
Endoplasmic Reticulum Stress in Tuberculosis: Molecular Bases and Pathophysiological Implications in the Immunopathogenesis of the Disease

 , , and
, , and 
Abstract
1. Introduction
2. General Aspects
3. Mechanisms of Endoplasmic Reticulum Stress
4. Pathophysiology of Tb, Defense Mechanisms, and Relationship with Reticulum Stress
5. Immune Evasion Strategies Related to Reticulum Stress
6. Influence of ER on the Cell Death Process Against M. tuberculosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO (Ed.) Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019; 397p. Available online: https://www.who.int/publications/i/item/9789241565714 (accessed on 19 April 2023).
- Alves, A.C.F.P.B.; Prado, A.I.F.; Takenami, I. Imunologia da tuberculose: Uma revisão narrativa da literatura. Arq. Asma Alerg. Imunol. 2022, 6, 239–250. [Google Scholar] [CrossRef]
- de Oliveira, G.M.; Petroni, T.F. Avaliação de indicadores epidemiológicos da tuberculose no Brasil. Rev. Saúde UniToledo 2017, 1, 134–146. [Google Scholar]
- Silva, D.R.; Rabahi, M.F.; Sant’Anna, C.C.; da Silva-Junior, J.L.R.; Capone, D.; Bombarda, S.; de Miranda, S.S.; da Rocha, J.L.; Dalcolmo, M.M.P.; Rick, M.F.; et al. Consenso sobre o diagnóstico da tuberculose da Sociedade Brasileira de Pneumologia e Tisiologia. J. Bras. Pneumol. 2021, 47, e20210054. [Google Scholar] [CrossRef] [PubMed]
- Ministério da Saúde; Secretaria de Vigilância em Saúde; Departamento de Vigilância das Doenças Transmissíveis. Manual de Recomendações Para O Controle da Tuberculose No Brasil; MS: Brasília, Brazil, 2019. Available online: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/svsa/tuberculose/manual-de-recomendacoes-e-controle-da-ttuberculose-no-brasil-2a-ed.pdf/@@download/file (accessed on 19 April 2023).
- Matteelli, A.; Rendon, A.; Tiberi, S.; Al-Abri, S.; Voniatis, C.; Carvalho, A.C.C.; Centis, R.; D’Ambrosio, L.; Visca, D.; Spanevello, A.; et al. Tuberculosis elimination: Where are we now? Eur. Respir. Rev. 2018, 27, 180035. [Google Scholar] [CrossRef] [PubMed]
- WHO Global Tuberculosis Report. 2020. Available online: https://apps.who.int/iris/handle/10665/336069 (accessed on 19 April 2023).
- WHO Global Tuberculosis Report. 2021. Available online: https://apps.who.int/iris/handle/10665/346387 (accessed on 19 April 2023).
- Bertolozzi, M.R.; Takahashi, R.F.; França, F.O.S.; Hino, P. The incidence of tuberculosis and its relation to social inequalities: Integrative Review Study on PubMed Base. Esc. Anna Nery 2020, 24, e20180367. [Google Scholar] [CrossRef]
- de Paiva, J.P.S.; Magalhães, M.A.F.M.; Leal, T.C.; da Silva, L.F.; da Silva, L.G.; do Carmo, R.F.D.; de Souza, C.D.F. Time trend, social vulnerability, and identification of risk areas for tuberculosis in Brazil: An ecological study. PLoS ONE 2022, 17, e0247894. [Google Scholar] [CrossRef]
- Pedrazzoli, D.; Boccia, D.; Dodd, P.J.; Lönnroth, K.; Dowdy, D.W.; Siroka, A.; Kimerling, M.E.; White, R.G.; Houben, R.M.G.J. Modelling the social and structural determinants of tuberculosis: Opportunities and challenges. Int. J. Tuberc. Lung Dis. 2017, 21, 957–964. [Google Scholar] [CrossRef]
- Paiva, J.P.S.; Brito, A.B.; Bezerra-Santos, M.; Carmo, R.F.; Souza, C.D.F. Temporal trend of Tuberculosis incidence in northeastern Brazilian municipalities according to Social Vulnerability Index parameters: An ecological study. J. Bras. Pneumol. 2023, 49, e20220353. [Google Scholar] [CrossRef]
- Soeiro, V.M.D.S.; Vasconcelos, V.V.; Caldas, A.J.M. A comorbidade tuberculose-diabetes no Brasil, 2012–2018: Análise espacial exploratória e modelagem estatística. Rev. Panam. Salud. Publica. 2022, 46, e51. (In Portuguese) [Google Scholar] [CrossRef]
- Ministério da Saúde; Secretaria de Vigilância em Saúde; Departamento de Doenças de Condições Crônicas e Infecções Sexualmente Transmissíveis—DCCI. Boletim Epidemiológico de Tuberculose; MS: Brasília, Brazil, 2020. Available online: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/boletins/epidemiologicos/especiais/2021/boletim-tuberculose-2021_24.03 (accessed on 19 April 2023).
- Brasil. Ministério da Saúde. Boletim Epidemiológico Especial Tuberculose. 2021. Available online: https://www.gov.br/saude/pt-br/media/pdf/2021/marco/24/boletim-tuberculose-2021_24.03#:~:text=Em%202020%2C%20o%20Brasil%20registrou,óbitos%20por%20100%20mil%20habitantes (accessed on 19 April 2023).
