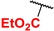3.5. Synthesis and Characterization
General Procedure for the Barton−Zard Reaction of β-Fluoro-β-nitrostyrenes with 2-isocyanoacetamide.
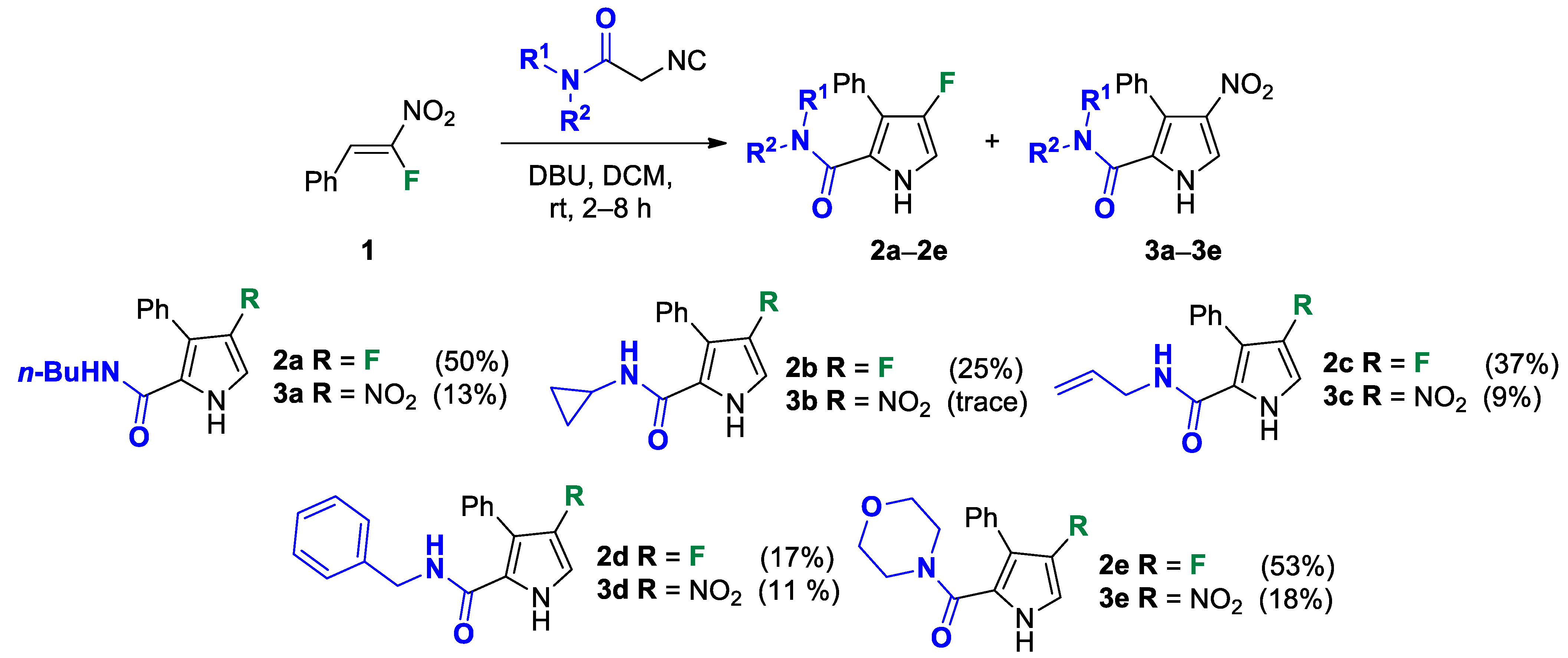
A solution of a selected 2-isocyanoacetamide (5.6 mmol, 2 mol equiv) and DBU (0.837 mL, 5.6 mmol, 2 mol equiv) in DCM (28 mL) was prepared and loaded into a round-bottom flask equipped with a dropping funnel. Then, a solution of (Z)-(2-fluoro-2-nitrovinyl)benzene (0.468 g, 2.8 mmol, 1 mol equiv) in DCM (57 mL) was added dropwise to the vigorously stirred reaction mixture at room temperature over 2 h. After completion of the reaction (TLC monitoring), a 5% aqueous solution of HCl (100 mL) was added to the reaction mixture to hydrolyze the residual 2-isocyanoacetamide. The resulting mixture was vigorously stirred for 2 h. Next, the organic layer was separated and the water layer was extracted with DCM (2 × 50 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered and concentrated under a vacuum. The residue was separated by column chromatography on silica gel using gradient elution to obtain the desired pyrrole 2 and pyrrole 3 as a side-product.
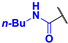
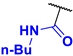
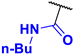
N-butyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide (2a). Eluent: Hex/EtOAc (9:1), Hex/EtOAc (6:1), Hex/EtOAc (3:1). Yield: 0.357 g (50%). Orange solid; mp 110–112 °C. 1H NMR (400 MHz, CDCl3): δ 10.62 (br s, 1H), 7.50–7.38 (m, 5H), 6.75 (t, J = 3.5 Hz, 1H), 5.70 (t, J = 4.9 Hz, 1H), 3.25 (q, J = 6.8 Hz, 2H), 1.31 (dt, J = 14.8, 7.0 Hz, 2H), 1.21–1.08 (m, 2H), 0.83 (t, J = 7.3 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 161.3 (d, 4JCF = 2.5 Hz), 149.6 (d, 1JCF = 242.9 Hz), 131.2 (d, 3JCF = 2.0 Hz), 130.5, 129.2, 128.3, 118.7 (d, 3JCF = 2.3 Hz), 113.0 (d, 2JCF = 12.9 Hz), 105.1 (d, 2JCF = 27.0 Hz), 39.0, 31.2, 19.9, 13.7. 19F NMR (376 MHz, CDCl3): δ −169.02 (s, 1F). HRMS (ESI) m/z: [M + H]+ calcd. for C15H18FN2O+ 261.1398; found 261.1400.
N-butyl-4-nitro-3-phenyl-1H-pyrrole-2-carboxamide (3a). Eluent: Hex/EtOAc (9:1), Hex/EtOAc (6:1), Hex/EtOAc (3:1). Yellow solid; mp 113–115 °C. Yield: 0.101 g (13%). 1H NMR (400 MHz, CDCl3): δ 11.28 (br s, 1H), 7.81 (s, 1H), 7.60–7.46 (m, 3H), 7.45–7.34 (m, 2H), 5.43 (br s, 1H), 3.17 (q, J = 6.2 Hz, 2H), 1.26–1.13 (m, 2H), 1.09–0.96 (m, 2H), 0.79 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.3, 135.9, 131.4, 130.1, 129.4, 129.2, 123.7, 122.0, 120.9, 39.2, 30.9, 19.8, 13.6. HRMS (ESI) m/z: [M + H]+ calcd. for C15H18N3O3+ 288.1343; found 288.1343.
