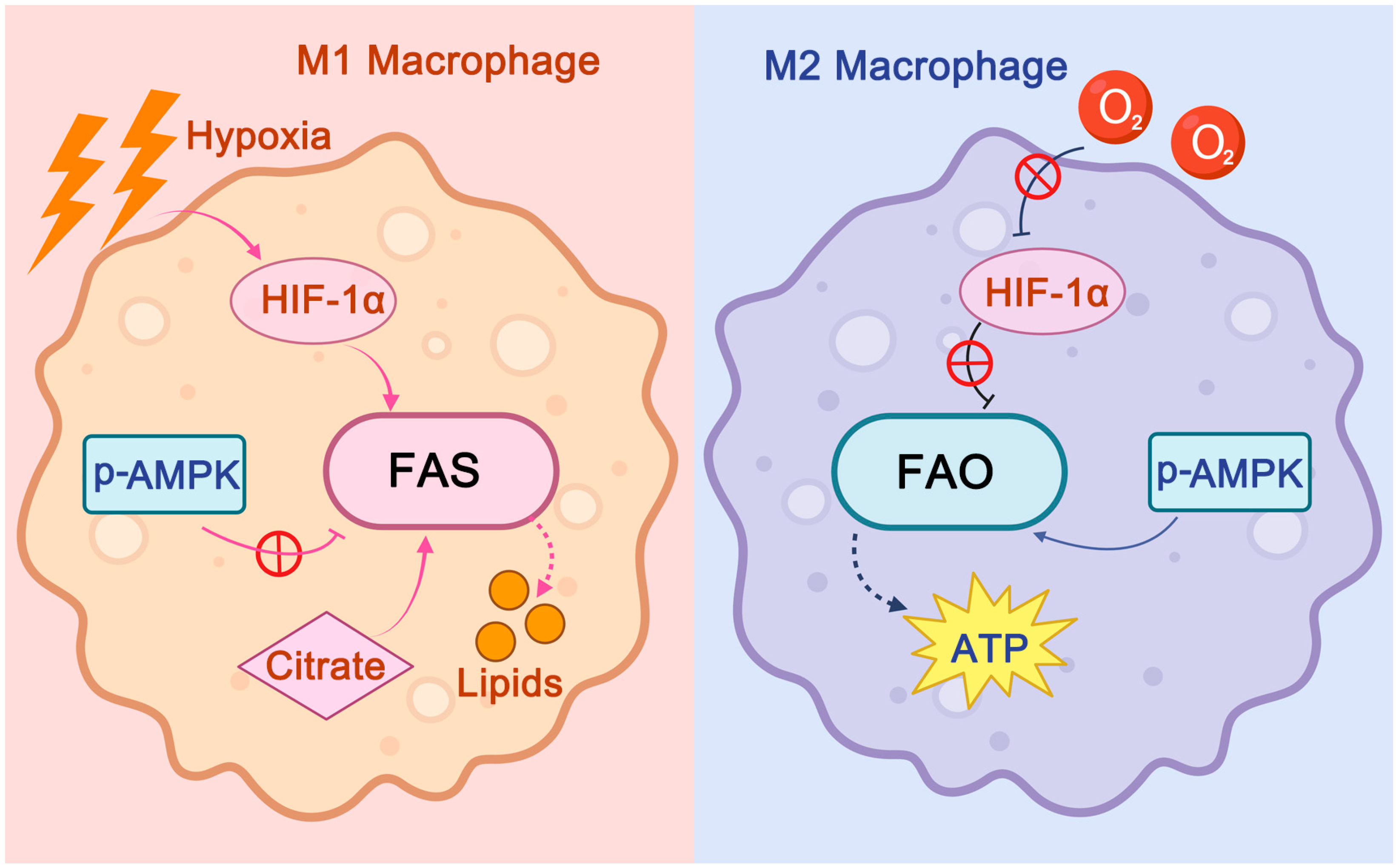
2.3. Atherosclerosis
2.4. Hypertension and Pulmonary Arterial Hypertension
3. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name |
| TRMs | Tissue-Resident Macrophages |
| CRMs | Cardiac Resident Macrophages |
| CCR2 | C-C Chemokine Receptor Type 2 |
| IL-1β | Interleukin-1β |
| TNF-α | Tumor Necrosis Factor-alpha |
| IL-10 | Interleukin-10 |
| TGF-β | Transforming Growth Factor-beta |
| PDGF-C | Platelet Derived Growth Factor C |
| MI | Myocardial Infarction |
| HF | Heart Failure |
| HFrEF | Heart Failure with Reduced Ejection Fraction |
| HFpEF | Heart Failure with Preserved Ejection Fraction |
| AS | Atherosclerosis |
| IGF-1 | Insulin-like growth factor 1 |
| OXPHOS | Oxidative Phosphorylation |
| TCA | Tricarboxylic Acid Cycle |
| FAS | Fatty Acid Synthesis |
| FAO | Fatty Acid Oxidation |
| HIF-1α | Hypoxia-Inducible Factor-1α |
| AMPK | AMP-Activated Protein Kinase |
| ROS | Reactive Oxygen Species |
| CCL2 | C-C Motif Chemokine Ligand 2 |
| CX3CR1 | C-X3-C Motif Chemokine Receptor 1 |
| LYVE1 | Lymphatic Vessel Endothelial Hyaluronan Receptor 1 |
| MHC-II | Major Histocompatibility Complex Class II |
| PH | Pulmonary arterial hypertension |
References
- Moskalik, A.; Niderla-Bielińska, J.; Ratajska, A. Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail. Rev. 2022, 27, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Anitua, E.; Troya, M.; Alkhraisat, M.H. Immunoregulatory role of platelet derivatives in the macrophage-mediated immune response. Front. Immunol. 2024, 15, 1399130. [Google Scholar] [CrossRef]
- Hu, S.; Yang, M.; Huang, S.; Zhong, S.; Zhang, Q.; Ding, H.; Xiong, X.; Hu, Z.; Yang, Y. Different roles of resident and non-resident macrophages in cardiac fibrosis. Front Cardiovasc. Med. 2022, 9, 818188. [Google Scholar] [CrossRef] [PubMed]
- Isidoro, C.A.; Deniset, J.F. The role of macrophage subsets in and around the heart in modulating cardiac homeostasis and pathophysiology. Front. Immunol. 2023, 14, 1111819. [Google Scholar] [CrossRef]
- Theofilis, P.; Oikonomou, E.; Tsioufis, K.; Tousoulis, D. The role of macrophages in atherosclerosis: Pathophysiologic mechanisms and treatment considerations. Int. J. Mol. Sci. 2023, 24, 9568. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Hou, P.; Fang, J.; Liu, Z.; Shi, Y.; Agostini, M.; Bernassola, F.; Bove, P.; Candi, E.; Rovella, V.; Sica, G.; et al. Macrophage polarization and metabolism in atherosclerosis. Cell Death Dis. 2023, 14, 691. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Liu, C.; Hu, J. Cardiac resident macrophages: Key regulatory mediators in the aftermath of myocardial infarction. Front. Immunol. 2023, 14, 1207100. [Google Scholar] [CrossRef]