- Dubé, J.Y.; Fava, V.M.; Schurr, E.; Behr, M.A. Underwhelming or Misunderstood? Genetic Variability of Pattern Recognition Receptors in Immune Responses and Resistance to Mycobacterium tuberculosis. Front. Immunol. 2021, 12, 714808. [Google Scholar] [CrossRef]
- Gupta, N.; Garg, S.; Vedi, S.; Kunimoto, D.Y.; Kumar, R.; Agrawal, B. Future Path Toward TB Vaccine Development: Boosting BCG or Re-educating by a New Subunit Vaccine. Front. Immunol. 2018, 9, 2371. [Google Scholar] [CrossRef] [PubMed]
- de Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front. Pediatr. 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Anton, C.; Machado, F.D.; Ramirez, J.M.; Bernardi, R.M.; Palominos, P.E.; Brenol, C.V.; Mello, F.C.D.Q.; Silva, D.R. Infecção latente por tuberculose em pacientes com doenças reumatológicas. J. Bras. Pneumol. 2019, 45, e20190023. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Naidu, G.; Sharma, V.; Jha, S.; Dhooria, A.; Dhir, V.; Bhatia, P.; Sharma, V.; Bhattad, S.; Chengappa, K.G.; et al. Deficiency of Adenosine Deaminase 2 in Adults and Children: Experience From India. Arthritis Rheumatol. 2021, 73, 276–285. [Google Scholar] [CrossRef]
- Williams, O.; Fatima, S. Granuloma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar] [PubMed]
- Rijnink, W.F.; Ottenhoff, T.H.M.; Joosten, S.A. B-Cells and Antibodies as Contributors to Effector Immune Responses in Tuberculosis. Front. Immunol. 2021, 12, 640168. [Google Scholar] [CrossRef]
- Namgaladze, D.; Khodzhaeva, V.; Brüne, B. ER-Mitochondria Communication in Cells of the Innate Immune System. Cells 2019, 8, 1088. [Google Scholar] [CrossRef]
- Galluzzi, L.; Diotallevi, A.; Magnani, M. Endoplasmic reticulum stress and unfolded protein response in infection by intracellular parasites. Future Sci. OA 2017, 3, FSO198. [Google Scholar] [CrossRef]
- Harada, M.; Takahashi, N.; Azhary, J.M.; Kunitomi, C.; Fujii, T.; Osuga, Y. Endoplasmic reticulum stress: A key regulator of the follicular microenvironment in the ovary. Mol. Hum. Reprod. 2021, 27, gaaa088. [Google Scholar] [CrossRef]
- Sharma, T.; Grover, S.; Arora, N.; P, M.; Ehtesham, N.Z.; Hasnain, S.E. PGRS Domain of Rv0297 of Mycobacterium tuberculosis Is Involved in Modulation of Macrophage Functions to Favor Bacterial Persistence. Front. Cell Infect. Microbiol. 2020, 10, 451. [Google Scholar] [CrossRef]
- Lee, K.I.; Choi, S.; Choi, H.G.; Gurmessa, S.K.; Dang, T.B.; Back, Y.W.; Park, H.; Kim, H. Recombinant Rv1654 protein of Mycobacterium tuberculosis induces mitochondria-mediated apoptosis in macrophage. Microbiol. Immunol. 2021, 65, 178–188. [Google Scholar] [CrossRef]
- Marchi, S.; Morroni, G.; Pinton, P.; Galluzzi, L. Control of host mitochondria by bacterial pathogens. Trends Microbiol. 2022, 30, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Zhang, Y.; Lei, Z.; Lu, Z.; Tan, S.; Ge, P.; Chai, Q.; Zhao, M.; Zhang, X.; Li, B.; et al. A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nat. Commun. 2023, 14, 1430. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wu, X.; Zhang, X.; Liu, X.; Deng, G. Heme oxygenase-1 modulates ferroptosis by fine-tuning levels of intracellular iron and reactive oxygen species of macrophages in response to Bacillus Calmette-Guerin infection. Front. Cell Infect. Microbiol. 2022, 12, 1004148. [Google Scholar] [CrossRef]
- Chen, X.Q.; Shen, T.; Fang, S.J.; Sun, X.M.; Li, G.Y.; Li, Y.F. Protein homeostasis in aging and cancer. Front. Cell Dev. Biol. 2023, 11, 1143532. [Google Scholar] [CrossRef]
- Merighi, A.; Lossi, L. Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death. Int. J. Mol. Sci. 2022, 23, 15186. [Google Scholar] [CrossRef]
- Moon, S.; Jung, H.S. Endoplasmic Reticulum Stress and Dysregulated Autophagy in Human Pancreatic Beta Cells. Diabetes Metab. J. 2022, 46, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.M.; Li, B.; He, X.N.; Guo, J.; Lei, X.; Jin, F.; Jin, Y. Endoplasmic Reticulum and Its Significance in Periodontal Disease. Chin. J. Dent. Res. 2021, 24, 79–84. [Google Scholar] [CrossRef]
- Yperman, K.; Kuijpers, M. Neuronal endoplasmic reticulum architecture and roles in axonal physiology. Mol. Cell Neurosci. 2023, 125, 103822. [Google Scholar] [CrossRef]
- Voronin, M.V.; Abramova, E.V.; Verbovaya, E.R.; Vakhitova, Y.V.; Seredenin, S.B. Chaperone-Dependent Mechanisms as a Pharmacological Target for Neuroprotection. Int. J. Mol. Sci. 2023, 24, 823. [Google Scholar] [CrossRef]
