1. Introduction
Systemic Lupus Erythematosus (SLE) is a chronic autoimmune disease that causes connective tissue inflammation, often leading to muscle pain, weakness, and rashes, as well as complications such as nephritis, pericarditis, or peripheral neuropathy. While much progress has been made in understanding the pathogenesis of SLE over the past few decades, there are still many unanswered questions with regard to the factors that lead to its development. Nonetheless, numerous genetic and environmental factors are postulated to play a major role [
1,
2,
3,
4,
5,
6,
7,
8]. One condition with a strong correlation to SLE is deficiency in the complement system molecule C1q [
3,
5,
6,
7,
8]. Although it is a rare multifactorial disease characterized by inflammation in several organ systems, there is abundant clinical evidence showing that homozygous deficiency in any of the classical complement pathway proteins—C1q, C1r, C1s, C4, and C2—predisposes an individual to SLE and other autoimmune diseases [
3]. These proteins function as opsonins and trigger membrane attack complex (MAC) formation through interactions with corresponding cell-surface receptors. Among them, C1q takes center stage in significance, as the recognition component of the C1 complex and the pathway initiator. C1q is a glycoprotein composed of 18 polypeptide chains organized into six globular heads and a collagenous tail. The head and tail domains of the C1q bind to the gC1qR and cC1qR cell-surface receptors, respectively, to function [
8]. Hereditary homozygous deficiency due to mutations in the C1q gene—predominantly a mutation in the A-chain, in which the C to T transition in codon 186 of exon 2 results in Gln-to-Stop (Q186X) substitution—is a powerful susceptibility factor for the development of not only SLE, but also other diseases such as angioedema and rheumatoid arthritis [
5,
6]. In fact, more than 90% of individuals with an inherited C1q deficiency go on to develop SLE. This deficiency is postulated to contribute to SLE development due to the consequent failure of the adequate clearance of apoptotic cells and immune complexes [
7,
8,
9,
10,
11,
12,
13,
14,
15]. However, due to the strong correlation between C1q deficiency and SLE and the existence of alternative and redundant immune mechanisms to account for apoptotic cell clearance, we postulate that C1q and its corresponding receptors play a much more prevalent role in lupus immunoregulation and autoimmunity [
16].
In addition to C1q deficiency, lupus is also hallmarked by an enhanced immune response in CD4+ T-cells. This contributes to a loss of self-tolerance in SLE patients, or the failure of the immune system to not react against one’s own antigens [
3,
17]. CD4+ T-cells, also known as helper T-cells (Th), are thymus-derived lymphocytes that are an essential part of the adaptive immune response. They target specific pathogenic antigens recognized as non-self by the T-cell receptor (TCR) by binding to major histocompatibility complex (MHC) Class II molecules on the foreign antigen-presenting cells. After being activated, naive helper T-cells differentiate into several effector cell types with diverse functions, including the activation of other innate and adaptive immune cells, the creation of memory and plasma B-cells, and the inhibition of exaggerated immune reactions when appropriate [
18,
19]. They do this through cytokine secretion, which is triggered by surface receptor–ligand binding. These pro- or anti-inflammatory cytokines then continue to cascade through interactions with T-cell receptors or the receptors of other surrounding immune and somatic cells [
19].
Autoimmune disorders such as lupus develop due to the loss of tolerance to self-antigens. This is driven by the erroneous activation of autoreactive T-cells or the loss of function in regulator T-cells (Treg), a subset of the CD4+ T-cells [
20]. In SLE, anti-self Th cells breakdown immunity. This is evidenced by the exaggerated inflammatory response in lupus, involving the elevated proliferation of CD4+ T-cells, excessive activation of B-cells and dendritic cells, and altered cytokine secretion at inflammation sites [
21]. Additionally, previous studies have shown that culturing CD4+ T-cells with C1q inhibits proliferation in a dose-dependent manner, suggesting that a lack of C1q-mediated T-cell immunosuppression could contribute to SLE pathogenesis [
21,
22]. Further studies on the role of CD4+ T-cells in the development of SLE and autoimmunity could, therefore, lead to the identification of new therapeutic targets for SLE.