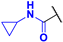
N-cyclopropyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide (2b). Eluent: Hex/EtOAc (4:1). Yield: 0.170 g (25%). Sandy solid; mp 182–184 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.42 (s, 1H), 7.43–7.36 (m, 4H), 7.35–7.27 (m, 1H), 7.16–7.05 (m, 1H), 6.90 (t, J = 3.4 Hz, 1H), 2.67 (dt, J = 10.4, 3.5 Hz, 1H), 0.65–0.56 (m, 2H), 0.37–0.30 (m, 2H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 161.9, 148.8 (d, 1JCF = 239.7 Hz), 131.3 (d, 3JCF = 2.5 Hz), 129.7, 128.2, 127.0, 119.7 (d, 3JCF = 3.2 Hz), 112.5 (d, 2JCF = 11.7 Hz), 104.3 (d, 2JCF = 26.9 Hz), 22.4, 5.9. 19F NMR (376 MHz, DMSO-d6): δ −171.74 (s, 1F). HRMS (ESI) m/z: [M + H]+ calcd. for C14H14FN2O+ 245.1085; found 245.1086.
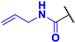
N-allyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide (2c). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T. Polarity of the elution system was gradually increased from Hex/EtOAc (9:1) to Hex/EtOAc (1:1). Yield: 0.260 g (37%). Dark yellow solid; mp 104–106 °C. 1H NMR (400 MHz, CDCl3): δ 10.96 (s, 1H), 7.54–7.36 (m, 5H), 6.79 (t, J = 3.6 Hz, 1H), 5.88 (t, J = 5.6 Hz, 1H), 5.73 (ddt, J = 22.4, 10.5, 5.3 Hz, 1H), 5.03 (dd, J = 10.4, 1.2 Hz, 1H), 4.96 (dd, J = 17.2, 1.3 Hz, 1H), 3.96–3.84 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 161.3 (d, 4JCF = 2.4 Hz), 149.4 (d, 1JCF = 242.8 Hz), 133.5, 131.0 (d, 3JCF = 1.8 Hz), 130.3, 129.1, 128.2, 118.2 (d, 3JCF = 2.2 Hz), 115.8, 113.3 (d, 2JCF = 12.8 Hz), 105.6 (d, 2JCF = 26.9 Hz), 41.5. 19F NMR (376 MHz, CDCl3): δ −169.18 (s, 1F). HRMS (ESI) m/z: [M + H]+ calcd. for C14H14FN2O+ 245.1085; found 245.1086.
N-allyl-4-nitro-3-phenyl-1H-pyrrole-2-carboxamide (3c). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T. Polarity of the elution system was gradually increased from Hex/EtOAc (9:1) to Hex/EtOAc (1:1). Yield: 0.074 g (9%). Yellow solid; mp 158–160 °C. 1H NMR (400 MHz, CDCl3): δ 11.70 (br s, 1H), 7.83 (s, 1H), 7.58–7.46 (m, 3H), 7.45–7.36 (m, 2H) 5.71–5.49 (m, 2H), 5.00 (d, J = 10.2 Hz, 1H), 4.82 (d, J = 17.1 Hz, 1H), 3.81 (s, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.4, 135.8, 132.8, 131.2, 130.0, 129.4, 129.3, 123.4, 122.4, 121.3, 116.2, 41.7. HRMS (ESI) m/z: [M + H]+ calcd. for C14H14N3O3+ 272.1030; found 272.1030.
Procedure for the Scaled-Up Preparation of 2d.
A solution of N-benzyl-2-isocyanoacetamide (4.607 g, 26.4 mmol, 2 mol equiv) and DBU (3.955 mL, 26.4 mmol, 2 mol equiv) in DCM (134 mL) was prepared and loaded into a round-bottom flask equipped with a dropping funnel. Then, a solution of (Z)-(2-fluoro-2-nitrovinyl)benzene (2.210 g, 13.2 mmol, 1 mol equiv) in DCM (267 mL) was added dropwise to the vigorously stirred reaction mixture at room temperature over 6 h. After completion of the reaction (TLC monitoring), 5% aqueous solution of HCl (400 mL) was added to the reaction mixture to hydrolyze the residual N-benzyl-2-isocyanoacetamide. The resulting mixture was vigorously stirred for 2 h. Next, the organic layer was separated and the water layer was extracted with DCM (2 × 200 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered and concentrated under a vacuum. The residue was separated by column chromatography on silica gel using SepaBean™ machine T to obtain the desired pyrrole 2d and pyrrole 3d as a side-product. The polarity of the elution system was gradually increased from Hex/EtOAc (9:1) to Hex/EtOAc (1:1), and then EtOAc was used.
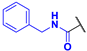
N-benzyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide (2d). Yield: 0.658 g (17%). Viscous oil. 1H NMR (400 MHz, CDCl3): δ 10.10 (s, 1H), 7.46–7.22 (m, 8H), 7.10 (m, 2H), 6.71 (t, J = 3.4 Hz, 1H), 6.05 (t, J = 5.6 Hz, 1H), 4.45 (d, J = 5.8 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 161.1 (d, 4JCF = 2.6 Hz), 149.7 (d, 1JCF = 243.5 Hz), 137.8, 130.9 (d, 3JCF = 1.7 Hz), 130.4, 129.2, 128.7, 128.4, 127.5, 127.5, 118.5 (d, 3JCF = 2.1 Hz), 113.4 (d, 2JCF = 12.9 Hz), 105.3 (d, 2JCF = 27.1 Hz), 43.5. 19F NMR (376 MHz, CDCl3): δ −168.68 (pseudo-t, 3JHF = 4JHF = 3.0 Hz, 1F). HRMS (ESI) m/z: [M + H]+ calcd. for C18H16FN2O+ 295.1241; found 295.1243.
N-benzyl-4-nitro-3-phenyl-1H-pyrrole-2-carboxamide (3d). Yield: 0.448 g (11%). Orange viscous oil. 1H NMR (400 MHz, CDCl3) δ 11.72 (s, 1H), 7.69 (d, J = 3.8 Hz, 1H), 7.47–7.24 (m, 8H), 7.00–6.93 (m, 2H), 5.84 (t, J = 5.4 Hz, 1H), 4.34 (d, J = 5.6 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3) δ 160.0, 136.4, 135.3, 130.6, 129.5, 128.8, 128.7, 128.3, 127.3, 126.7, 122.8, 122.0, 120.9, 43.3. HRMS (ESI) m/z: [M + H]+ calcd. for C18H16N3O3+ 322.1186; found 322.1188.
Procedure for the Scaled-Up Preparation of 2e.
A solution of 2-isocyano-1-morpholinoethanone (5.486 g, 35.6 mmol, 2 mol equiv) and DBU (5.322 mL, 35.6 mmol, 2 mol equiv) in DCM (180 mL) was prepared and loaded into a round-bottom flask equipped with a dropping funnel. Then, a solution of (Z)-(2-fluoro-2-nitrovinyl)benzene (2.974 g, 17.8 mmol, 1 mol equiv) in DCM (359 mL) was added dropwise to the vigorously stirred reaction mixture at room temperature over 8 h. After completion of the reaction (TLC monitoring), 5% aqueous solution of HCl (500 mL) was added to the reaction mixture to hydrolyze the residual 2-isocyano-1-morpholinoethanone. The resulting mixture was vigorously stirred for 2 h. Next, the organic layer was separated and the water layer was extracted with DCM (2 × 250 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered and concentrated under a vacuum. The residue was separated by column chromatography on silica gel using SepaBean™ machine T to obtain the desired pyrrole 2d and pyrrole 3d as a side-product. The polarity of the elution system was gradually increased from Hex/EtOAc (2:1) to Hex/EtOAc (1:4).