- Tedesco, B.; Vendredy, L.; Timmerman, V.; Poletti, A. The chaperone-assisted selective autophagy complex dynamics and dysfunctions. Autophagy 2023, 19, 1619–1641. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, K.; Jin, Y.; Sheng, X. Endoplasmic reticulum proteostasis control and gastric cancer. Cancer Lett. 2019, 449, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef]
- Lam, M.; Marsters, S.A.; Ashkenazi, A.; Walter, P. Misfolded proteins bind and activate death receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. Elife 2020, 9, e52291. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Liang, X.; Liu, J.; Liu, X.; Jin, Y.; Xu, M.; Han, Z.; Wang, K.; Zhang, C.; Zou, F.; Zhou, L. LINP1 represses unfolded protein response by directly inhibiting eIF2α phosphorylation to promote cutaneous squamous cell carcinoma. Exp. Hematol. Oncol. 2023, 12, 31. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Correia de Sousa, M.; Delangre, E.; Türkal, M.; Foti, M.; Gjorgjieva, M. Endoplasmic Reticulum Stress in Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 4914. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Shi, D.; Zhou, L.; Shi, H.; Zhang, J.; Zhang, J.; Zhang, L.; Liu, D.; Feng, T.; Zeng, M.; Chen, J.; et al. Autophagy is induced by swine acute diarrhea syndrome coronavirus through the cellular IRE1-JNK-Beclin 1 signaling pathway after an interaction of viral membrane-associated papain-like protease and GRP78. PLoS Pathog. 2023, 19, e1011201. [Google Scholar] [CrossRef]
- Chadwick, S.R.; Lajoie, P. Endoplasmic Reticulum Stress Coping Mechanisms and Lifespan Regulation in Health and Diseases. Front Cell Dev. Biol. 2019, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.M.; Jeon, Y.J. Proteostasis In The Endoplasmic Reticulum: Road to Cure. Cancers 2019, 11, 1793. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef]
- Yoo, Y.M.; Joo, S.S. Melatonin Can Modulate Neurodegenerative Diseases by Regulating Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2023, 24, 2381. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Song, N.J.; Riesenberg, B.P.; Li, Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2020, 10, 3154. [Google Scholar] [CrossRef]
- Choi, J.A.; Song, C.H. Insights Into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Front. Immunol. 2020, 10, 3147. [Google Scholar] [CrossRef]
- Amankwah, Y.S.; Collins, P.; Fleifil, Y.; Unruh, E.; Ruiz Márquez, K.J.; Vitou, K.; Kravats, A.N. Grp94 Works Upstream of BiP in Protein Remodeling Under Heat Stress. J. Mol. Biol. 2022, 434, 167762. [Google Scholar] [CrossRef]
- Thaxton, J.E.; Wallace, C.; Riesenberg, B.; Zhang, Y.; Paulos, C.M.; Beeson, C.C.; Liu, B.; Li, Z. Modulation of Endoplasmic Reticulum Stress Controls CD4+ T-cell Activation and Antitumor Function. Cancer Immunol. Res. 2017, 5, 666–675. [Google Scholar] [CrossRef]
- Jiang, Y.; Tao, Z.; Chen, H.; Xia, S. Endoplasmic Reticulum Quality Control in Immune Cells. Front. Cell Dev. Biol. 2021, 9, 740653. [Google Scholar] [CrossRef]
- Stadhouders, R.; Lubberts, E.; Hendriks, R.W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J. Autoimmun. 2018, 87, 1–15. [Google Scholar] [CrossRef]
- van Hamburg, J.P.; Tas, S.W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 2018, 87, 69–81. [Google Scholar] [CrossRef]
- Di Gioacchino, M.; Della Valle, L.; Allegra, A.; Pioggia, G.; Gangemi, S. AllergoOncology: Role of immune cells and immune proteins. Clin. Transl. Allergy 2022, 12, e12133. [Google Scholar] [CrossRef]
- So, J.S. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 2018, 41, 705–716. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Liao, Y.P.; Luo, L.; Xia, T.; Nel, A.E. Use of a Liver-Targeting Immune-Tolerogenic mRNA Lipid Nanoparticle Platform to Treat Peanut-Induced Anaphylaxis by Single- and Multiple-Epitope Nucleotide Sequence Delivery. ACS Nano 2023, 17, 4942–4957. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Chang, S.; Paton, A.W.; Paton, J.C.; Gabrilovich, D.I.; Ploegh, H.L.; Del Valle, J.R.; Hu, C.-C.A. Phosphorylation of IRE1 at S729 regulates RIDD in B cells and antibody production after immunization. J. Cell Biol. 2018, 217, 1739–1755. [Google Scholar] [CrossRef]