Most functions of the immune system, including the activation of immune cells and the maintenance of self-tolerance, are ultimately regulated by the release of specific pro-inflammatory and anti-inflammatory cytokines, which are small molecules that mediate both the innate and adaptive immune responses [
23]. The major cytokines secreted by CD4+ T-cells and considered essential for normative immune function are interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), transforming growth factor beta (TGF-β), and several interleukins (ILs) [
18]. Of these, those also associated with the development of SLE are IL-2, IL-4, IL-6, IL-10, IL-17, IFN-γ, TNF-α, and TGF-β [
23]. While IL-2, IL-6, IFN-γ, and TNF-α are thought to be pro-inflammatory cytokines [
24], IL-4 is known to have anti-inflammatory functions. Treg cells secrete IL-10 and TGF-β to suppress autoreactive helper cell activation, preventing autoimmunity [
19,
24]. Additionally, interestingly, while IFN-γ is centrally recognized for its pro-inflammatory effect and phagocytic activation, some studies have also shown that IFN-γ suppresses the inflammatory response, promoting Treg differentiation and apoptotic mediator expression [
18,
25,
26]. Therefore, it is believed to have both pro-inflammatory and anti-inflammatory effects [
23].
In the present work, we investigated IL-6 and TNF-α as pro-inflammatory cytokines and IL-10 and IFN-γ as anti-inflammatory cytokines, all of which are elevated in serum samples of SLE patients and associated with disease activity [
23]. The first of these, IL-6, is secreted by macrophages in response to the binding of pathogens to cell-surface receptors, such as Toll-like receptors. This induces the cellular transduction pathways leading to TNF-α and further IL-6 production, establishing a positive feedback loop of immune stimulation. Some known functions of IL-6 are acute phase inflammatory protein synthesis, neutrophil production in the bone marrow, and Treg cell suppression, thereby promoting immunogenicity and decreasing tolerance [
27]. Mouse model experiments also found that IL-6 can specifically exacerbate T-cell driven immunity by inducing IL-17 and promoting T-cell proliferation [
28]. TNF-α works in conjunction with IL-6 in many of these pathways, including the acute phase response, and can trigger local inflammatory symptoms such as heat, swelling, etc. Additionally, TNF-α assists with neutrophil migration and induced phagocytosis [
29]. In contrast, IL-10 inhibits the synthesis of pro-inflammatory cytokines, including IL-2, IL-3, TNF-α, and granulocyte–macrophage colony-stimulating factor (GM-CSF), a hematopoietic glycoprotein produced by macrophages and Th1 cells assisting with white blood cell production. IL-10 has also been shown to suppress immune cell capacity to present antigens and trigger an immune response [
30]. As mentioned earlier, IFN- γ has pleiotropic functions. Under normal immune conditions, it serves to activate macrophages and induces the expression of MHC Class II proteins [
31,
32,
33]. However, it has also been shown to contradictorily ameliorate autoimmune disorders like SLE by suppressing the inflammatory response, increasing the expression of apoptotic mediators to cause cell death, and suppressing the differentiation of Th17 cells and osteoclasts [
25].
With both C1q deficiency and irregular CD4 + T-cell signaling playing major roles in SLE development, we investigated the interaction of these two factors to gain new insight into lupus progression. These experiments aim to re-affirm C1q’s inhibition of T-cell proliferation and explore the mechanism of this function via the relationship between C1q and its receptors, gC1qR and cC1qR, and the secretion of the identified central cytokines. The results of these studies will not only give better insight into the role of C1q in the development of SLE, but could also be used to design novel therapeutic options for lupus treatment.
