Molecular Dynamics-Guided Repositioning of FDA-Approved Drugs for PD-L1 Inhibition with In Vitro Anticancer Potential


Abstract
1. Introduction
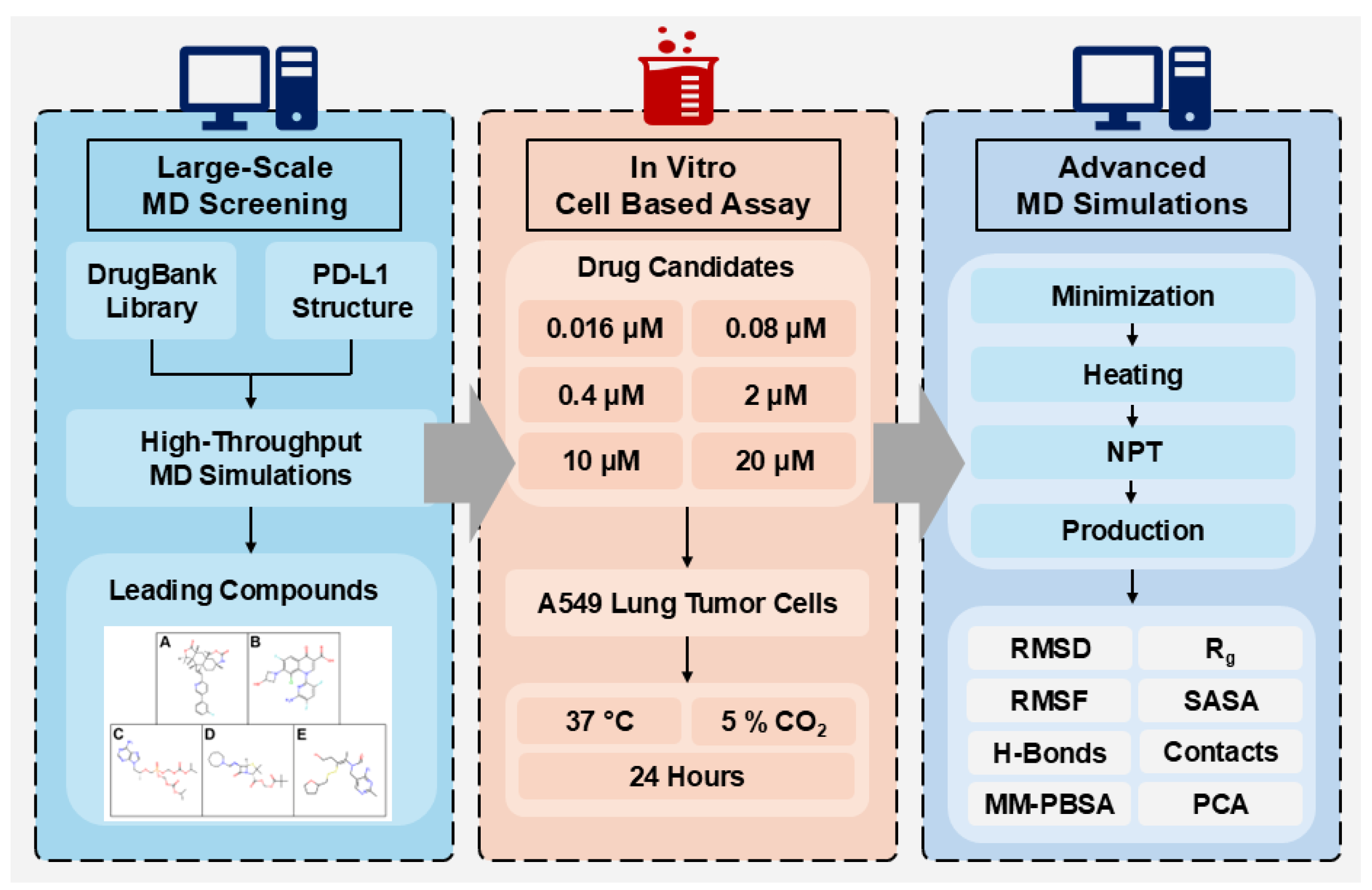
2. Results and Discussions
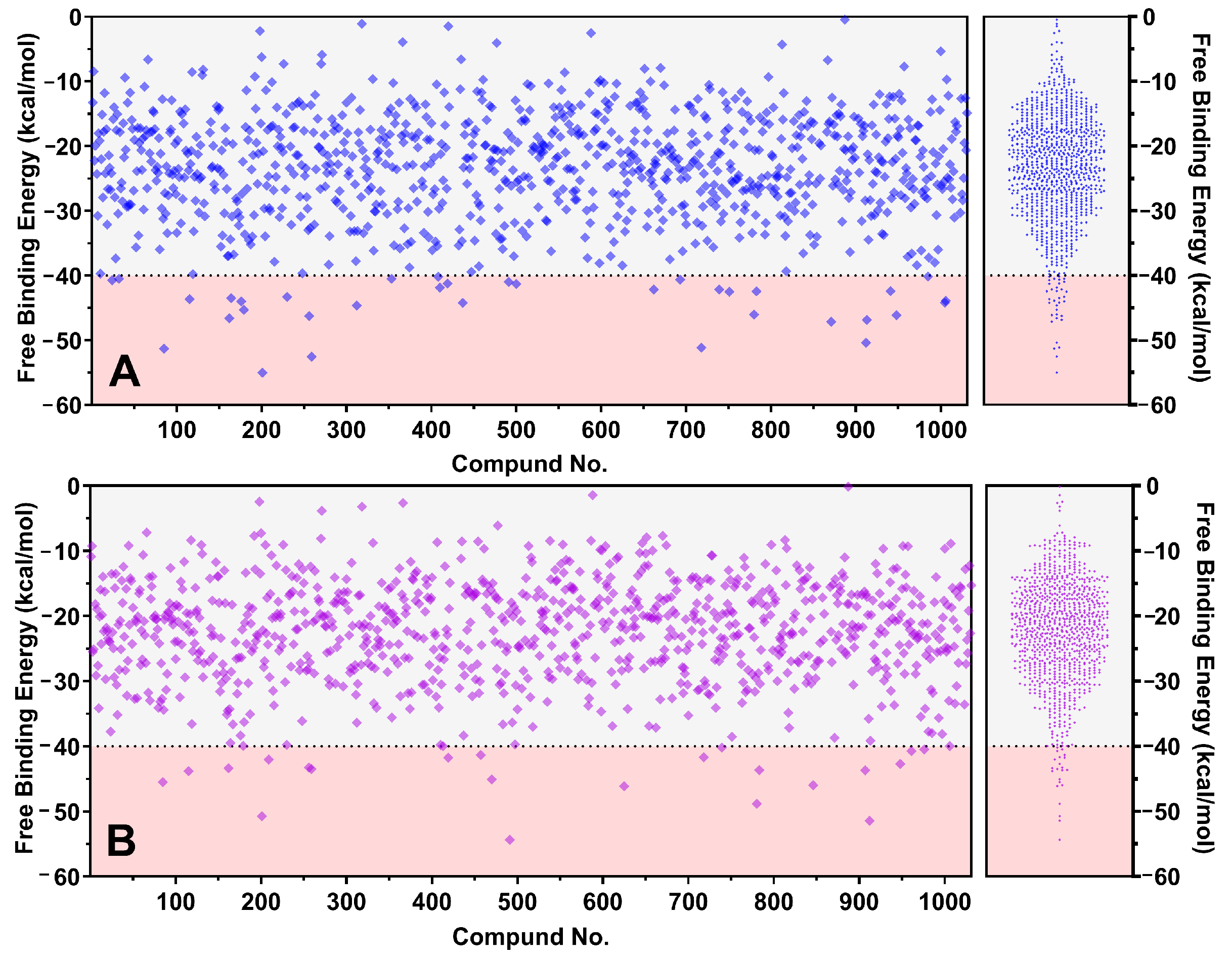
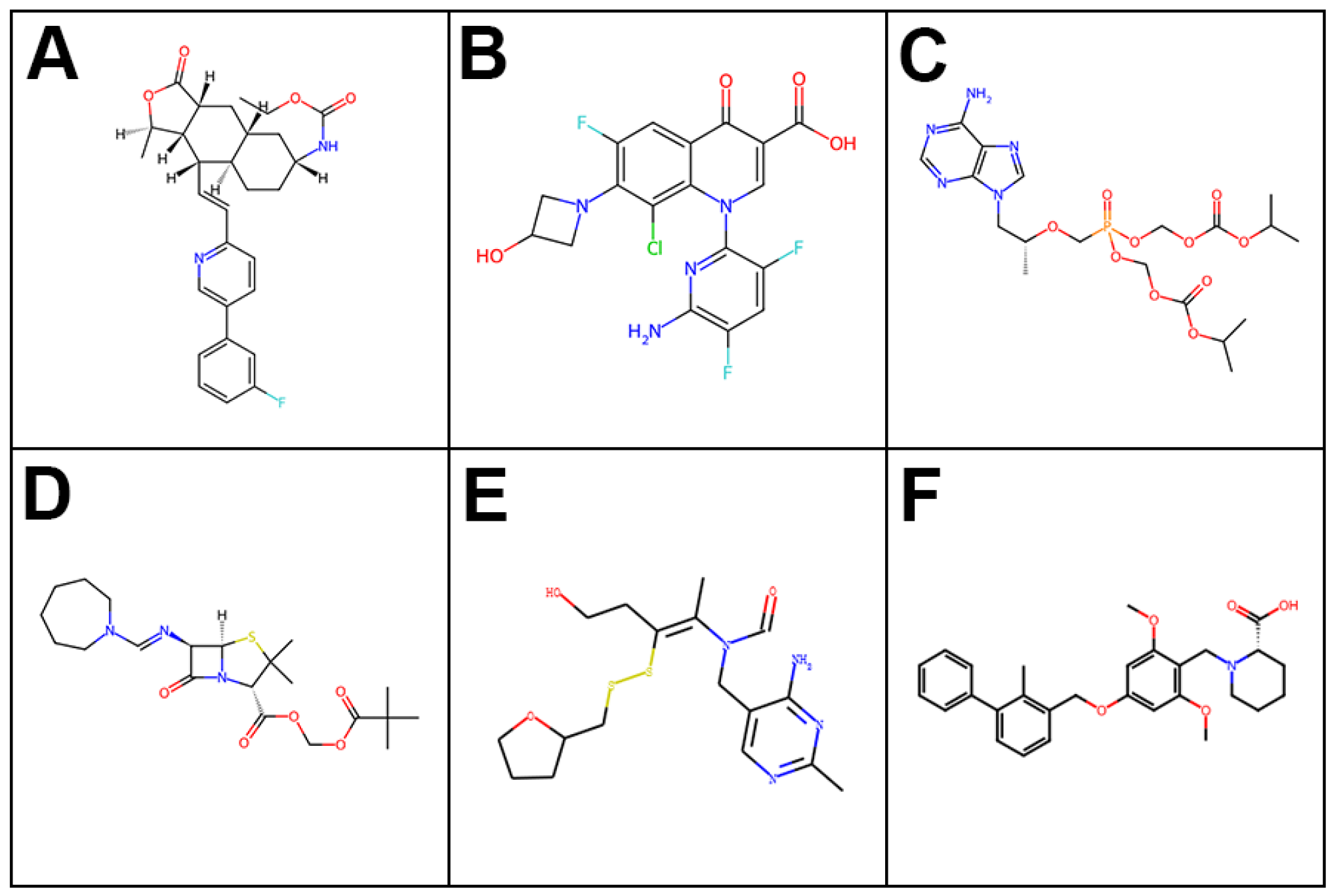
2.1. Large-Scale Molecular Dynamics Screening
2.2. MTT Cell Assays for Leading FDA-Approved Drugs
2.3. Post-MD Analysis of Leading Candidates
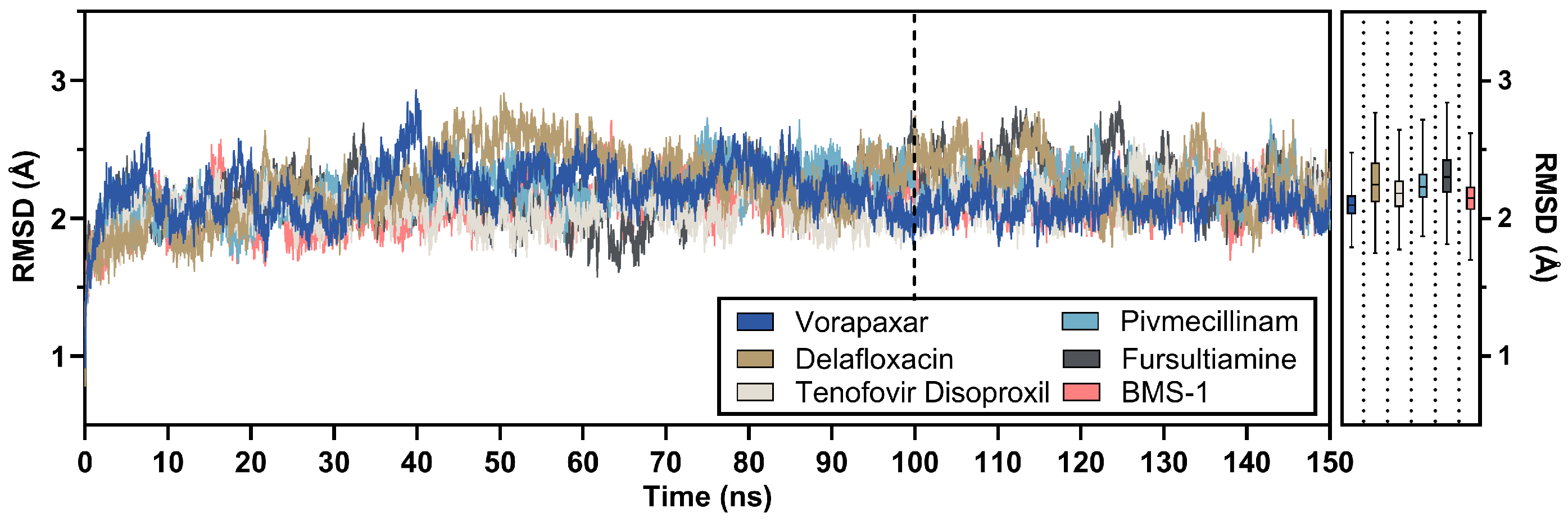
2.3.1. RMSD Analysis
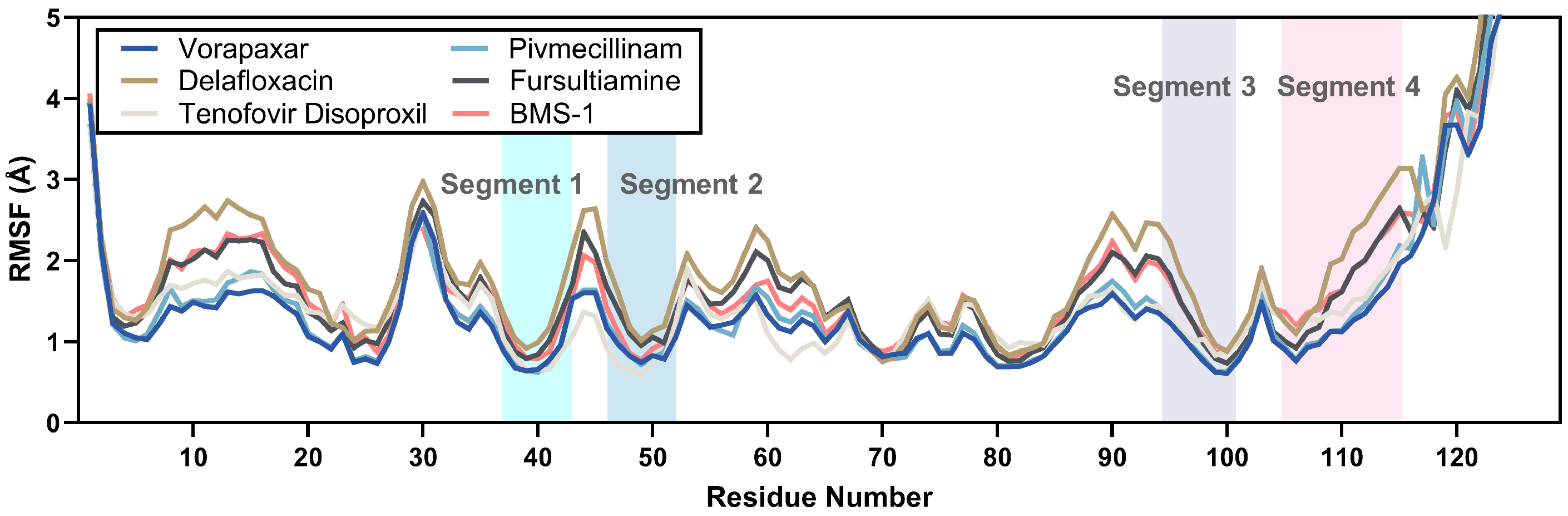
2.3.2. RMSF Analysis
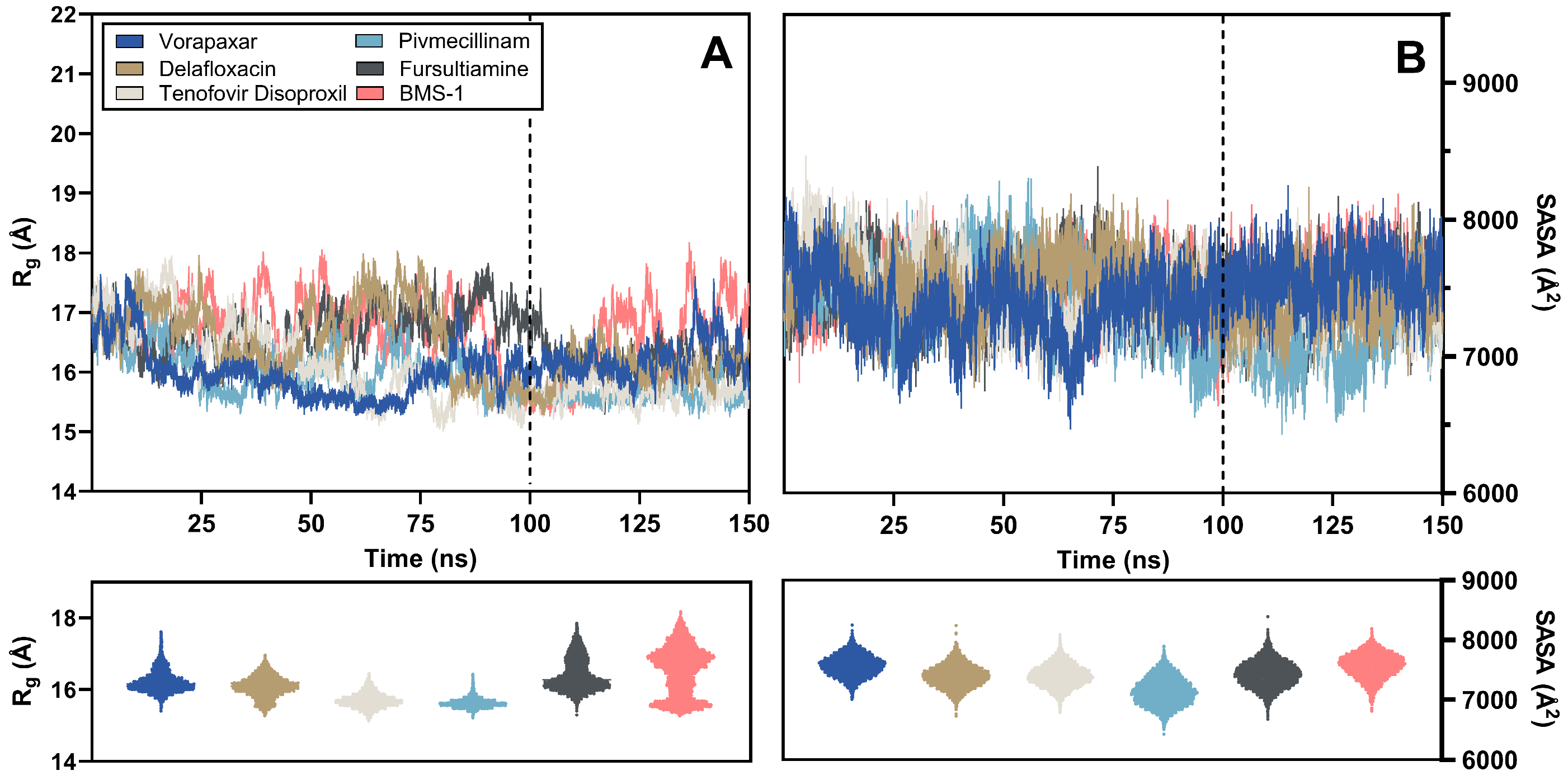
2.3.3. Rg and SASA Analysis
2.3.4. Hydrogen Bonds Analysis
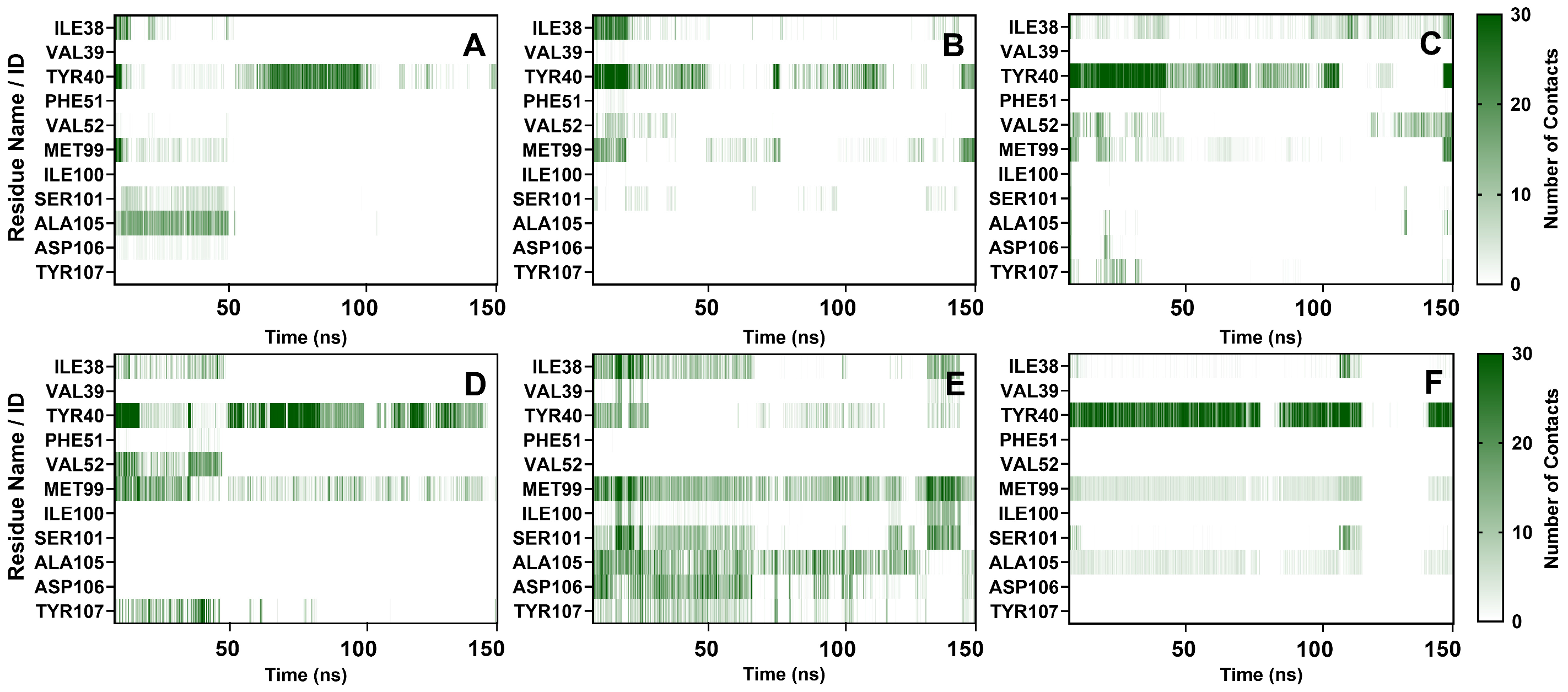
2.3.5. Contact Numbers Analysis
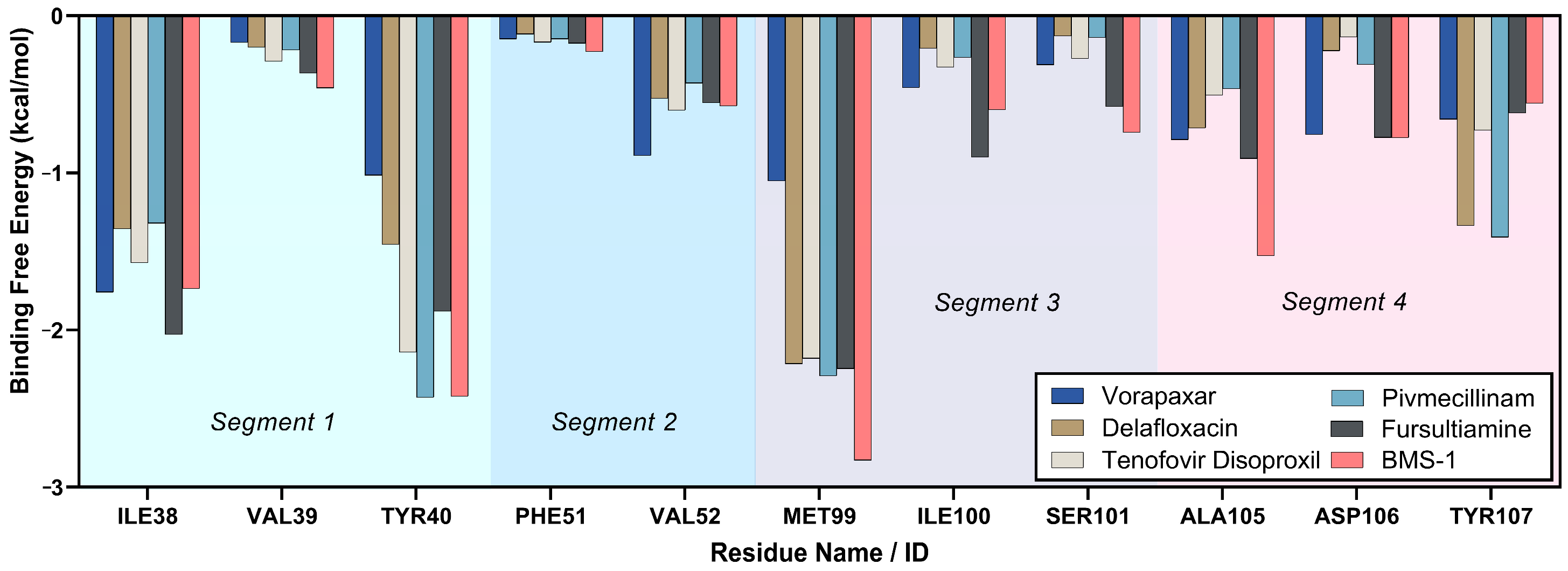
2.3.6. Binding Free Energy Analysis
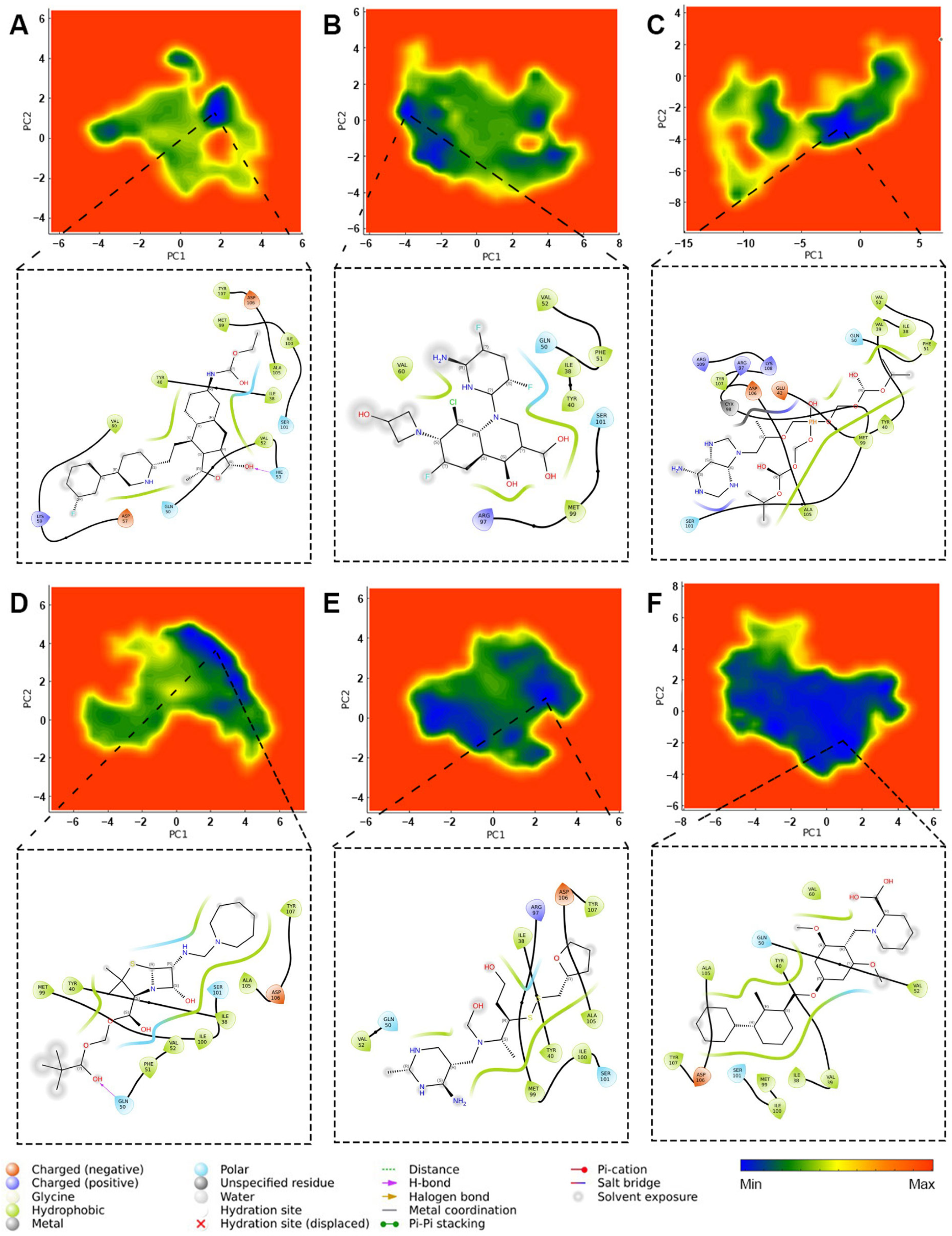
2.3.7. Principal Component Analysis
2.4. Lead Selection and Reference Comparison
3. Materials and Methods
3.1. Protein and Drug Structure Preparation
3.2. Large-Scale Pre-Screening Methodology
3.3. Reagents and Cell Culture
3.4. MTT Assay Methodology
3.5. Advanced Long-Term MD Simulations
3.6. Binding Free Energy Calculations
3.7. Principal Component Analysis Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef]
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination Strategies with PD-1/PD-L1 Blockade: Current Advances and Future Directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50%. JCO 2021, 39, 2339–2349. [Google Scholar] [CrossRef]