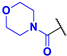
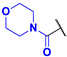
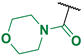
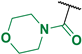
(4-Fluoro-3-phenyl-1H-pyrrol-2-yl)(morpholino)methanone (2e). Yield: 2.604 g (53%). Yellow solid; mp 209–211 °C. 1H NMR (400 MHz, CDCl3): δ 10.02 (s, 1H), 7.46–7.30 (m, 5H), 6.73 (t, J = 3.1 Hz, 1H), 3.33 (br s, 8H). 13C{1H} NMR (100 MHz, CDCl3): δ 164.0 (d, 4JCF = 2.0 Hz), 149.0 (d, 1JCF = 243.5 Hz), 131.7 (d, 3JCF = 2.4 Hz), 129.3, 129.0, 127.6, 118.0 (d, 3JCF = 2.9 Hz), 113.7 (d, 2JCF = 12.1 Hz), 105.0 (d, 2JCF = 27.5 Hz), 66.1, 66.1. 19F NMR (376 MHz, CDCl3): δ −170.39 (pseudo-t, 3JHF = 4JHF = 3.1 Hz, 1F). HRMS (ESI) m/z: [M + H]+ calcd. for C15H16FN2O2+ 275.1190; found 275.1190.
Morpholino(4-nitro-3-phenyl-1H-pyrrol-2-yl)methanone (3e). Yield: 0.980 g (18%). Dark yellow solid; mp 213–215 °C. 1H NMR (400 MHz, CDCl3): δ 11.61 (s, 1H), 7.84 (d, J = 3.6 Hz, 1H), 7.49–7.35 (m, 5H), 3.76–2.55 (m, 8H). 13C{1H} NMR (100 MHz, CDCl3): δ 162.9, 134.6, 131.1, 130.6, 128.8, 128.7, 123.2, 123.0, 121.3, 65.8, 65.7. HRMS (ESI) m/z: [M + H]+ calcd. for C15H16N3O4+ 302.1135; found 302.1136.
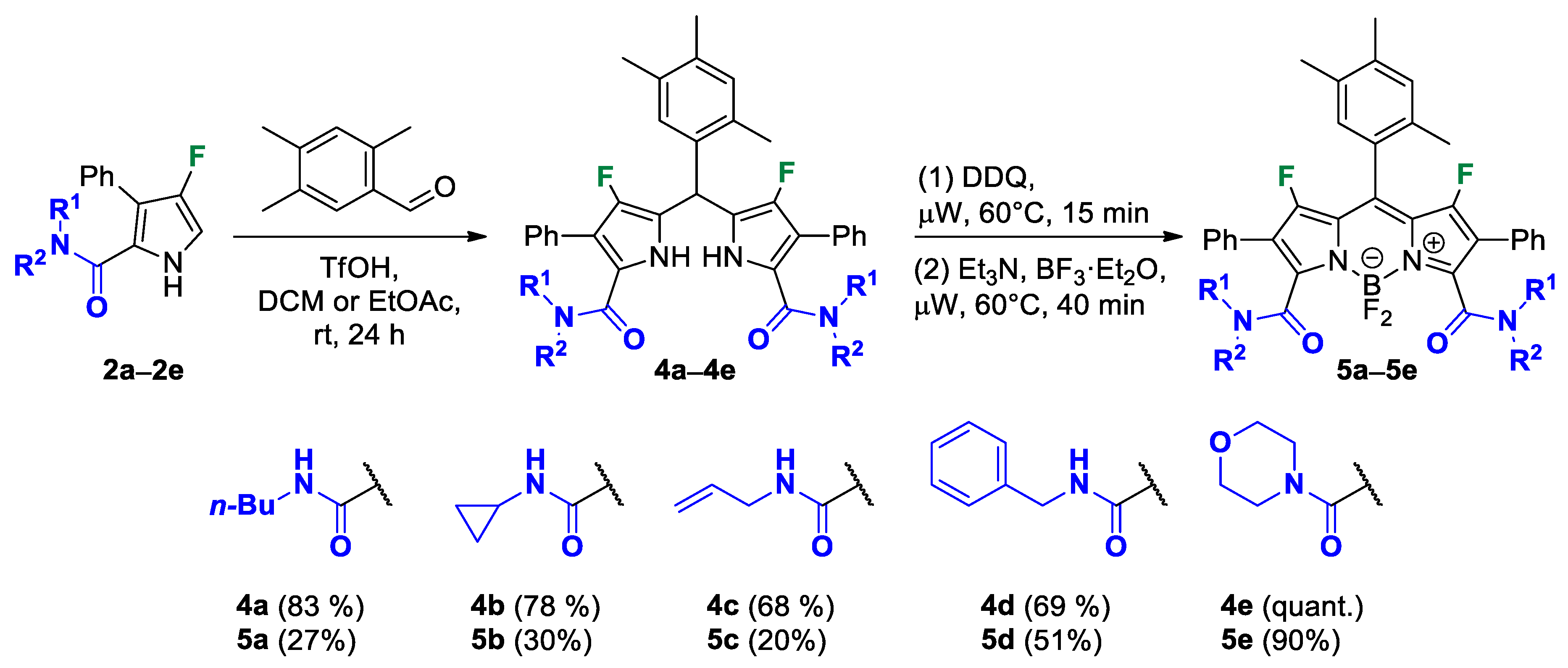
General Procedure for the Preparation of Dipyrromethanes 4.
In a typical experiment, a selected pyrrole 2 (0.4–1.4 mmol, 1 mol equiv) and a corresponding aldehyde (0.4–1.4 mmol, 1 mol equiv) were loaded into a suitable vial and dissolved in DCM (2–7 mL). In the case of obtaining dipyrromethane 4b, EtOAc was used instead of DCM. Then triflic acid (0.4–1.4 mmol, 1 mol equiv) was added to the reaction mixture and the resulting mixture was stirred for 24 h. Progress of the reaction was monitored by TLC or 19F NMR spectroscopy. After completion of the reaction, the reaction mixture was diluted with EtOAc (50–100 mL) and extracted with water (3 × (50–100) mL). Then, the organic layer was separated, dried over Na2SO4, filtered and concentrated under a vacuum. The pure product was isolated by column chromatography on silica gel using appropriate elution mixtures.
5,5′-((2,4,5-Trimethylphenyl)methylene)bis(N-butyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide) (4a). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T. Polarity of the elution system was gradually increased from DCM/EtOAc (99:1) to DCM/EtOAc (4:1). Yield: 0.135 g (83%). Orange solid; mp 208–210 °C. 1H NMR (400 MHz, CDCl3): δ 10.14 (br s, 2H), 7.46–7.33 (m, 10H), 7.14 (s, 1H), 6.94 (s, 1H), 5.87 (s, 1H), 5.64 (t, J = 5.4 Hz, 2H), 3.22–3.08 (m, 4H), 2.30 (s, 3H), 2.19 (s, 3H), 2.17 (s, 3H), 1.28–1.15 (m, 4H), 1.14–1.01 (m, 4H), 0.77 (t, J = 7.3 Hz, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.8 (d, 4JCF = 2.5 Hz), 146.3 (d, 1JCF = 244.4 Hz), 135.8, 134.7, 134.2, 133.1, 132.3, 131.2 (d, 3JCF = 1.9 Hz), 130.5, 129.5, 129.1, 128.2, 117.3 (d, 2JCF = 21.4 Hz), 117.1 (d, 3JCF = 2.2 Hz), 113.3 (d, 2JCF = 12.4 Hz), 39.0, 34.3, 31.1, 20.0, 19.7, 19.4, 19.0, 13.7. 19F NMR (376 MHz, CDCl3): δ −168.15 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C40H45F2N4O2+ 651.3505; found 651.3505.