3. Discussion
The role of the complement system, and the role of C1q in particular, in the pathogenesis of SLE is very well established [
1,
2,
3,
4,
5,
6,
7,
8]. However, previous research has singularly focused on proving the postulate that deficiency in C1q leads to the inadequate removal of apoptotic cells, thereby contributing to autoimmune responses and lupus progression. Although C1q may indeed play a role in the removal of apoptotic debris, there are other well-orchestrated mechanisms that would ensure that apoptotic cleanup is duly regulated, even in the absence of C1q [
12]. In addition, although C1q expression on the surface of a wide range of cell types has been demonstrated [
12], the functions of cell-surface C1q and of cells that express both C1q and the gC1qR receptor are still incompletely understood. This study therefore focused on examining the role of cell-surface-expressed C1q and its function in SLE in order to understand why a deficiency in C1q predisposes one to develop autoimmune diseases such as lupus. Since cytokines play an essential role in regulating inflammation, we investigated whether the influence of C1q on the secretion and suppression of specific cytokines could be, in part, responsible for the loss of self-tolerance that largely contributes to the development and progression of SLE.
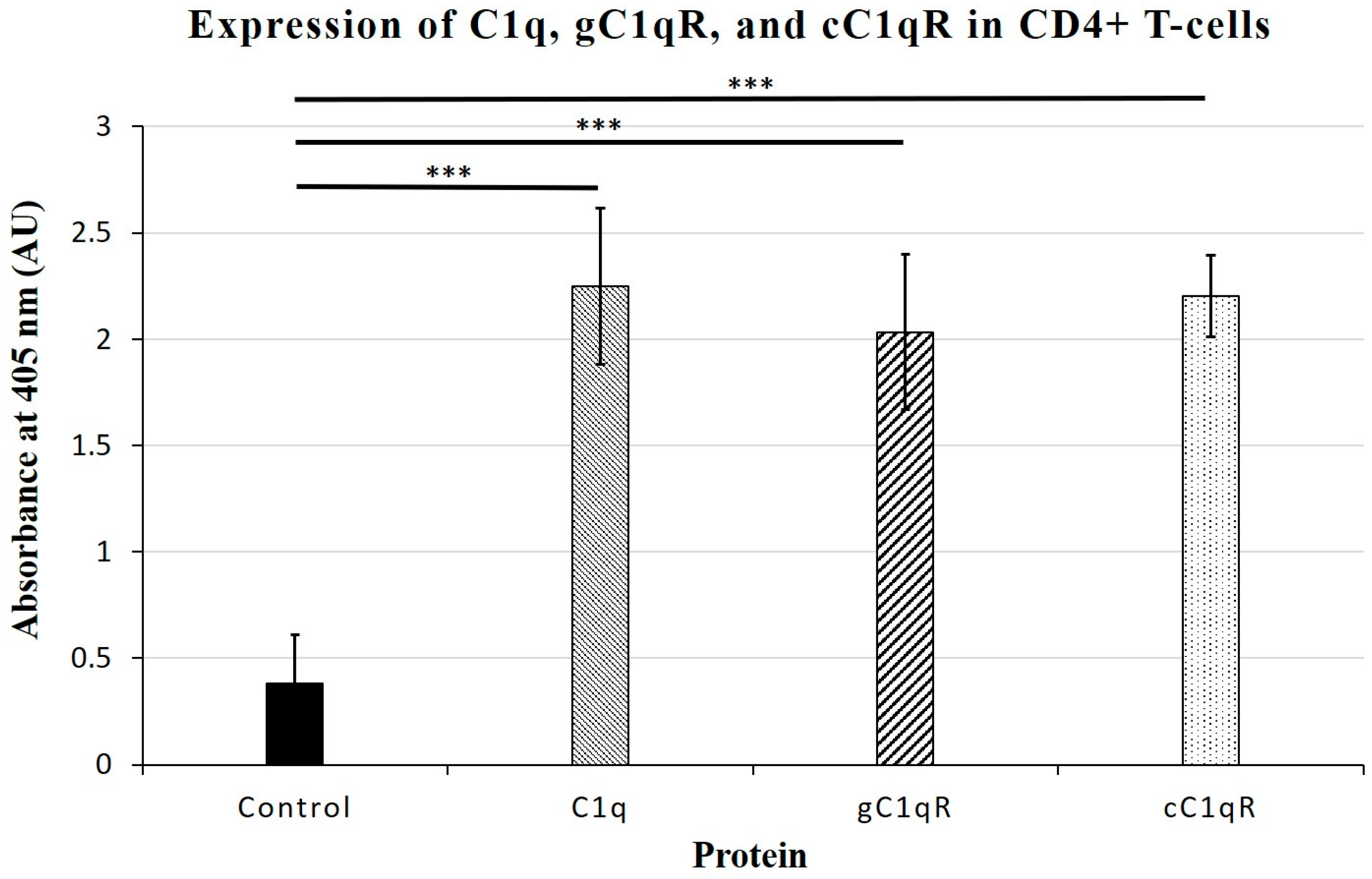
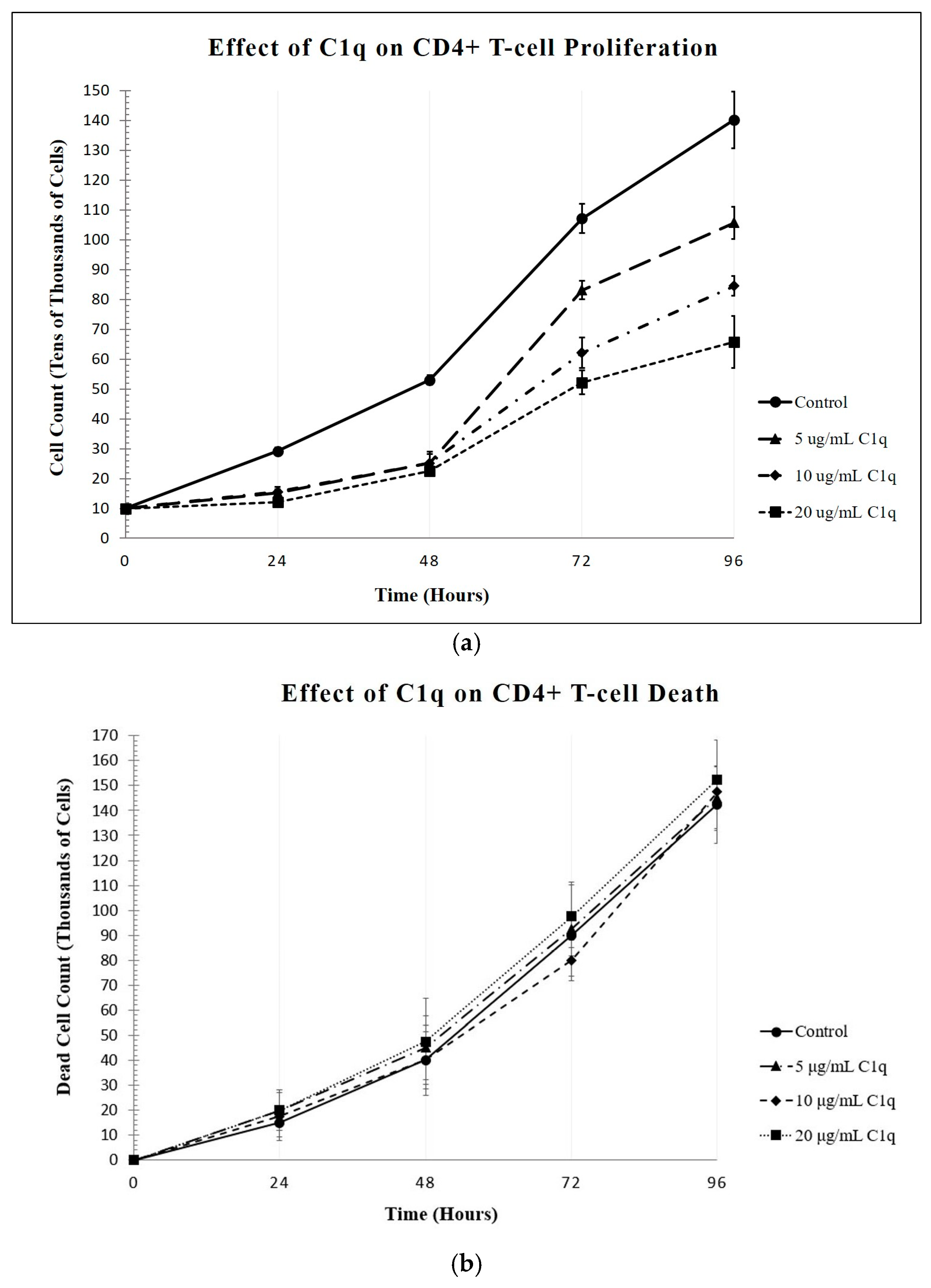
Our studies show that CD4+ T-cells expressed cell-surface C1q, in addition to the receptors for its globular and collagen-like regions, gC1qR and cC1qR, respectively. Therefore, under the assumption that the logic of “structure-serves-function” holds true, this finding likely signifies that C1q and its receptors play essential roles in the regulation of the T-cell immune response. Employing a cell proliferation assay, we show here that the culturing of T-cells with C1q did, in fact, significantly inhibit the proliferation of the T-cells, which is consistent with our previously published observations [
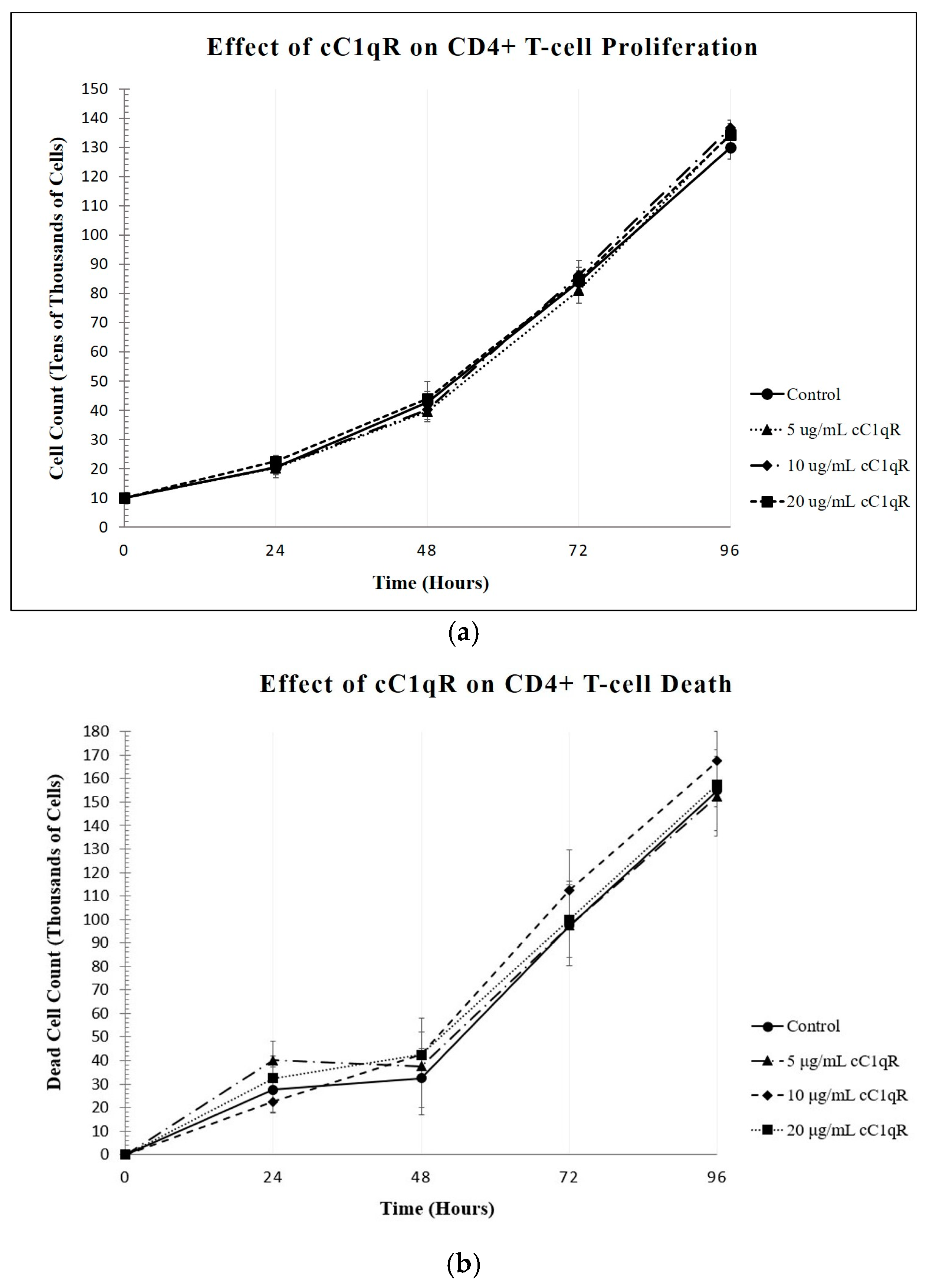