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer with PD-L1 of at Least 50%: A Multicentre, Open-Label, Global, Phase 3, Randomised, Controlled Trial. Lancet 2021, 397, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated Overall Survival and PD-L1 Subgroup Analysis of Patients With Extensive-Stage Small-Cell Lung Cancer Treated With Atezolizumab, Carboplatin, and Etoposide (IMpower133). JCO 2021, 39, 619–630. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Herbst, R.S.; Giaccone, G.; De Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1–Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. JCO 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Wang, T.-W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1–PD-1 Improves Senescence Surveillance and Ageing Phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef]
- Avram, S.; Bologa, C.G.; Holmes, J.; Bocci, G.; Wilson, T.B.; Nguyen, D.-T.; Curpan, R.; Halip, L.; Bora, A.; Yang, J.J.; et al. DrugCentral 2021 Supports Drug Discovery and Repositioning. Nucleic Acids Res. 2021, 49, D1160–D1169. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Lu, C.; Xu, J.; Meng, Y.; Wang, P.; Fu, X.; Zeng, X.; Su, Y. Drug Repositioning Based on the Heterogeneous Information Fusion Graph Convolutional Network. Brief. Bioinform. 2021, 22, bbab319. [Google Scholar] [CrossRef]
- Hua, Y.; Dai, X.; Xu, Y.; Xing, G.; Liu, H.; Lu, T.; Chen, Y.; Zhang, Y. Drug Repositioning: Progress and Challenges in Drug Discovery for Various Diseases. Eur. J. Med. Chem. 2022, 234, 114239. [Google Scholar] [CrossRef]