5,5′-((2,4,5-Trimethylphenyl)methylene)bis(N-cyclopropyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide) (4b). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T. Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (2.3:1). Yield: 0.087 g (78%). Orange solid; mp 160–162 °C. 1H NMR (400 MHz, CDCl3): δ 10.74 (br s, 2H), 7.36 (s, 10H), 7.25 (s, 1H), 6.94 (s, 1H), 5.94 (s, 1H), 5.73 (d, J = 3.3 Hz, 2H), 2.70 (dt, J = 10.6, 3.5 Hz, 2H), 2.32 (s, 3H), 2.18 (s, 3H), 2.14 (s, 3H), 0.57 (q, J = 6.2 Hz, 4H), 0.16 (q, J = 6.6 Hz, 4H). 13C{1H} NMR (100 MHz, CDCl3): δ 162.1, 146.2 (d, 1JCF = 244.2 Hz), 135.6, 134.5, 134.4, 133.2, 132.1, 131.1 (d, 3JCF = 1.5 Hz), 130.4, 129.8, 128.9, 128.2, 118.0 (d, 2JCF = 20.8 Hz), 116.9 (d, 3JCF = 2.4 Hz), 113.5 (d, 2JCF = 12.0 Hz), 34.3, 22.4, 19.6, 19.4, 19.1, 6.5, 6.5. 19F NMR (376 MHz, CDCl3): δ −167.94 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C38H37F2N4O2+ 619.2879; found 619.2881.
5,5′-((2,4,5-Trimethylphenyl)methylene)bis(N-allyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide) (4c). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T. Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (3:1). Yield: 0.103 g (68%). Sandy solid; mp 237–239 °C. 1H NMR (400 MHz, CDCl3): δ 11.07 (br s, 2H), 7.43–7.30 (m, 11H), 6.95 (s, 1H), 5.94 (s, 1H), 5.78 (t, J = 5.3 Hz, 2H), 5.61 (ddt, J = 22.6, 10.7, 5.5 Hz, 2H), 4.96 (d, J = 10.3 Hz, 2H), 4.85 (d, J = 17.2 Hz, 2H), 3.86–3.69 (m, 4H), 2.37 (s, 3H), 2.20 (s, 3H), 2.18 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.7 (d, 4JCF = 2.6 Hz), 146.4 (d, 1JCF = 244.0 Hz), 135.5, 134.6, 134.5, 133.5, 133.0, 132.0, 131.2 (d, 3JCF = 1.8 Hz), 130.4, 129.9, 129.1, 128.1, 118.3 (d, 2JCF = 20.2 Hz), 116.5 (d, 3JCF = 2.5 Hz), 116.2, 113.5 (d, 2JCF = 12.0 Hz), 41.8, 34.4, 19.6, 19.4, 19.1. 19F NMR (376 MHz, CDCl3): δ −168.08 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C38H37F2N4O2+ 619.2879; found 619.2879.
5,5′-((2,4,5-Trimethylphenyl)methylene)bis(N-benzyl-4-fluoro-3-phenyl-1H-pyrrole-2-carboxamide) (4d). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T. Polarity of the elution system was gradually increased from DCM/EtOAc (99:1) to DCM/EtOAc (9:1). Yield: 0.179 g (56%). Sandy solid; mp 126–128 °C. 1H NMR (400 MHz, CDCl3): δ 11.46 (br s, 2H), 7.56 (s, 1H), 7.33–7.23 (m, 12H), 7.19 (t, J = 7.4 Hz, 4H), 7.02 (s, 1H), 7.02–6.96 (m, 4H), 6.10 (s, 1H), 6.06 (t, J = 5.2 Hz, 2H), 4.48–4.31 (m, 4H), 2.47 (s, 3H), 2.24 (s, 3H), 2.22 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.8 (d, 4JCF = 2.4 Hz), 146.4 (d, 1JCF = 244.3 Hz), 137.2, 135.4, 134.8, 134.4, 133.1, 131.9, 130.9 (d, 3JCF = 1.9 Hz), 130.3, 130.1, 128.9, 128.6, 127.9, 127.7, 127.4, 118.6 (d, 2JCF = 20.2 Hz), 116.5 (d, 3JCF = 2.6 Hz), 113.6 (d, 2JCF = 12.0 Hz), 43.7, 34.4, 19.5, 19.4, 19.2. 19F NMR (376 MHz, CDCl3): δ −167.90 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C46H41F2N4O2+ 719.3192; found 719.3192.
(5,5′-((2,4,5-Trimethylphenyl)methylene)bis(4-fluoro-3-phenyl-1H-pyrrole-5,2-diyl))bis(morpholinomethanone) (4e). Eluent: Hex/EtOAc (1:1), EtOAc. Yield: 0.261 g (84%). Orange solid; mp 180–182 °C. 1H NMR (400 MHz, CDCl3): δ 9.40 (br s, 2H), 7.42–7.27 (m, 10H), 7.05 (s, 1H), 6.95 (s, 1H), 5.89 (s, 1H), 3.21 (br s, 16H), 2.29 (s, 3H), 2.18 (s, 3H), 2.14 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 163.6, 145.5 (d, 1JCF = 244.0 Hz), 136.0, 134.8, 134.0, 133.2, 132.5, 131.5, 129.3, 129.1, 128.9, 127.7, 117.0 (d, 2JCF = 21.6 Hz), 116.5 (d, 3JCF = 3.6 Hz), 114.3 (d, 2JCF = 10.3 Hz), 65.9, 65.9, 34.1, 19.6, 19.4, 19.0. 19F NMR (376 MHz, CDCl3): δ −169.47 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C40H41F2N4O4+ 679.3090; found 679.3090.