22]. Since the live cell counts decreased while the dead cell counts remained the same, C1q appears to inhibit the proliferation of these cells without damaging them. Furthermore, this method was used to show that treating CD4+ T-cells with gC1qR also inhibited proliferation in a similar fashion. In contrast, cC1qR did not significantly impact T-cell proliferation. This is postulated to be a result of the fact that cC1qR binds predominantly to a region in the collagen tail of the C1q A-chain, through which the molecule is anchored to the cell membrane, thereby making the cC1qR site unavailable for binding. Alternatively, this could also signify that the binding of cC1qR to surface C1q in T-cells may have a heretofore unidentified function that relates to another means of lupus development.
These findings raise an interesting proposition for the mechanism by which T-cells independently regulate population growth intercellularly. If T-cells have both gC1qR and the exposed globular heads of C1q on their surfaces, gC1qR may be released into the pericellular milieu as the population density increases and cell crowding occurs, which we have previously demonstrated [
22]. The surface C1q of one T-cell will then easily come into contact with the globular receptor (gC1qR) of another, and this binding could trigger an anti-proliferative pathway and control the T-cell population size. More importantly, it also suggests that genetic C1q deficiency could cause a loss of this self-regulation, as the deficient T-cells which lack soluble and/or cell-surface-expressed C1q would proliferate uncontrollably, and the inhibitory effects of surface C1q on other T-cells would decrease. Overactivity and loss of tolerance could, in turn, easily lead to the central symptoms seen in SLE. Furthermore, the presence of abnormally high levels of C1q would be expected to diminish the proliferation of CD4+ T-cells, and thus hinder a healthy immune response. Similarly, a C1q molecule displayed on any malignant cell could suppress the proliferative potential of a T-cell by binding to gC1qR in a manner akin to PD-L1 and PD-1 interactions.