- Yap, J.; Irei, J.; Lozano-Gerona, J.; Vanapruks, S.; Bishop, T.; Boisvert, W.A. Macrophages in cardiac remodelling after myocardial infarction. Nat. Rev. Cardiol. 2023, 20, 373–385. [Google Scholar] [CrossRef]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.R.; Mohan, J.; Kopecky, B.J.; Guo, S.; Du, L.; Leid, J.; Feng, G.; Lokshina, I.; Dmytrenko, O.; Luehmann, H.; et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 2021, 54, 2072–2088.e7. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, H.; Tang, B.; Luo, Y.; Yang, Y.; Zhong, X.; Chen, S.; Xu, X.; Huang, S.; Liu, C. Macrophages in cardiovascular diseases: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 130. [Google Scholar] [CrossRef]
- Liu, D.; Yan, W.; Huang, J.; Zhao, J.; Kilby, H.; Christopher, T.A.; Lopez, B.; Tao, L.; Ma, X.; Gu, G.; et al. Macrophages in ischemic heart failure: Yesterday, today, and tomorrow. Cardiol. Discov. 2021, 1, 128–134. [Google Scholar] [CrossRef]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.-M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Li, Q.-L.; Tang, T.-T.; Cheng, X. Emerging role of macrophage-fibroblast interactions in cardiac homeostasis and remodeling. JACC Basic Transl. Sci. 2024, 10, 113–127. [Google Scholar] [CrossRef]
- Bianchi, S.; Torge, D.; Rinaldi, F.; Piattelli, M.; Bernardi, S.; Varvara, G. Platelets’ role in dentistry: From oral pathology to regenerative potential. Biomedicines 2022, 10, 218. [Google Scholar] [CrossRef]
- Sagris, M.; Antonopoulos, A.S.; Theofilis, P.; Oikonomou, E.; Siasos, G.; Tsalamandris, S.; Antoniades, C.; Brilakis, E.S.; Kaski, J.C.; Tousoulis, D. Risk factors profile of young and older patients with myocardial infarction. Cardiovasc. Res. 2022, 118, 2281–2292. [Google Scholar] [CrossRef]
- Algoet, M.; Janssens, S.; Himmelreich, U.; Gsell, W.; Pusovnik, M.; Van den Eynde, J.; Oostetrlinck, W. Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc. Med. 2023, 33, 357–366. [Google Scholar] [CrossRef]
- Canty, J.M. Myocardial injury, troponin release, and cardiomyocyte death in brief ischemia, failure, and ventricular remodeling. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H1–H15. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Sun, X.; Sun, Y.; Jiao, B.; Yao, J.; Hu, Y.; Deng, Q.; Dong, J.; Wang, W.; Wang, Y. Transcriptional regulation of macrophages in heart failure. Front. Cardiovasc. Med. 2023, 10, 1148041. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.-M.; Weinheimer, C.J.; et al. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef]
- Penberthy, K.K.; Ravichandran, K.S. Apoptotic cell recognition receptors and scavenger receptors. Immunol. Rev. 2016, 269, 44–59. [Google Scholar] [CrossRef]
- Kubota, A.; Frangogiannis, N.G. Macrophages in myocardial infarction. Am. J. Physiol. Cell Physiol. 2022, 323, C1304–C1324. [Google Scholar] [CrossRef]
- González, A.; Richards, A.M.; de Boer, R.A.; Thum, T.; Arfsten, H.; Hülsmann, M.; Falcao-Pires, I.; Díez, J.; Foo, R.S.; Chan, M.Y.; et al. Cardiac remodelling—Part 1: From cells and tissues to circulating biomarkers. A review from the Study Group on Biomarkers of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2022, 24, 927–943. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Kataria, R.; Zannad, F.; Sauer, A.J.; Damman, K.; Sharma, K.; Shah, S.J.; Van Spall, H.G.C. Heart failure with preserved ejection fraction: Recent concepts in diagnosis, mechanisms and management. Heart 2022, 108, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Jia, H.; Feng, M.; Ren, H.; Gao, J.; Liu, Y.; Zhang, L.; Zhou, M.-S. Cardiac macrophages in maintaining heart homeostasis and regulating ventricular remodeling of heart diseases. Front. Immunol. 2024, 15, 1467089. [Google Scholar] [CrossRef]