- He, B.; Hou, F.; Ren, C.; Bing, P.; Xiao, X. A Review of Current In Silico Methods for Repositioning Drugs and Chemical Compounds. Front. Oncol. 2021, 11, 711225. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wu, G.; Zhao, Q.; Li, Y.; Wang, J. Computational Drug Repositioning Based on Multi-Similarities Bilinear Matrix Factorization. Brief. Bioinform. 2021, 22, bbaa267. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Meng, Y.; Peng, L.; Cai, L.; Tang, X.; Liang, Y.; Tian, G.; Yang, J. Computational Drug Repositioning Using Similarity Constrained Weight Regularization Matrix Factorization: A Case of COVID-19. J. Cell. Mol. Medi 2022, 26, 3772–3782. [Google Scholar] [CrossRef]
- Mahgoub, M.A.; Alnaem, A.; Fadlelmola, M.; Abo-idris, M.; Makki, A.A.; Abdelgadir, A.A.; Alzain, A.A. Discovery of Novel Potential Inhibitors of TMPRSS2 and Mpro of SARS-CoV-2 Using E-Pharmacophore and Docking-Based Virtual Screening Combined with Molecular Dynamic and Quantum Mechanics. J. Biomol. Struct. Dyn. 2023, 41, 6775–6788. [Google Scholar] [CrossRef]
- Cava, C.; Bertoli, G.; Castiglioni, I. Potential Drugs Against COVID-19 Revealed By Gene Expression Profile, Molecular Docking and Molecular Dynamic Simulation. Future Virol. 2021, 16, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Azizian, H.; Pedrood, K.; Moazzam, A.; Valizadeh, Y.; Khavaninzadeh, K.; Zamani, A.; Mohammadi-Khanaposhtani, M.; Mojtabavi, S.; Faramarzi, M.A.; Hosseini, S.; et al. Docking Study, Molecular Dynamic, Synthesis, Anti-α-Glucosidase Assessment, and ADMET Prediction of New Benzimidazole-Schiff Base Derivatives. Sci. Rep. 2022, 12, 14870. [Google Scholar] [CrossRef]