- Cairrão, F.; Santos, C.C.; Le Thomas, A.; Marsters, S.; Ashkenazi, A.; Domingos, P.M. Pumilio protects Xbp1 mRNA from regulated Ire1-dependent decay. Nat. Commun. 2022, 13, 1587. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Joe, Y.; Surh, Y.J.; Chung, H.T. Differential Regulation of Toll-Like Receptor-Mediated Cytokine Production by Unfolded Protein Response. Oxidative Med. Cell. Longev. 2018, 2018, 9827312. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Somma, D.; Kerrigan, D.; Herrington, F.; Keeshan, K.; Nibbs, R.J.B.; Carmody, R.J. The IκB-protein BCL-3 controls Toll-like receptor-induced MAPK activity by promoting TPL-2 degradation in the nucleus. Proc. Natl. Acad. Sci. USA 2019, 116, 25828–25838. [Google Scholar] [CrossRef]
- Sicari, D.; Delaunay-Moisan, A.; Combettes, L.; Chevet, E.; Igbaria, A. A guide to assessing endoplasmic reticulum homeostasis and stress in mammalian systems. FEBS J. 2020, 287, 27–42. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef] [PubMed]
- García-González, P.; Cabral-Miranda, F.; Hetz, C.; Osorio, F. Interplay Between the Unfolded Protein Response and Immune Function in the Development of Neurodegenerative Diseases. Front. Immunol. 2018, 9, 2541. [Google Scholar] [CrossRef]
- Di Conza, G.; Ho, P.C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef]
- Rico-Llanos, G.; Porras-Perales, Ó.; Escalante, S.; Vázquez-Calero, D.B.; Valiente, L.; Castillo, M.I.; Pérez-Tejeiro, J.M.; Baglietto-Vargas, D.; Becerra, J.; Reguera, J.M.; et al. Cellular stress modulates severity of the inflammatory response in lungs via cell surface BiP. Front. Immunol. 2022, 13, 1054962. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Yong, J.; Kaufman, R.J. The Impact of the ER Unfolded Protein Response on Cancer Initiation and Progression: Therapeutic Implications. Adv. Exp. Med. Biol. 2020, 1243, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liang, X.; Yu, D. Low expression of endoplasmic reticulum stress-related gene SERP1 is associated with poor prognosis and immune infiltration in skin cutaneous melanoma. Aging 2021, 13, 23036–23071. [Google Scholar] [CrossRef]
- Luhr, M.; Torgersen, M.L.; Szalai, P.; Hashim, A.; Brech, A.; Staerk, J.; Engedal, N. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J. Biol. Chem. 2019, 294, 8197–8217. [Google Scholar] [CrossRef]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-level inhibition of coronavirus replication by chemical ER stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Schmitz, M.L.; Ziebuhr, J.; Kracht, M. Reply to: The stress-inducible ER chaperone GRP78/BiP is upregulated during SARS-CoV-2 infection and acts as a pro-viral protein. Nat. Commun. 2022, 13, 6550. [Google Scholar] [CrossRef]
- Kusaczuk, M. Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells. 2019, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Zangerolamo, L.; Vettorazzi, J.F.; Rosa, L.R.O.; Carneiro, E.M.; Barbosa, H.C.L. The bile acid TUDCA and neurodegenerative disorders: An overview. Life Sci. 2021, 272, 119252. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Tornese, P.; Cocco, A.; Albanese, A. Tauroursodeoxycholic acid: A potential therapeutic tool in neurodegenerative diseases. Transl. Neurodegener. 2022, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Chaudhary, P.; Sandhir, R.; Bharadwaj, A.; Gupta, R.K.; Khatri, R.; Bajaj, A.C.; Baburaj, T.P.; Kumar, S.; Pal, M.S.; et al. Heat-induced endoplasmic reticulum stress in soleus and gastrocnemius muscles and differential response to UPR pathway in rats. Cell Stress Chaperones 2021, 26, 323–339. [Google Scholar] [CrossRef]
- Tamura, Y.; Matsunaga, Y.; Kitaoka, Y.; Hatta, H. Effects of Heat Stress Treatment on Age-dependent Unfolded Protein Response in Different Types of Skeletal Muscle. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2017, 72, 299–308. [Google Scholar] [CrossRef]
- Gómez-Sierra, T.; Jiménez-Uribe, A.P.; Ortega-Lozano, A.J.; Ramírez-Magaña, K.J.; Pedraza-Chaverri, J. Antioxidants affect endoplasmic reticulum stress-related diseases. Vitam Horm. 2023, 121, 169–196. [Google Scholar] [CrossRef]
- Cao, J.; Wang, C.; Hao, N.; Fujiwara, T.; Wu, T. Endoplasmic Reticulum Stress and Reactive Oxygen Species in Plants. Antioxidants 2022, 11, 1240. [Google Scholar] [CrossRef]
- Gansemer, E.R.; Rutkowski, D.T. Pathways Linking Nicotinamide Adenine Dinucleotide Phosphate Production to Endoplasmic Reticulum Protein Oxidation and Stress. Front. Mol. Biosci. 2022, 9, 858142. [Google Scholar] [CrossRef]