- Zaman, R.; Epelman, S. Resident cardiac macrophages: Heterogeneity and function in health and disease. Immunity 2022, 55, 1549–1563. [Google Scholar] [CrossRef]
- Yerra, V.G.; Advani, A. Role of CCR2-positive macrophages in pathological ventricular remodelling. Biomedicines 2022, 10, 661. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, K.; Chen, F.; Liu, Q.; Ni, J.; Cao, W.; Hua, Y.; He, F.; Liu, Z.; Li, L.; et al. Role of the CCL2-CCR2 axis in cardiovascular disease: Pathogenesis and clinical implications. Front. Immunol. 2022, 13, 975367. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Zile, M.R. From systemic inflammation to myocardial fibrosis: The heart failure with preserved ejection fraction paradigm revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef]
- González, G.E.; Rhaleb, N.-E.; D’Ambrosio, M.A.; Nakagawa, P.; Liao, T.-D.; Peterson, E.L.; Leung, P.; Dai, X.; Janic, B.; Liu, Y.-H.; et al. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1287–H1296. [Google Scholar] [CrossRef] [PubMed]
- Honold, L.; Nahrendorf, M. Resident and monocyte-derived macrophages in cardiovascular disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef]
- Chen, B.; Frangogiannis, N.G. The role of macrophages in nonischemic heart failure. JACC Basic Transl. Sci. 2018, 3, 245–248. [Google Scholar] [CrossRef]
- An, T.; Guo, M.; Fan, C.; Huang, S.; Liu, H.; Liu, K.; Wang, Z. sFgl2-Treg positive feedback pathway protects against atherosclerosis. Int. J. Mol. Sci. 2023, 24, 2338. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Xu, D. New insights into macrophage subsets in atherosclerosis. J. Mol. Med. 2022, 100, 1239–1251. [Google Scholar] [CrossRef]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.-E.; Zernecke, A. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol. 2018, 330, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Zaitsev, K.; Kim, K.-W.; Ivanov, S.; Saunders, B.T.; Schrank, P.R.; Kim, K.; Elvington, A.; Kim, S.H.; Tucker, C.G.; et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 2020, 21, 1194–1204. [Google Scholar] [CrossRef]
- Williams, J.W.; Martel, C.; Potteaux, S.; Esaulova, E.; Ingersoll, M.A.; Elvington, A.; Saunders, B.T.; Huang, L.-H.; Habenicht, A.J.; Zinselmeyer, B.H.; et al. Limited macrophage positional dynamics in progressing or regressing murine atherosclerotic plaques—Brief report. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1702–1710. [Google Scholar] [CrossRef]