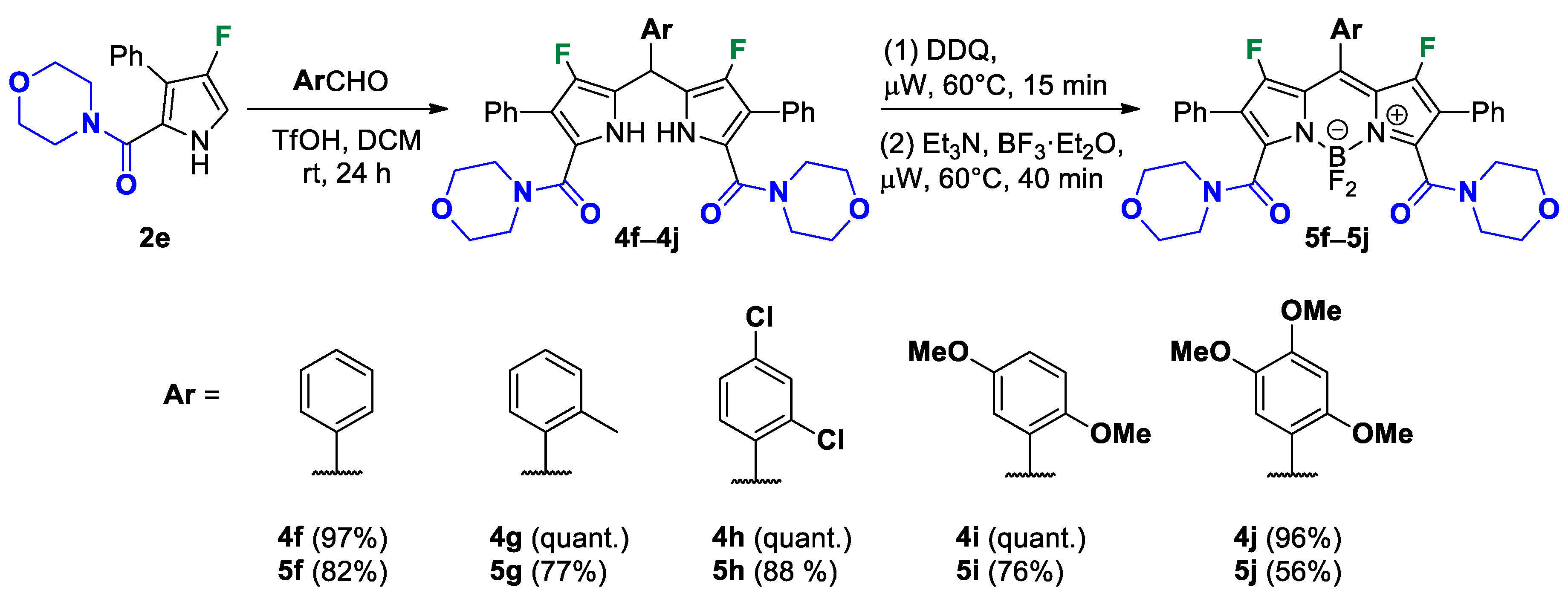
(5,5′-(Phenylmethylene)bis(4-fluoro-3-phenyl-1H-pyrrole-5,2-diyl))bis(morpholinomethanone) (4f). Eluent: Hex/EtOAc (1:1), EtOAc. Yield: 0.125 g (97%). Colourless solid; mp 194–196 °C. 1H NMR (400 MHz, CDCl3): δ 10.77 (s, 2H), 7.41–7.28 (m, 10H), 7.13 (d, J = 7.2 Hz, 2H), 7.10–7.00 (m, 3H), 5.90 (s, 1H), 3.22 (br s, 16H). 13C{1H} NMR (100 MHz, CDCl3): δ 163.9 (d, 4JCF = 2.1 Hz), 145.9 (d, 1JCF = 243.9 Hz), 139.5, 131.6 (d, 3JCF = 2.5 Hz), 129.4, 128.9, 128.6, 127.7, 127.6, 127.2, 117.4 (d, 2JCF = 22.4 Hz), 117.0 (d, 3JCF = 3.6 Hz), 113.7 (d, 2JCF = 11.7 Hz), 65.9, 65.9, 36.1. 19F NMR (376 MHz, CDCl3): δ −171.02 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C37H35F2N4O4+ 637.2621; found 637.2621.
(5,5′-(O-tolylmethylene)bis(4-fluoro-3-phenyl-1H-pyrrole-5,2-diyl))bis(morpholinomethanone) (4g). Eluent: Hex/EtOAc (1:1), EtOAc. Yield: 0.130 g (quant.). Pale pink solid; mp 184–186 °C. 1H NMR (400 MHz, CDCl3): δ 10.47 (s, 2H), 7.42–7.25 (m, 11H), 7.10 (d, J = 7.1 Hz, 1H), 7.04 (t, J = 7.2 Hz, 1H), 6.91 (t, J = 7.3 Hz, 1H), 5.97 (s, 1H), 3.22 (br s, 16H), 2.33 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 163.8 (d, 4JCF = 2.1 Hz), 145.4 (d, 1JCF = 244.0 Hz), 137.4, 136.1, 131.6 (d, 3JCF = 2.2 Hz), 130.9, 129.3, 128.8, 127.9, 127.5, 127.4, 126.4, 117.0 (d, 2JCF = 21.3 Hz), 116.8 (d, 3JCF = 3.1 Hz), 113.7 (d, 2JCF = 11.7 Hz), 65.9, 65.9, 34.0, 19.6. 19F NMR (376 MHz, CDCl3): δ −170.16 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C38H37F2N4O4+ 651.2777; found 651.2777.
(5,5′-((2,4-Dichlorophenyl)methylene)bis(4-fluoro-3-phenyl-1H-pyrrole-5,2-diyl))bis(morpholinomethanone) (4h). Eluent: Hex/EtOAc (1:1), EtOAc. Yield: 0.141 g (quant.). Powdery solid; mp 195–197 °C. 1H NMR (400 MHz, CDCl3): δ 10.87 (s, 2H), 7.42–7.25 (m, 12H), 6.93 (dd, J = 8.4, 2.0 Hz, 1H), 6.19 (s, 1H), 3.22 (br s, 16H). 13C{1H} NMR (100 MHz, CDCl3): δ 163.7 (d, 4JCF = 2.0 Hz), 145.7 (d, 1JCF = 245.0 Hz), 135.7, 134.3, 133.6, 131.4 (d, 3JCF = 2.3 Hz), 130.7, 129.6, 129.0, 128.9, 127.6, 127.3, 117.1 (d, 3JCF = 3.7 Hz), 115.6 (d, 2JCF = 21.4 Hz), 113.5 (d, 2JCF = 11.6 Hz), 65.8, 65.8, 34.4. 19F NMR (376 MHz, CDCl3): δ −169.19 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C37H33Cl2F2N4O4+ 705.1841; found 705.1841.
(5,5′-((2,5-Dimethoxyphenyl)methylene)bis(4-fluoro-3-phenyl-1H-pyrrole-5,2-diyl))bis(morpholinomethanone) (4i). Eluent: Hex/EtOAc (1:1), EtOAc. Yield: 0.138 g (quant.). Sandy solid; mp 212–214 °C. 1H NMR (400 MHz, CDCl3): δ 10.38 (s, 2H), 7.39–7.23 (m, 10H), 6.87 (d, J = 2.9 Hz, 1H), 6.83 (d, J = 8.9 Hz, 1H), 6.71 (dd, J = 8.9, 3.0 Hz, 1H), 6.04 (s, 1H), 3.77 (s, 3H), 3.61 (s, 3H), 3.19 (br s, 16H). 13C{1H} NMR (100 MHz, CDCl3): δ 163.7 (d, 4JCF = 2.0 Hz), 153.6, 151.1, 145.2 (d, 1JCF = 244.0 Hz), 131.7 (d, 3JCF = 2.2 Hz), 129.3, 129.2, 128.7, 127.3, 116.7 (d, 2JCF = 22.1 Hz), 116.5, 116.5 (d, 3JCF = 3.9 Hz), 113.6 (d, 2JCF = 11.7 Hz), 112.4, 111.7, 65.9, 65.9, 56.6, 55.5, 32.1. 19F NMR (376 MHz, CDCl3): δ −170.19 (s, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C39H39F2N4O6+ 697.2832; found 697.2832.