- Kumar, N.; Khan, N.; Cleveland, D.; Geiger, J.D. A common approach for fighting tuberculosis and leprosy: Controlling endoplasmic reticulum stress in myeloid-derived suppressor cells. Immunotherapy 2021, 13, 1555–1563. [Google Scholar] [CrossRef]
- Lou, X.; Gao, D.; Yang, L.; Wang, Y.; Hou, Y. Endoplasmic reticulum stress mediates the myeloid-derived immune suppression associated with cancer and infectious disease. J. Transl. Med. 2023, 21, 1. [Google Scholar] [CrossRef]
- Stanley, S.; Liu, Q.; Fortune, S.M. Mycobacterium tuberculosis functional genetic diversity, altered drug sensitivity, and precision medicine. Front. Cell Infect Microbiol. 2022, 12, 1007958. [Google Scholar] [CrossRef]
- Eoh, H.; Liu, R.; Lim, J.; Lee, J.J.; Sell, P. Central carbon metabolism remodeling as a mechanism to develop drug tolerance and drug resistance in Mycobacterium tuberculosis. Front. Cell Infect. Microbiol. 2022, 12, 958240. [Google Scholar] [CrossRef]
- Borah, P.; Deb, P.K.; Venugopala, K.N.; Al-Shar’i, N.A.; Singh, V.; Deka, S.; Srivastava, A.; Tiwari, V.; Mailavaram, R.P. Tuberculosis: An Update on Pathophysiology, Molecular Mechanisms of Drug Resistance, Newer Anti-TB Drugs, Treatment Regimens and Host- Directed Therapies. Curr. Top. Med. Chem. 2021, 21, 547–570. [Google Scholar] [CrossRef] [PubMed]
- Mengesha, D.; Manyazewal, T.; Woldeamanuel, Y. Five-year trend analysis of tuberculosis in Bahir Dar, Northwest Ethiopia, 2015–2019. Int. J. Mycobacteriology 2021, 10, 437–441. [Google Scholar] [CrossRef]
- Maison, D.P. Tuberculosis pathophysiology and anti-VEGF intervention. J. Clin. Tuberc. Other Mycobact. Dis. 2022, 27, 100300. [Google Scholar] [CrossRef]
- Harding, J.S.; Herbath, M.; Chen, Y.; Rayasam, A.; Ritter, A.; Csoka, B.; Hasko, G.; Michael, I.P.; Fabry, Z.; Nagy, A.; et al. VEGF-A from Granuloma Macrophages Regulates Granulomatous Inflammation by a Non-angiogenic Pathway during Mycobacterial Infection. Cell Rep. 2019, 27, 2119–2131.e6. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Rani, A.; Alam, A.; Zarin, S.; Pandey, S.; Singh, H.; Hasnain, S.E.; Ehtesham, N.Z. Macrophage: A Cell With Many Faces and Functions in Tuberculosis. Front. Immunol. 2022, 13, 747799. [Google Scholar] [CrossRef] [PubMed]
- Shim, D.; Kim, H.; Shin, S.J. Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tuberculosis Control as Targets for Host-Directed Therapy. Front. Immunol. 2020, 11, 910. [Google Scholar] [CrossRef]
- Bussi, C.; Gutierrez, M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019, 43, 341–361. [Google Scholar] [CrossRef]
- Chandra, P.; Grigsby, S.J.; Philips, J.A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2022, 20, 750–766. [Google Scholar] [CrossRef]
- Dabla, A.; Liang, Y.C.; Rajabalee, N.; Irwin, C.; Moonen, C.G.J.; Willis, J.V.; Berton, S.; Sun, J. TREM2 Promotes Immune Evasion by Mycobacterium tuberculosis in Human Macrophages. mBio 2022, 13, e0145622. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, M.V.S.; Arnett, E.; Azad, A.K.; Guirado, E.; Ni, B.; Gerberick, A.D.; He, L.-Z.; Keler, T.; Thomas, L.J.; Lafuse, W.P.; et al. M. tuberculosis-Initiated Human Mannose Receptor Signaling Regulates Macrophage Recognition and Vesicle Trafficking by FcRγ-Chain, Grb2, and SHP-1. Cell Rep. 2017, 21, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449. [Google Scholar] [CrossRef]
- Maulahela, H.; Simadibrata, M.; Nelwan, E.J.; Rahadiani, N.; Renesteen, E.; Suwarti, S.W.T.; Anggraini, Y.W. Recent advances in the diagnosis of intestinal tuberculosis. BMC Gastroenterol. 2022, 22, 89. [Google Scholar] [CrossRef] [PubMed]
- Araújo, T.; Silva, A.; Laranjo, P.; Shigaeva, Y.; Bernardo, T. Delayed Diagnosis of Intestinal Tuberculosis: A Case Report. Cureus 2022, 14, e30600. [Google Scholar] [CrossRef]
- Yang, Q.; Han, J.; Shen, J.; Peng, X.; Zhou, L.; Yin, X. Diagnosis and treatment of tuberculosis in adults with HIV. Medicine 2022, 101, e30405. [Google Scholar] [CrossRef]
- Bell, L.C.K.; Noursadeghi, M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat. Rev. Microbiol. 2018, 16, 80–90. [Google Scholar] [CrossRef]
- Sia, J.K.; Rengarajan, J. Immunology of Mycobacterium tuberculosis Infections. Microbiol. Spectr. 2019, 7, 10–1128. [Google Scholar] [CrossRef]
- Ferluga, J.; Yasmin, H.; Al-Ahdal, M.N.; Bhakta, S.; Kishore, U. Natural and trained innate immunity against Mycobacterium tuberculosis. Immunobiology 2020, 225, 151951. [Google Scholar] [CrossRef]