- Flynn, M.C.; Pernes, G.; Lee, M.K.S.; Nagareddy, P.R.; Murphy, A.J. Monocytes, macrophages, and metabolic disease in atherosclerosis. Front. Pharmacol. 2019, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Meshkani, R.; Vakili, S. Tissue-resident macrophages: Key players in the pathogenesis of type 2 diabetes and its complications. Clin. Chim. Acta 2016, 462, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Miteva, K.; Madonna, R.; De Caterina, R.; Van Linthout, S. Innate and adaptive immunity in atherosclerosis. Vasc. Pharmacol. 2018, 107, 67–77. [Google Scholar] [CrossRef]
- Lee-Rueckert, M.; Lappalainen, J.; Kovanen, P.T.; Escola-Gil, J.C. Lipid-laden macrophages and inflammation in atherosclerosis and cancer: An integrative view. Front. Cardiovasc. Med. 2022, 9, 777822. [Google Scholar] [CrossRef]
- Nahrendorf, M. Myeloid cells in cardiovascular organs. J. Intern. Med. 2019, 285, 491–502. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Corr, E.M.; Erbay, E.; Moore, K.J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018, 19, 526–537. [Google Scholar] [CrossRef]
- Andrejeva, G.; Rathmell, J.C. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef]
- Lachmandas, E.; Boutens, L.; Ratter, J.M.; Hijmans, A.; Hooiveld, G.J.; Joosten, L.A.; Rodenburg, R.J.; Fransen, J.A.M.; Houtkooper, R.H.; van Crevel, R.; et al. Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol. 2016, 2, 16246. [Google Scholar] [CrossRef]
- Liu, L.; Lu, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G.; et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc. Natl. Acad. Sci. USA 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Netea-Maier, R.T.; Riksen, N.P.; Joosten, L.A.; Netea, M.G. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab. 2017, 26, 142–156. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-C.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.; Curtis, A.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef]
- Tannahill, G.; Curtis, A.; Adamik, J.; Palsson-McDermott, E.; McGettrick, A.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.-C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Gupta, S.; Sarangi, P.P. Inflammation-driven metabolic regulation and adaptation in macrophages. Clin. Immunol. 2023, 246, 109216. [Google Scholar] [CrossRef]
- Yurdagul, A. Crosstalk between macrophages and vascular smooth muscle cells in atherosclerotic plaque stability. Arter. Thromb. Vasc. Biol. 2022, 42, 372–380. [Google Scholar] [CrossRef]
- Shen, L.; Chen, W.; Ding, J.; Shu, G.; Chen, M.; Zhao, Z.; Xia, S.; Ji, J. The role of metabolic reprogramming of oxygen-induced macrophages in the dynamic changes of atherosclerotic plaques. FASEB J. 2023, 37, e22791. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Hou, R.; Xie, L.; Niu, M.; Pan, X.; Zhu, X. Macrophage metabolic reprogramming and atherosclerotic plaque microenvironment: Fostering each other? Clin. Transl. Med. 2023, 13, e1257. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.B. Cardiac macrophages and emerging roles for their metabolism after myocardial infarction. J. Clin. Investig. 2023, 133, e171953. [Google Scholar] [CrossRef]