(5,5′-((2,4,5-Trimethoxyphenyl)methylene)bis(4-fluoro-3-phenyl-1H-pyrrole-5,2-diyl))bis(morpholinomethanone) (4j). Eluent: Hex/EtOAc (1:1), EtOAc, EtOAc/EtOH (98:2). Yield: 0.140 g (96%). Pale orange solid; mp 192–194 °C. 1H and 19F NMR spectra of the sample are complex due to the presence of two sets of signals corresponding to two stable rotamers in a ratio of 75:25. In 1H spectra some similar signals overlap. 1H NMR (400 MHz, CDCl3): δ 11.27 (br s, 1H), 10.04 (br s, 1H), 7.43–7.20 (m, 10H), 7.05 (s, 1H), 6.60 (s, 1H), 5.95 (s, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.78 (s, 3H), 3.20 (br s, 16H). 13C{1H} NMR (100 MHz, CDCl3): δ 163.8 (d, 4JCF = 1.0 Hz), 151.1, 149.3, 145.3 (d, 1JCF = 243.8 Hz), 143.5, 131.7 (d, 3JCF = 2.5 Hz), 128.9, 128.8, 128.6, 127.4, 127.3, 119.4, 117.1 (d, 2JCF = 21.4 Hz), 116.3 (d, 3JCF = 3.6 Hz), 114.1, 113.2 (d, 2JCF = 11.5 Hz), 66.0, 65.8, 57.3, 56.6, 56.2. 19F NMR (376 MHz, CDCl3): major rotamer δ −170.4 (s, 1F); minor rotamer δ -171.9 (s, 1F). HRMS (ESI) m/z: [M + H]+ calcd. for C40H41F2N4O7+ 727.2938; found 727.2938.
General Procedure for the Preparation of BODIPYs 5.
In a typical experiment, a selected dipyrromethane 4 (0.15 mmol, 1 mol equiv) and DDQ (0.102 g, 0.45 mmol, 3 mol equiv) were loaded into a screw cap vial (30 mL) for the microwave reactor Nova-2S (PreeKem) and dissolved in MeCN (7.5 mL). The reaction mixture was heated under microwave irradiation at 60 °C for 15 min with stirring. After completion of the oxidation step (monitoring by TLC or 19F NMR spectroscopy), the reaction mixture was cooled to room temperature. Then, triethylamine (0.209 mL, 1.5 mmol, 10 mol equiv) was added and the resulting mixture was stirred for 3 min. Next, BF3⋅Et2O (0.278 mL, 2.25 mmol, 15 mol equiv) was added to the reaction mixture. The vial with the resulting mixture was purged with argon and then heated under microwave irradiation at 60 °C for 40 min with stirring. After completion of the reaction (monitoring by TLC or 19F NMR spectroscopy), the reaction mixture was diluted with EtOAc (100 mL) and extracted with water (5 × 100 mL). Then, the organic layer was separated, dried over Na2SO4, filtered and concentrated under a vacuum. The pure product was isolated by column chromatography on silica gel using appropriate eluents.
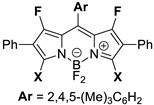
3,7-Bis(butylcarbamoyl)-1,5,5,9-tetrafluoro-2,8-diphenyl-10-(2,4,5-trimethylphenyl)-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5a). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (96:4). Yield: 0.031 g (27%). Red solid; mp 219–221 °C. 1H NMR (400 MHz, CDCl3): δ 7.46–7.40 (m, 4H), 7.38–7.27 (m, 6H), 7.09 (s, 1H), 7.02 (s, 1H), 6.64 (br s, 2H), 3.45 (q, J = 6.8 Hz, 4H), 2.29 (s, 3H), 2.25 (s, 6H), 1.60 (dt, J = 15.0, 7.5 Hz, 4H), 1.44–1.33 (m, 4H), 0.94 (t, J = 7.4 Hz, 6H). 13C NMR (100MHz, CDCl3): δ 160.6, 159.4 (d, 1JCF = 287.5 Hz), 147.5, 146.7, 139.0, 134.4, 132.6, 131.8, 128.8, 128.6, 128.6, 128.4, 128.0 (d, 3JCF = 1.8 Hz), 127.3, 121.0 (d, 2JCF = 11.2 Hz), 118.3 (d, 2JCF = 7.4 Hz), 40.1, 31.2, 20.2, 19.8, 19.4, 19.2, 13.8. 19F NMR (376 MHz, CDCl3): δ −132.19 (s, 2F), −136.36–−136.81 (m, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C40H42BF4N4O2+ 697.3331; found 697.3331.
3,7-Bis(cyclopropylcarbamoyl)-1,5,5,9-tetrafluoro-2,8-diphenyl-10-(2,4,5-trimethylphenyl)-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5b). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (96:4). Yield: 0.028 g (30%). Red solid; mp 195–197 °C. 1H NMR (400 MHz, CDCl3): δ 7.46–7.40 (m, 4H), 7.40–7.28 (m, 6H), 7.09 (s, 1H), 6.99 (s, 1H), 6.70 (s, 1H), 6.69 (s, 1H), 2.89 (dt, J = 10.5, 3.5 Hz, 2H), 2.29 (s, 3H), 2.24 (s, 3H), 2.23 (s, 3H), 0.91–0.85 (m, 4H), 0.75–0.68 (m, 4H). 13C{1H} NMR (100 MHz, CDCl3): δ 162.0, 159.4 (d, 1JCF = 282.3 Hz), 147.2, 146.9, 139.1, 134.4, 132.5, 131.9, 128.8, 128.7, 128.6, 128.5, 127.9 (d, 3JCF = 2.0 Hz), 127.2, 121.0 (d, 2JCF = 12.7 Hz), 118.4 (d, 2JCF = 8.8 Hz), 23.1, 19.8, 19.3, 19.1, 6.8. 19F NMR (376 MHz, CDCl3): δ −131.80 (s, 2F), −135.74–−136.17 (m, 2F). HRMS (ESI) m/z: [M + Na]+ calcd. for C38H33BF4N4NaO2+ 687.2525; found 687.2525.
3,7-Bis(allylcarbamoyl)-1,5,5,9-tetrafluoro-2,8-diphenyl-10-(2,4,5-trimethylphenyl)-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5c). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (96:4). Yield: 0.018 g (20%). Red solid; mp 253–255 °C (with decomp.). 1H NMR (400 MHz, CDCl3): δ 7.43 (d, J = 6.9 Hz, 4H), 7.39–7.28 (m, 6H), 7.10 (s, 1H), 7.03 (s, 1H), 6.73 (br s, 2H), 5.90 (ddt, J = 22.6, 10.8, 5.7 Hz, 2H), 5.28 (dd, J = 17.2, 1.0 Hz, 2H), 5.18 (dd, J = 10.3, 0.9 Hz, 2H), 4.09 (t, J = 5.7 Hz, 4H), 2.30 (s, 3H), 2.26 (s, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.5, 159.5 (d, 1JCF = 285.0 Hz), 147.1, 147.0, 139.1, 134.4, 133.1, 132.6, 131.9, 128.8, 128.7, 128.6, 128.5, 127.9 (d, 3JCF = 1.5 Hz), 127.3, 121.1 (d, 2JCF = 13.5 Hz), 118.55 (d, 2JCF = 9.7 Hz), 117.4, 42.7, 19.9, 19.4, 19.2. 19F NMR (376 MHz, CDCl3): δ −131.91 (s, 2F), −135.64–−136.76 (m, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C38H34BF4N4O2+ 665.2705; found 665.2706.