- Cui, Y.; Zhao, D.; Barrow, P.A.; Zhou, X. The endoplasmic reticulum stress response: A link with tuberculosis? Tuberculosis 2016, 97, 52–56. [Google Scholar] [CrossRef]
- Lim, Y.J.; Yi, M.H.; Choi, J.A.; Lee, J.; Han, J.Y.; Jo, S.H.; Oh, S.M.; Cho, H.J.; Kim, D.W.; Kang, M.W.; et al. Roles of endoplasmic reticulum stress-mediated apoptosis in M1-polarized macrophages during mycobacterial infections. Sci. Rep. 2016, 6, 37211. [Google Scholar] [CrossRef] [PubMed]
- de Waal, A.M.; Hiemstra, P.S.; Ottenhoff, T.H.; Joosten, S.A.; van der Does, A.M. Lung epithelial cells interact with immune cells and bacteria to shape the microenvironment in tuberculosis. Thorax 2022, 77, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Miramontes, C.V.; Rodríguez-Carlos, A.; Marin-Luévano, S.P.; Trejo Martínez, L.A.; de Haro Acosta, J.; Enciso-Moreno, J.Á.; Rivas-Santiago, B. Nicotine promotes the intracellular growth of Mycobacterium tuberculosis in epithelial cells. Tuberculosis 2021, 127, 102026. [Google Scholar] [CrossRef]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef] [PubMed]
- Kireev, F.D.; Lopatnikova, J.A.; Laushkina, Z.A.; Sennikov, S.V. Autoantibodies to Tumor Necrosis Factor in Patients with Active Pulmonary Tuberculosis. Front. Biosci. 2022, 27, 133. [Google Scholar] [CrossRef]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef]
- Reichmann, M.T.; Tezera, L.B.; Vallejo, A.F.; Vukmirovic, M.; Xiao, R.; Reynolds, J.; Jogai, S.; Wilson, S.; Marshall, B.; Jones, M.G.; et al. Integrated transcriptomic analysis of human tuberculosis granulomas and a biomimetic model identifies therapeutic targets. J. Clin. Investig. 2021, 131, e148136. [Google Scholar] [CrossRef]
- Elkington, P.; Polak, M.E.; Reichmann, M.T.; Leslie, A. Understanding the tuberculosis granuloma: The matrix revolutions. Trends Mol. Med. 2022, 28, 143–154. [Google Scholar] [CrossRef]
- Xu, P.; Tang, J.; He, Z.G. Induction of Endoplasmic Reticulum Stress by CdhM Mediates Apoptosis of Macrophage During Mycobacterium tuberculosis Infection. Front. Cell Infect. Microbiol. 2022, 12, 877265. [Google Scholar] [CrossRef]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016, 4, 4–5. [Google Scholar] [CrossRef]
- López Ramírez, G.M.; Rom, W.N.; Ciotoli, C.; Talbot, A.; Martiniuk, F.; Cronstein, B.; Reibman, J. Mycobacterium tuberculosis alters expression of adhesion molecules on monocytic cells. Infect Immun. 1994, 62, 2515–2520. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Kim, M.J.; Blumenthal, A.; Koo, J.; Ehrt, S.; Wainwright, H.; Bekker, L.-G.; Kaplan, G.; Nathan, C.; Tabas, I.; et al. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE 2010, 5, e12772. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, Y.; Liu, S.; Zhang, X.L. PE_PGRS: Vital proteins in promoting mycobacterial survival and modulating host immunity and metabolism. Cell Microbiol. 2021, 23, e13290. [Google Scholar] [CrossRef]
- Bosserman, R.E.; Nicholson, K.R.; Champion, M.M.; Champion, P.A. A New ESX-1 Substrate in Mycobacterium marinum That Is Required for Hemolysis but Not Host Cell Lysis. J. Bacteriol. 2019, 201, e00760-18. [Google Scholar] [CrossRef]
- Guo, Q.; Bi, J.; Wang, H.; Zhang, X. Mycobacterium tuberculosis ESX-1-secreted substrate protein EspC promotes mycobacterial survival through endoplasmic reticulum stress-mediated apoptosis. Emerg. Microbes Infect. 2021, 10, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wang, X.; Yi, L.; Yang, B.; Wei, P.; Ruan, H.; Wang, J.; Yang, X.; Zhang, H. Enhanced Serum IgG Detection Potential Using 38KD-MPT32-MPT64, CFP10-Mtb81-EspC Fusion Protein and Lipoarabinomannan (LAM) for Human Tuberculosis. Pathogens 2022, 11, 1545. [Google Scholar] [CrossRef] [PubMed]
- Mertaniasih, N.M.; Surya Suameitria Dewi, D.N.; Soedarsono, S.; Kurniati, A.; Rohman, A.; Nuha, Z.; Matsumoto, S. The espD full gene as a potential biomarker in active pulmonary tuberculosis. Int. J. Mycobacteriology 2021, 10, 421–427. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Y.; Sun, J.; Shen, J.; Liu, F.; Ning, B.; Lu, Z.; Wei, L.; Jiang, X. Tanshinone IIA alleviates NLRP3 inflammasome-mediated pyroptosis in Mycobacterium tuberculosis-(H37Ra-) infected macrophages by inhibiting endoplasmic reticulum stress. J. Ethnopharmacol. 2022, 282, 114595. [Google Scholar] [CrossRef]