- Groh, L.; Keating, S.T.; Joosten, L.A.; Netea, M.G.; Riksen, N.P. Monocyte and macrophage immunometabolism in atherosclerosis. Semin. Immunopathol. 2018, 40, 203–214. [Google Scholar] [CrossRef]
- Vassiliou, E.; Farias-Pereira, R. Impact of lipid metabolism on macrophage polarization: Implications for inflammation and tumor immunity. Int. J. Mol. Sci. 2023, 24, 12032. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Ma, H.; Yang, Q.; Shang, Q.; Song, L.; Zheng, Z.; Zhang, S.; Pan, Y.; Huang, P.; et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, HIF-1α upregulation and autophagy. Free Radic. Biol. Med. 2020, 161, 339–350. [Google Scholar] [CrossRef]
- Yang, S.; Yuan, H.-Q.; Hao, Y.-M.; Ren, Z.; Qu, S.-L.; Liu, L.-S.; Wei, D.-H.; Tang, Z.-H.; Zhang, J.-F.; Jiang, Z.-S. Macrophage polarization in atherosclerosis. Clin. Chim. Acta 2020, 501, 142–146. [Google Scholar] [CrossRef]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front. Immunol. 2020, 10, 2993. [Google Scholar] [CrossRef]
- Seo, J.W.; Park, K.S.; Lee, G.B.; Park, S.-e.; Choi, J.-H.; Moon, M.H. Comprehensive lipid profiling recapitulates enhanced lipolysis and fatty acid metabolism in intimal foamy macrophages from murine atherosclerotic aorta. Immune Netw. 2023, 23, e28. [Google Scholar] [CrossRef]
- Rudemiller, N.P.; Crowley, S.D. Interactions between the immune and the renin-angiotensin systems in hypertension. Hypertension 2016, 68, 289–296. [Google Scholar] [CrossRef]
- Moore, J.P.; Vinh, A.; Tuck, K.L.; Sakkal, S.; Krishnan, S.M.; Chan, C.T.; Lieu, M.; Samuel, C.S.; Diep, H.; Kemp-Harper, B.K.; et al. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H906–H917. [Google Scholar] [CrossRef] [PubMed]
- Elmarakby, A.A.; Quigley, J.E.; Olearczyk, J.J.; Sridhar, A.; Cook, A.K.; Inscho, E.W.; Pollock, D.M.; Imig, J.D. Chemokine receptor 2b inhibition provides renal protection in angiotensin II-salt hypertension. Hypertension 2007, 50, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Barhoumi, T.; Todryk, S. Role of monocytes/macrophages in renin-angiotensin system-induced hypertension and end organ damage. Front. Physiol. 2023, 14, 1199934. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, S.; Hu, H.; Steven, S.; Schönfelder, T.; Fraccarollo, D.; Mikhed, Y.; Brähler, M.; Knorr, M.; Brandt, M.; Karbach, S.H.; et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J. Biol. Chem. 2014, 289, 27540–27550. [Google Scholar] [CrossRef]
- Ricardo, S.D.; Van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef]
- Zhang, M.-Q.; Wang, C.-C.; Pang, X.-B.; Shi, J.-Z.; Li, H.-R.; Xie, X.-M.; Wang, Z.; Zhang, H.-D.; Zhou, Y.-F.; Chen, J.-W. Role of macrophages in pulmonary arterial hypertension. Front. Immunol. 2023, 14, 1152881. [Google Scholar] [CrossRef]
- Chen, S.; Yan, D.; Qiu, A. The role of macrophages in pulmonary hypertension: Pathogenesis and targeting. Int. Immunopharmacol. 2020, 88, 106934. [Google Scholar] [CrossRef]