3,7-Bis(benzylcarbamoyl)-1,5,5,9-tetrafluoro-2,8-diphenyl-10-(2,4,5-trimethylphenyl)-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5d). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (99:1) to DCM/EtOAc (17:1). Yield: 0.016 g (51%), 0.045 g (46%). Red solid; mp 223–225 °C. 1H NMR (400 MHz, CDCl3): δ 7.41–7.27 (m, 20H), 7.09 (s, 1H), 7.01 (s, 1H), 6.88 (br s, 2H), 4.64 (d, J = 5.7 Hz, 4H), 2.29 (s, 3H), 2.25 (s, 3H), 2.24 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.7, 159.3 (d, 1JCF = 286.7 Hz), 147.1 (d, 3JCF = 1.9 Hz), 147.0, 139.1, 137.1, 134.4, 132.6, 131.8, 128.8, 128.7, 128.6, 128.4, 128.2, 128.0 (d, 3JCF = 3.8 Hz), 127.8, 127.8, 127.2, 121.1 (d, 2JCF = 11.8 Hz), 118.4 (d, 2JCF = 9.4 Hz), 44.3, 19.8, 19.3, 19.1.19F NMR (376 MHz, CDCl3): δ −131.99 (s, 2F), −136.55–−137.14 (m, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C46H38BF4N4O2+ 765.3018; found 765.3020.
1,5,5,9-Tetrafluoro-3,7-di(morpholine-4-carbonyl)-2,8-diphenyl-10-(2,4,5-trimethylphenyl)-5H-dipyrrolo[1,2-c:2′,1’-f][1,3,2]diazaborinin-4-ium-5-uide (5e). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (3:1). Yield: 0.098 g (90%). Red solid; mp 298–300 °C. The NMR spectra of the sample are complex due to the presence of two sets of signals corresponding to two stable rotamers in ratio 1:1, where many similar signals overlap. 1H NMR (400 MHz, CDCl3): δ 7.43 (d, J = 7.4 Hz, 4H), 7.41–7.29 (m, 6H), 7.10 (s, 1H), 7.02 (s, 1H), 3.92–3.26 (m, 12H), 3.04–2.91 (m, 2H), 2.85–2.67 (m, 2H), 2.29 (s, 3H), 2.27 (s, 3H), 2.24 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.8, 160.7, 158.8 (d, 1JCF = 285.7 Hz), 158.4 (d, 1JCF = 284.8 Hz), 147.1, 146.7, 146.1, 139.2, 134.3, 132.9, 131.9, 129.2, 129.0, 128.7, 127.9, 127.7 (d, 3JCF = 2.5 Hz), 127.1, 121.4 (d, 2JCF = 12.3 Hz), 116.2 (d, 2JCF = 9.2 Hz), 66.1, 66.0, 47.0, 42.2, 42.2, 19.8, 19.3, 19.2. 19F NMR (376 MHz, CDCl3): δ −131.57 (s, 1F), −133.29 (s, 1F), −142.27–−143.52 (m, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C40H38BF4N4O4+ 725.2917; found 725.2917.
1,5,5,9-Tetrafluoro-3,7-di(morpholine-4-carbonyl)-2,8,10-triphenyl-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5f). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (2:1). Yield: 0.083 g (82%). Red solid; mp 316–318 °C. 1H NMR (400 MHz, CDCl3): δ 7.63–7.47 (m, 5H), 7.46–7.29 (m, 10H), 3.89–3.79 (m, 2H), 3.78–3.47 (m, 6H), 3.43–3.26 (m, 4H), 3.03–2.91 (m, 2H), 2.73 (t, J = 8.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 160.6, 158.6 (d, 1JCF = 286.6 Hz), 147.3, 145.9, 131.1, 129.8, 129.0, 128.8, 128.6, 128.5, 127.9, 127.6, 121.0 (d, 2JCF = 11.9 Hz), 116.4 (d, 2JCF = 9.5 Hz), 66.1, 66.0, 47.0, 42.2. 19F NMR (376 MHz, CDCl3): δ −130.54 (s, 2F), −142.77 (dd, J = 58.8, 28.7 Hz, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C37H32BF4N4O4+ 683.2447; found 683.2448.
1,5,5,9-Tetrafluoro-3,7-di(morpholine-4-carbonyl)-2,8-diphenyl-10-(o-tolyl)-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5g). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (2:1). Yield: 0.081g (77%). Red solid; mp 324–326 °C (with decomp.). The NMR spectra of the sample are complex due to the presence of two sets of signals corresponding to two stable rotamers in ratio 1:1, where many similar signals overlap. 1H NMR (400 MHz, CDCl3): δ 7.46–7.26 (m, 14H), 3.88–3.78 (m, 2H), 3.78–3.60 (m, 4H), 3.59–3.48 (m, 2H), 3.46–3.26 (m, 4H), 3.04–2.91 (m, 2H), 2.85–2.70 (m, 2H), 2.35 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.6, 160.6, 158.9 (d, 1JCF = 280.5 Hz), 158.8 (d, 1JCF = 289.3 Hz), 147.5, 147.2, 145.3, 135.6, 130.5, 130.4, 129.7, 129.0, 128.8, 128.1, 127.8, 127.6, 126.0, 121.3 (d, 2JCF = 12.3 Hz), 121.2 (d, 2JCF = 13.5 Hz), 116.3 (d, J = 8.6 Hz), 66.1, 66.0, 47.0, 42.2, 42.2, 19.8. 19F NMR (376 MHz, CDCl3): δ −131.79 (s, 1F), −133.28 (s, 1F), −142.92 (ddd, J = 39.5, 28.5, 10.0 Hz, 2F). HRMS (ESI) m/z: [M + H]+ calcd. for C38H34BF4N4O4+ 697.2604; found 697.2604.
10-(2,4-Dichlorophenyl)-1,5,5,9-tetrafluoro-3,7-di(morpholine-4-carbonyl)-2,8-diphenyl-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5h). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (3:1). The NMR spectra of the sample are complex due to the presence of two sets of signals corresponding to two stable rotamers in ratio 53:47, where many similar signals overlap. Yield: 0.098 g (88%). Violet solid; mp 186–188 °C. 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 1.9 Hz, 1H), 7.46–7.31 (m, 12H), 3.88–3.60 (m, 6H), 3.60–3.48 (m, 2H), 3.45–3.26 (m, 4H), 3.06–2.91 (m, 2H), 2.86–2.71 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.4, 158.6 (d, 1JCF = 283.8 Hz), 158.5 (d, 1JCF = 285.6 Hz), 148.3, 139.7, 137.3, 133.6, 130.7, 130.1, 129.1, 128.9, 127.9, 127.7, 127.6, 127.4 (d, 3JCF = 3.4 Hz), 127.3 (d, 3JCF = 3.9 Hz), 121.2 (d, 2JCF = 9.4 Hz), 121.1 (d, 2JCF = 13.1 Hz), 116.6 (d, 2JCF = 10.2 Hz), 116.5 (d, 2JCF = 9.4 Hz), 66.1, 66.0, 47.0, 42.0, 42.2. 19F NMR (376 MHz, CDCl3): −132.40 (s, 1F), −132.84 (s, 1F), −141.90–−142.60 (m, 1F), −143.21–−143.90 (m, 1F). HRMS (ESI) m/z: [M + H]+ calcd. for C37H30BCl2F4N4O4+ 751.1668; found 751.1669.