- Afzal, M.A.F.; Browning, A.R.; Goldberg, A.; Halls, M.D.; Gavartin, J.L.; Morisato, T.; Hughes, T.F.; Giesen, D.J.; Goose, J.E. High-Throughput Molecular Dynamics Simulations and Validation of Thermophysical Properties of Polymers for Various Applications. ACS Appl. Polym. Mater. 2021, 3, 620–630. [Google Scholar] [CrossRef]
- Sun, M.; Lv, S.; Pan, Y.; Song, Q.; Ma, C.; Yu, M.; Gao, X.; Guo, X.; Wang, S.; Gao, Z.; et al. Discovery of Daclatasvir as a Potential PD-L1 Inhibitor from Drug Repurposing. Bioorganic Chem. 2024, 153, 107874. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.A.; Badary, H.A.; Omran, D.; Shousha, H.I.; Abdelaziz, A.O.; El Tayebi, H.M.; Mandour, Y.M. Virtual Screening and Biological Evaluation of Potential PD-1/PD-L1 Immune Checkpoint Inhibitors as Anti-Hepatocellular Carcinoma Agents. ACS Omega 2023, 8, 33242–33254. [Google Scholar] [CrossRef]
- Park, K. A Review of Computational Drug Repurposing. Transl. Clin. Pharmacol. 2019, 27, 59. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Tjong, H.; Zhou, H.-X. G Br 6 NL: A Generalized Born Method for Accurately Reproducing Solvation Energy of the Nonlinear Poisson-Boltzmann Equation. J. Chem. Phys. 2007, 126, 195102. [Google Scholar] [CrossRef]
- Guo, Y.; Liang, J.; Liu, B.; Jin, Y. Molecular Mechanism of Food-Derived Polyphenols on PD-L1 Dimerization: A Molecular Dynamics Simulation Study. Int. J. Mol. Sci. 2021, 22, 10924. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rai, Y.; Bhatt, A.N. Anti-Cancer Drug-Mediated Increase in Mitochondrial Mass Limits the Application of Metabolic Viability-Based MTT Assay in Cytotoxicity Screening. Cytotechnology 2024, 76, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Garcia-de-Alba, C. Repurposing A549 Adenocarcinoma Cells: New Options for Drug Discovery. Am. J. Respir. Cell Mol. Biol. 2021, 64, 405–406. [Google Scholar] [CrossRef]
- Smoktunowicz, N.; Platé, M.; Stern, A.O.; D’Antongiovanni, V.; Robinson, E.; Chudasama, V.; Caddick, S.; Scotton, C.J.; Jarai, G.; Chambers, R.C. TGFβ Upregulates PAR-1 Expression and Signalling Responses in A549 Lung Adenocarcinoma Cells. Oncotarget 2016, 7, 65471–65484. [Google Scholar] [CrossRef] [PubMed]
- Do, J.Y.; Kim, J.; Kim, M.-J.; Lee, J.Y.; Park, S.-Y.; Yanai, R.; Lee, I.-K.; Park, S.; Park, D.H. Fursultiamine Alleviates Choroidal Neovascularization by Suppressing Inflammation and Metabolic Reprogramming. Investig. Ophthalmol. Vis. Sci. 2020, 61, 24. [Google Scholar] [CrossRef]
- Cihlar, T.; Birkus, G.; Greenwalt, D.E.; Hitchcock, M.J.M. Tenofovir Exhibits Low Cytotoxicity in Various Human Cell Types: Comparison with Other Nucleoside Reverse Transcriptase Inhibitors. Antivir. Res. 2002, 54, 37–45. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, Y. Comparative Analysis of the Techniques for the Determination of Binding Affinity between a Small Molecule Inhibitor and a Protein Target. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ganesan, A.; Ahmed, M.; Okoye, I.; Arutyunova, E.; Babu, D.; Turnbull, W.L.; Kundu, J.K.; Shields, J.; Agopsowicz, K.C.; Xu, L.; et al. Comprehensive in Vitro Characterization of PD-L1 Small Molecule Inhibitors. Sci. Rep. 2019, 9, 12392. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, Ł.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew. Chem. Int. Ed. 2017, 56, 13732–13735. [Google Scholar] [CrossRef]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2024, 52, D1265–D1275. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A Web Server for Ligand Binding Site Prediction and Visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, T.A. CASTp: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2003, 31, 3352–3355. [Google Scholar] [CrossRef]
- Shahwan, M.; Hassan, N.; Ashames, A.; Alrouji, M.; Alhumaydhi, F.; Al Abdulmonem, W.; Muhsinah, A.B.; Furkan, M.; Khan, R.H.; Shamsi, A.; et al. PF543-like Compound, a Promising Sphingosine Kinase 1 Inhibitor: Structure-Based Virtual Screening and Molecular Dynamic Simulation Approaches. Int. J. Biol. Macromol. 2023, 245, 125466. [Google Scholar] [CrossRef]
- Mochizuki, M. Growth Characteristics of Canine Pathogenic Viruses in MDCK Cells Cultured in RPMI 1640 Medium without Animal Protein. Vaccine 2006, 24, 1744–1748. [Google Scholar] [CrossRef]
- Maurya, D.K.; Nandakumar, N.; Devasagayam, T.P.A. Anticancer Property of Gallic Acid in A549, a Human Lung Adenocarcinoma Cell Line, and Possible Mechanisms. J. Clin. Biochem. Nutr. 2010, 48, 85–90. [Google Scholar] [CrossRef]