- Feng, Y.; Li, M.; Yangzhong, X.; Zhang, X.; Zu, A.; Hou, Y.; Li, L.; Sun, S. Pyroptosis in inflammation-related respiratory disease. J. Physiol. Biochem. 2022, 78, 721–737. [Google Scholar] [CrossRef]
- Li, L.; Abudureheman, Z.; Zhong, X.; Gao, L.; Gong, H.; He, C.; Yang, B.; Ren, J.; Alimu, A.; Yilamujiang, S.; et al. Astaxanthin Prevents Tuberculosis-Associated Inflammatory Injury by Inhibiting the Caspase 4/11-Gasdermin-Pyroptosis Pathway. Evid Based Complement. Altern. Med. 2022, 2022, 4778976. [Google Scholar] [CrossRef]
- Chirakos, A.E.; Balaram, A.; Conrad, W.; Champion, P.A. Modeling Tubercular ESX-1 Secretion Using Mycobacterium marinum. Microbiol. Mol. Biol. Rev. 2020, 84, e00082-19. [Google Scholar] [CrossRef]
- Naeem, M.A.; Ahmad, W.; Tyagi, R.; Akram, Q.; Younus, M.; Liu, X. Stealth Strategies of Mycobacterium tuberculosis for Immune Evasion. Curr. Issues Mol. Biol. 2021, 41, 597–616. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.D. Mechanisms of M. tuberculosis Immune Evasion as Challenges to TB Vaccine Design. Cell Host Microbe 2018, 24, 34–42. [Google Scholar] [CrossRef]
- Jagannath, C.; McBride, J.W.; Vergne, I. Editorial: The Autophagy Pathway: Bacterial Pathogen Immunity and Evasion. Front. Immunol. 2021, 12, 768935. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Tiwari, M.; Tiwari, V. Molecular mechanisms of bacteria induced autophagy and its escape strategies. Future Microbiol. 2020, 15, 303–306. [Google Scholar] [CrossRef]
- Siqueira, M.D.S.; Ribeiro, R.M.; Travassos, L.H. Autophagy and Its Interaction With Intracellular Bacterial Pathogens. Front. Immunol. 2018, 9, 935. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Divangahi, M. Targeting immunometabolism in host defence against Mycobacterium tuberculosis. Immunology 2021, 162, 145–159. [Google Scholar] [CrossRef]
- Dos Santos, C.C.; Walburg, K.V.; van Veen, S.; Wilson, L.G.; Trufen, C.E.M.; Nascimento, I.P.; Ottenhoff, T.H.M.; Leite, L.C.C.; Haks, M.C. Recombinant BCG-LTAK63 Vaccine Candidate for Tuberculosis Induces an Inflammatory Profile in Human Macrophages. Vaccines 2022, 10, 831. [Google Scholar] [CrossRef]
- Lam, A.; Prabhu, R.; Gross, C.M.; Riesenberg, L.A.; Singh, V.; Aggarwal, S. Role of apoptosis and autophagy in tuberculosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L218–L229. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, A.; Rashid, M.; Amnekar, R.V.; Gupta, S.; Kaur, J. A Phagosomally Expressed Gene, rv0428c, of Mycobacterium tuberculosis Demonstrates Acetyl Transferase Activity and Plays a Protective Role Under Stress Conditions. Protein J. 2022, 41, 260–273. [Google Scholar] [CrossRef]
- Ramon-Luing, L.A.; Olvera, Y.; Flores-Gonzalez, J.; Palacios, Y.; Carranza, C.; Aguilar-Duran, Y.; Vargas, M.A.; Gutierrez, N.; Medina-Quero, K.; Chavez-Galan, L. Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens 2022, 11, 492. [Google Scholar] [CrossRef] [PubMed]
- Loxton, A.G.; van Rensburg, I.C. FasL regulatory B-cells during Mycobacterium tuberculosis infection and TB disease. J. Mol. Biol. 2021, 433, 166984. [Google Scholar] [CrossRef] [PubMed]
- Loxton, A.G. Bcells and their regulatory functions during Tuberculosis: Latency and active disease. Mol. Immunol. 2019, 111, 145–151. [Google Scholar] [CrossRef]
- Han, L.; Lu, Y.; Wang, X.; Zhang, S.; Wang, Y.; Wu, F.; Zhang, W.; Wang, X.; Zhang, L. Regulatory role and mechanism of the inhibition of the Mcl-1 pathway during apoptosis and polarization of H37Rv-infected macrophages. Medicine 2020, 99, e22438. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.; Pattnaik, K.; Biswas, M.; Jagadeb, M.; Behera, A.; Sonawane, A. Mycobacterium tuberculosis LprE Suppresses TLR2-Dependent Cathelicidin and Autophagy Expression to Enhance Bacterial Survival in Macrophages. J. Immunol. 2019, 203, 2665–2678. [Google Scholar] [CrossRef]
- Le, Y.; Cao, W.; Zhou, L.; Fan, X.; Liu, Q.; Liu, F.; Gai, X.; Chang, C.; Xiong, J.; Rao, Y.; et al. Infection of Mycobacterium tuberculosis Promotes Both M1/M2 Polarization and MMP Production in Cigarette Smoke-Exposed Macrophages. Front. Immunol. 2020, 11, 1902. [Google Scholar] [CrossRef]
- Deng, S.; Shen, S.; Liu, K.; El-Ashram, S.; Alouffi, A.; Cenci-Goga, B.T.; Ye, G.; Cao, C.; Luo, T.; Zhang, H.; et al. Integrated bioinformatic analyses investigate macrophage-M1-related biomarkers and tuberculosis therapeutic drugs. Front. Genet. 2023, 14, 1041892. [Google Scholar] [CrossRef]