10-(2,4-Dimethoxyphenyl)-1,5,5,9-tetrafluoro-3,7-di(morpholine-4-carbonyl)-2,8-diphenyl-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5i). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (98:2) to DCM/EtOAc (3:1). Yield: 0.086 g (76%). Violet solid; mp 325–327 °C (with decomp.). The NMR spectra of the sample are complex due to the presence of two sets of signals corresponding to two stable rotamers in ratio 1:1, where many similar signals overlap. 1H NMR (400 MHz, CDCl3): δ 7.47–7.29 (m, 10H), 7.04 (dd, J = 9.0, 3.0 Hz, 1H), 6.94 (d, J = 9.1 Hz, 1H), 6.86 (d, J = 2.1 Hz, 1H), 3.87–3.79 (m, 2H), 3.76 (s, 3H), 3.74 (s, 3H), 3.77–3.58 (m, 4H), 3.57–3.46 (m, 2H), 3.43–3.25 (m, 4H), 3.03–2.89 (m, 2H), 2.80–2.67 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 160.8, 160.7, 158.6 (d, 1JCF = 285.8 Hz), 153.4, 151.1, 146.8, 146.6, 142.4, 129.0, 128.7, 127.9, 127.8 (d, 3JCF = 3.2 Hz), 121.8 (d, 2JCF = 13.3 Hz), 121.3 (d, 2JCF = 13.3 Hz), 119.4, 117.4, 116.1 (d, 2JCF = 7.2 Hz), 116.0 (d, 2JCF = 8.7 Hz), 115.6, 112.0 (s), 66.1, 66.0, 56.1, 55.9, 47.0, 42.2, 42.1. 19F NMR (376 MHz, CDCl3): δ −131.24 (s, 1F), −133.17 (s, 1F), −141.53–−142.36 (m, 1F), −143.23–−143.96 (m, 1F). HRMS (ESI) m/z: [M + Na]+ calcd. for C39H35BF4N4NaO6+ 765.2478; found 765.2479.
1,5,5,9-Tetrafluoro-3,7-di(morpholine-4-carbonyl)-2,8-diphenyl-10-(2,4,5-trimethoxyphenyl)-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5j). The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from DCM/EtOAc (6:1) to DCM/EtOAc (3:1). Yield: 0.064 g (56%). Violet solid; mp 195–197 °C. The NMR spectra of the sample are complex due to the presence of two sets of signals corresponding to two stable rotamers in ratio 1:1, where many similar signals overlap. 1H NMR (400 MHz, CDCl3): δ 7.47–7.30 (m, 10H), 6.82 (d, J = 2.3 Hz, 1H), 6.59 (s, 1H), 3.97 (s, 3H), 3.79 (s, 3H), 3.88–3.68 (m, 4H), 3.77 (s, 3H), 3.67–3.56 (m, 2H), 3.55–3.44 (m, 2H), 3.41–3.23 (m, 4H), 3.00–2.87 (m, 2H), 2.74–2.62 (m, 2H). 13C{1H} (100 MHz, CDCl3): δ 160.9, 160.8, 158.3 (d, 1JCF = 283.2 Hz), 158.1 (d, 1JCF = 285.5 Hz), 152.8, 152.6, 146.1, 145.8, 143.1, 142.8, 129.0, 128.6 (d, 3JCF = 3.5 Hz), 128.0 (d, 3JCF = 2.9 Hz), 128.0, 127.9, 127.8, 122.0 (d, 2JCF = 12.4 Hz), 121.1 (d, 2JCF = 11.8 Hz), 116.0 (d, 2JCF = 9.6 Hz), 115.9 (d, 2JCF = 7.9 Hz), 113.9 (d, J = 3.6 Hz), 109.7, 96.4, 66.1, 66.0, 66.0, 56.6, 56.2, 56.2, 47.0, 42.2, 42.1. 19F NMR (376 MHz, CDCl3): δ −131.45 (s, 1F), −133.70 (s, 1F), −141.55–−142.33 (m, 1F), −143.09–−143.77 (m, 1F). HRMS (ESI) m/z: [M + Na]+ calcd. for C40H37BF4N4NaO7+ 795.2584; found 795.2585.
Procedure for the Preparation of BODIPYs 5k.
Diethyl 5,5′-((2,4,5-trimethylphenyl)methylene)bis(4-fluoro-3-phenyl-1H-pyrrole-2-carboxylate) (0.090 g, 0.15 mmol, 1 mol equiv) and DDQ (0.102 g, 0.45 mmol, 3 mol equiv) were loaded into a screw cap vial (30 mL) for the microwave reactor Nova-2S (PreeKem) and dissolved in MeCN (7.5 mL). The reaction mixture was heated under microwave irradiation at 80 °C for 2 h with stirring. After completion of the oxidation step (TLC monitoring), the reaction mixture was cooled to room temperature. Then, triethylamine (0.209 mL, 1.5 mmol, 10 mol equiv) was added and the resulting mixture was stirred for 3 min. Next, BF3⋅Et2O (0.278 mL, 2.25 mmol, 15 mol equiv) was added to the reaction mixture. The vial with the resulting mixture was purged with argon and then heated under microwave irradiation at 80 °C for 2 h with stirring.
After completion of the reaction (TLC monitoring), the reaction mixture was diluted with EtOAc (100 mL) and extracted with water (5 × 100 mL). Then, the organic layer was separated, dried over Na2SO4, filtered and concentrated under a vacuum. The pure product was isolated by column chromatography with gradient elution on silica gel using SepaBean™ machine T (Santai Science Inc., Montréal, QC, Canada). Polarity of the elution system was gradually increased from Hex to Hex/EtOAc (1:1).
3,7-Bis(ethoxycarbonyl)-1,5,5,9-tetrafluoro-2,8-diphenyl-10-(2,4,5-trimethylphenyl)-5H-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-4-ium-5-uide (5k). Yield: 0.055 g (57%). Red solid; mp 210–212 °C. 1H NMR (400 MHz, CDCl3): δ 7.42–7.27 (m, 10H), 7.10 (s, 1H), 7.04 (s, 1H), 4.41 (q, J = 7.1 Hz, 4H), 2.29 (s, 3H), 2.27 (s, 3H), 2.26 (s, 3H), 1.30 (t, J = 7.1 Hz, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 161.2, 159.3 (d, 1JCF = 285.5 Hz), 148.1, 144.2, 139.2, 134.3, 132.7, 131.9, 128.8, 128.6, 128.6, 128.5, 128.0 (d, 3JCF = 1.4 Hz), 127.3, 121.90 (d, 2JCF = 10.5 Hz), 118.3 (d, 2JCF = 10.2 Hz), 62.9, 19.8, 19.3, 19.1, 13.8. 19F NMR (376 MHz, CDCl3): δ −131.29 (s, 2F), −141.20–−141.92 (m, 1F), −142.12–−142.82 (m, 1F). HRMS (ESI) m/z: [M + Na]+ calcd. for C36H31BF4N2NaO4+ 665.2205; found 665.2207.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}