- Cheng, X.-D.; Gu, J.-F.; Yuan, J.-R.; Feng, L.; Jia, X.-B. Suppression of A549 Cell Proliferation and Metastasis by Calycosin via Inhibition of the PKC-α/ERK1/2 Pathway: An in Vitro Investigation. Mol. Med. Rep. 2016, 13, 3709–3710. [Google Scholar] [CrossRef]
- Jiang, D.; Kwon, H.-K.; Kwon, O.W.; Choi, Y. A Comparative Molecular Dynamics Study of Food-Derived Compounds as PD-L1 Inhibitors: Insights Across Six Flavonoid Subgroups. Molecules 2025, 30, 907. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Cerutti, D.S.; Cisneros, G.A.; Cruzeiro, V.W.D.; Forouzesh, N.; Giese, T.J.; Götz, A.W.; Gohlke, H.; et al. AmberTools. J. Chem. Inf. Model. 2023, 63, 6183–6191. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-Donor | H-Acceptor | Fraction | Total Fraction |
|---|---|---|---|
| Vorapaxar | TYR40 | 8.68% | 15.35% |
| ARG109 | Vorapaxar | 6.67% | |
| Delafloxacin | MET99 | 7.54% | 14.02% |
| ARG97 | Delafloxacin | 6.48% | |
| Tenofovir Disoproxil | TRY107 | 3.52% | 28.67% |
| ARG109 | Tenofovir Disoproxil | 25.15% | |
| Pivmecillinam | MET99 | 2.73% | 8.04% |
| GLN50 | Pivmecillinam | 5.31% | |
| Fursultiamine | MET99 | 5.08% | 10.03% |
| SER101 | Fursultiamine | 4.95% | |
| BMS-1 | GLN50 | 1.62% | 5.28% |
| GLN50 | BMS-1 | 3.66% |
| Vorapaxar | Delafloxacin | Tenofovir Disoproxil | Pivmecillinam | Fursultiamine | BMS-1 | |
|---|---|---|---|---|---|---|
| VDW | −23.92 ± 4.00 | −21.07 ± 3.62 | −28.31 ± 5.40 | −26.07 ± 4.46 | −25.58 ± 3.78 | −33.01 ± 4.12 |
| EEL | −7.65 ± 6.85 | 17.89 ± 14.61 | −9.19 ± 31.93 | −2.85 ± 3.75 | −3.76 ± 4.06 | −5.15 ± 4.54 |
| EGB | 16.90 ± 6.71 | −6.52 ± 14.77 | 24.10 ± 29.73 | 14.22 ± 4.12 | 13.42 ± 4.30 | 14.38 ± 4.06 |