- Nguyen, T.K.; D’Aigle, J.; Chinea, L.; Niaz, Z.; Hunter, R.L.; Hwang, S.-A.; Actor, J.K. Mycobacterial Trehalose 6,6′-Dimycolate–Induced M1-Type Inflammation. Am. J. Pathol. 2020, 190, 286–294. [Google Scholar] [CrossRef]
- Nguyen, T.K.T.; Niaz, Z.; d’Aigle, J.; Hwang, S.A.; Kruzel, M.L.; Actor, J.K. Lactoferrin reduces mycobacterial M1-type inflammation induced with trehalose 6,6’-dimycolate and facilitates the entry of fluoroquinolone into granulomas. Biochem. Cell Biol. 2021, 99, 73–80. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Liu, Q.; Long, R.; Feng, J.; Qin, H.; Li, M.; Liu, L.; Luo, J. Mycobacterium tuberculosis Heat-Shock Protein 16.3 Induces Macrophage M2 Polarization Through CCRL2/CX3CR1. Inflammation 2020, 43, 487–506. [Google Scholar] [CrossRef]

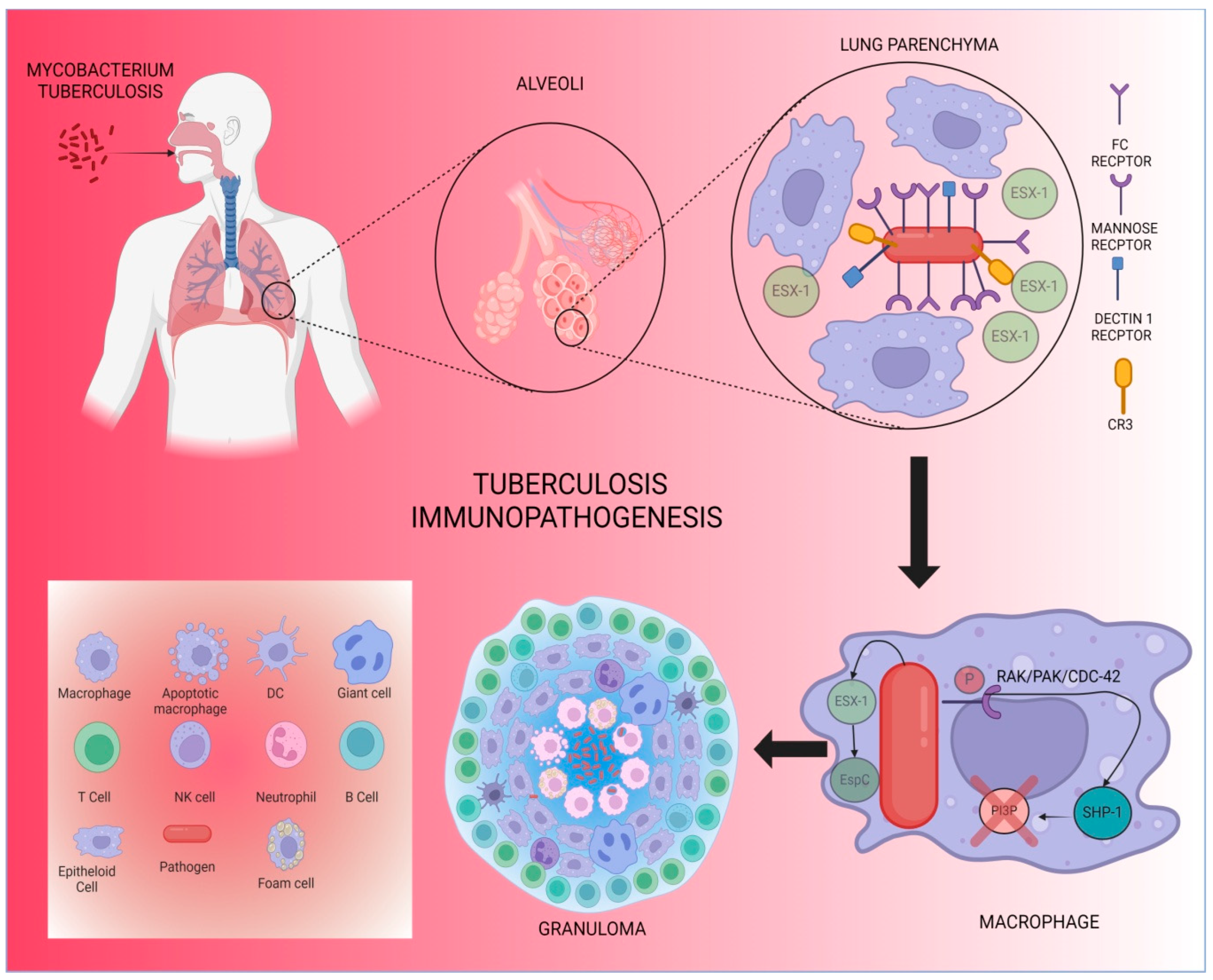
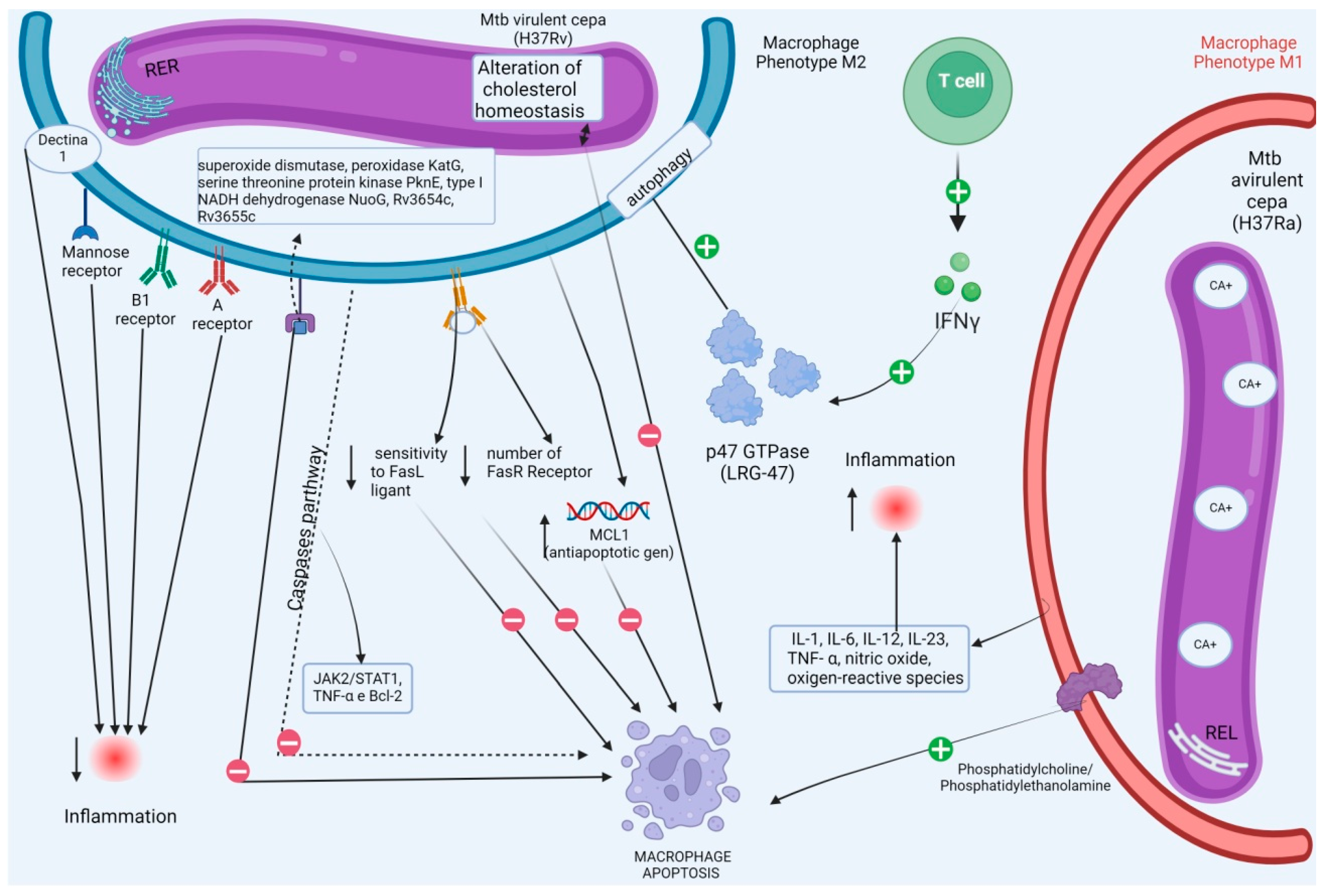

{kind=link}
{kind=link}
{kind=link}
| Aspects | Immunoprotective Effects of ER Stress | Immunopathogenic Effects of ER Stress | References |
|---|---|---|---|
| Immune signaling and cytokine activation | Infection by M. tuberculosis induces initial ER stress, activating sensors such as IRE1α, PERK, and ATF6, which promote the expression of IL-6, TNF-α, and IFN-γ via NF-kB and XBP1s, strengthening the TH1 response and M1 macrophage activation, essential for restricting bacterial replication in the early stages of infection. | The persistence of M. tuberculosis in the intracellular environment, by chronically deregulating the UPR pathway, tends to favor immunosuppression and induce an imbalance in Th1/Th2 responses, contributing to increased bacterial burden. | Cui et al., 2016 [107] Lim et al., 2016 [108] |
| Intracellular bacterial control | ESAT-6 activates the endoplasmic reticulum (ER) stress pathway, leading to the expression of key proteins such as GRP78, CHOP, and phosphorylation of eIF2α. The induction of these proteins is related to activation of the eIF2α/ATF4/CHOP pathway, resulting in increased Ca2+ concentration and ROS production, contributing to the microbicidal response. | One immune evasion strategy involves ER stress induction by CdhM, which may facilitate the release of M. tuberculosis from macrophages, promoting infection dissemination and prolonging its intracellular survival. | Xu et al., 2022 [116] Choi et al., 2010 [53] |
| Granuloma formation and stability | Cellular stress induced by M. tuberculosis infection leads to the coordinated release of chemokines (CCL2, CXCL10) and adhesion molecules (ICAM-1), favoring cell recruitment and the formation of structured granulomas that contain the infection. | M. tuberculosis can exploit chronic ER stress to induce the expression of CHOP/GADD153/Ire1α/ATF3 and promote uncontrolled cell death, destabilizing the granuloma, favoring its rupture, and facilitating bacillary dissemination. | Domingo-Gonzalez et al., 2016 [117] López Ramírez et al., 1994 [118] Seimon et al., 2010 [119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, J.; Martins, L.C.; Moura, J.; Pereira, A.; Vasconcelos, B.; Ferro, G.; Vasconcelos, P.; Quaresma, J. Endoplasmic Reticulum Stress in Tuberculosis: Molecular Bases and Pathophysiological Implications in the Immunopathogenesis of the Disease. Int. J. Mol. Sci. 2025, 26, 4522. https://doi.org/10.3390/ijms26104522
Sousa J, Martins LC, Moura J, Pereira A, Vasconcelos B, Ferro G, Vasconcelos P, Quaresma J. Endoplasmic Reticulum Stress in Tuberculosis: Molecular Bases and Pathophysiological Implications in the Immunopathogenesis of the Disease. International Journal of Molecular Sciences. 2025; 26(10):4522. https://doi.org/10.3390/ijms26104522
Chicago/Turabian StyleSousa, Jorge, Lívia Caricio Martins, Julia Moura, Amanda Pereira, Bárbara Vasconcelos, Gustavo Ferro, Pedro Vasconcelos, and Juarez Quaresma. 2025. "Endoplasmic Reticulum Stress in Tuberculosis: Molecular Bases and Pathophysiological Implications in the Immunopathogenesis of the Disease" International Journal of Molecular Sciences 26, no. 10: 4522. https://doi.org/10.3390/ijms26104522
APA StyleSousa, J., Martins, L. C., Moura, J., Pereira, A., Vasconcelos, B., Ferro, G., Vasconcelos, P., & Quaresma, J. (2025). Endoplasmic Reticulum Stress in Tuberculosis: Molecular Bases and Pathophysiological Implications in the Immunopathogenesis of the Disease. International Journal of Molecular Sciences, 26(10), 4522. https://doi.org/10.3390/ijms26104522