- Sansonetti, M.; Zeppilli, D.; Waller, V.; Remadna, L.; Pignel, J.; Tiret, L.; Serio, A.; Montagner, M. Macrophage-Based Therapeutic Approaches for Cardiovascular Diseases. Basic Res. Cardiol. 2024, 119, 1–33. [Google Scholar] [CrossRef]
- Cordes, T.; Michelucci, A.; Hiller, K. Itaconate Alters Succinate and Coenzyme A Metabolism via Inhibition of Mitochondrial Complex II and Methylmalonyl-CoA Mutase. Cell Metab. 2016, 24, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Fang, Y.; Han, Y.; Bai, X.; Jiang, W.; Zhang, J.; Chen, Q.; Liu, X.; Zhao, Z.; Du, X.; et al. Macrophage-Targeted Nanomedicine for the Diagnosis and Treatment of Atherosclerosis. Nat. Rev. Cardiol. 2022, 19, 43–60. [Google Scholar] [CrossRef]
- Kanemaru, K.; Cranley, J.; Muraro, D.; Viola, M.F.; Webster, G.A.; Michael, N.; Guzzetta, M.; Ortalli, A.; Shorthouse, D.; Andrews, T.S.; et al. Spatially Resolved Multiomics of Human Cardiac Niches. Nature 2023, 619, 365–372. [Google Scholar] [CrossRef]
- Ruiz-Orera, J.; Latorre-Pellicer, A.; Albà, M.M. Evolution of translational control and the emergence of genes and open reading frames in human and non-human primate hearts. Nat. Cardiovasc. Res. 2024, 3, 105–120. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Chen, W.; Xu, S.; Cheng, Y.; Chen, Y.; Zhao, H.; Zhang, J.; Chen, X.; Zhang, Q.; et al. Self-Assembling Human Heart Organoids for the Modeling of Cardiac Development and Congenital Heart Disease. Nat. Commun. 2021, 12, 1044. [Google Scholar] [CrossRef]
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 2020, 3, 720. [Google Scholar] [CrossRef]
- Khouzam, J.P.S.; Tivakaran, V.S. CRISPR-Cas9 applications in cardiovascular disease. Curr. Probl. Cardiol. 2021, 46, 100652. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Tawakol, A. Imaging atherosclerosis with positron emission tomography. Eur. Heart J. 2016, 37, 2974–2980. [Google Scholar] [CrossRef]
- Ribas, J.; Sadeghi, H.; Manbachi, A.; Leijten, J.; Brinegar, K.; Zhang, Y.S.; Ferreira, L.; Khademhosseini, A. Cardiovascular organ-on-a-chip platforms for drug discovery and development. Appl. Vitr. Toxicol. 2016, 2, 82–96. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, T.; Guo, M.; Wang, Z.; Liu, K. Tissue-Resident Macrophages in Cardiovascular Diseases: Heterogeneity and Therapeutic Potential. Int. J. Mol. Sci. 2025, 26, 4524. https://doi.org/10.3390/ijms26104524
An T, Guo M, Wang Z, Liu K. Tissue-Resident Macrophages in Cardiovascular Diseases: Heterogeneity and Therapeutic Potential. International Journal of Molecular Sciences. 2025; 26(10):4524. https://doi.org/10.3390/ijms26104524
Chicago/Turabian StyleAn, Tianhui, Mengyuan Guo, Zhaohui Wang, and Kun Liu. 2025. "Tissue-Resident Macrophages in Cardiovascular Diseases: Heterogeneity and Therapeutic Potential" International Journal of Molecular Sciences 26, no. 10: 4524. https://doi.org/10.3390/ijms26104524
APA StyleAn, T., Guo, M., Wang, Z., & Liu, K. (2025). Tissue-Resident Macrophages in Cardiovascular Diseases: Heterogeneity and Therapeutic Potential. International Journal of Molecular Sciences, 26(10), 4524. https://doi.org/10.3390/ijms26104524