| ESURF | −3.31 ± 0.46 | −2.67 ± 0.45 | −4.15 ± 0.73 | −3.32 ± 0.54 | −3.30 ± 0.45 | −3.92 ± 0.48 |
| ΔGgas | −31.56 ± 9.17 | −3.18 ± 15.61 | −37.50 ± 34.54 | −28.92 ± 5.76 | −29.35 ± 5.33 | −38.16 ± 6.38 |
| ΔGsolv | 13.59 ± 6.47 | −9.19 ± 14.77 | 19.95 ± 29.26 | 10.90 ± 3.95 | 10.12 ± 4.18 | 10.46 ± 3.97 |
| ΔGTOTAL | −17.97 ± 3.88 | −12.37 ± 4.00 | −17.55 ± 7.50 | −18.01 ± 3.94 | −19.23 ± 3.94 | −27.70 ± 4.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, D.; Yoou, M.-S.; Cho, S.; Choi, Y. Molecular Dynamics-Guided Repositioning of FDA-Approved Drugs for PD-L1 Inhibition with In Vitro Anticancer Potential. Int. J. Mol. Sci. 2025, 26, 4497. https://doi.org/10.3390/ijms26104497
Jiang D, Yoou M-S, Cho S, Choi Y. Molecular Dynamics-Guided Repositioning of FDA-Approved Drugs for PD-L1 Inhibition with In Vitro Anticancer Potential. International Journal of Molecular Sciences. 2025; 26(10):4497. https://doi.org/10.3390/ijms26104497
Chicago/Turabian StyleJiang, Dejun, Myoung-Schook Yoou, Sungjoon Cho, and Youngjin Choi. 2025. "Molecular Dynamics-Guided Repositioning of FDA-Approved Drugs for PD-L1 Inhibition with In Vitro Anticancer Potential" International Journal of Molecular Sciences 26, no. 10: 4497. https://doi.org/10.3390/ijms26104497
APA StyleJiang, D., Yoou, M.-S., Cho, S., & Choi, Y. (2025). Molecular Dynamics-Guided Repositioning of FDA-Approved Drugs for PD-L1 Inhibition with In Vitro Anticancer Potential. International Journal of Molecular Sciences, 26(10), 4497. https://doi.org/10.3390/ijms26104497