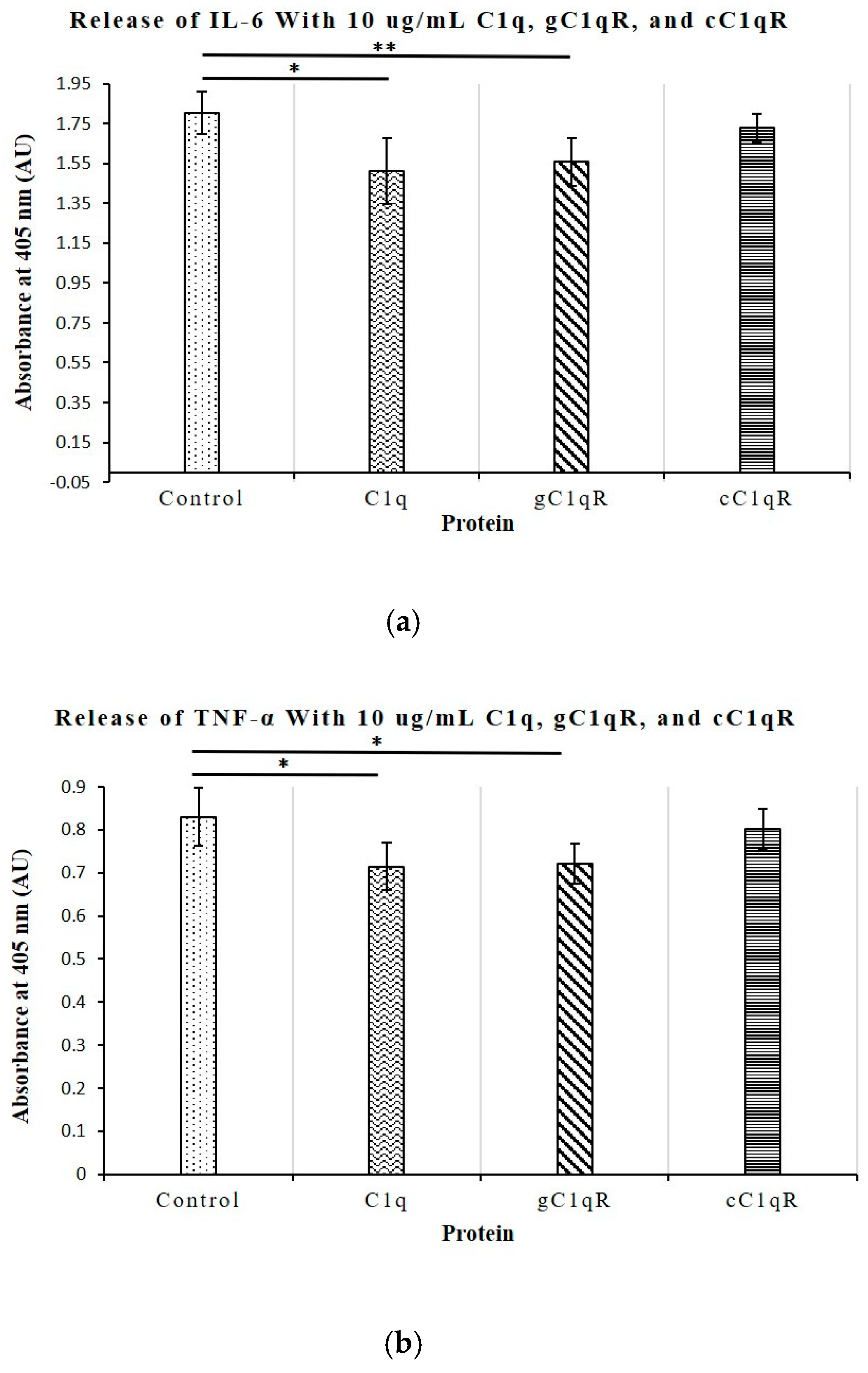
To better understand the role of C1q in T-cell proliferation, we also investigated its relationship with the secretion of the key cytokines IL-6, TNF-α, IL-10, and IFN-γ, all of which play a role in the development of SLE and are upregulated in diseased serum samples. Of these cytokines, only the levels of IL-6 and TNF-α, which are known to contribute to the induction of a hyperactive immune system, were significantly inhibited by C1q and gC1qR. None of the cytokine levels were affected by cC1qR. This observation mirrors the results of previous experiments, which demonstrated that the co-culturing of T cells with C1q or gC1qR limits T cell proliferation [
22]. The lack of effect observed with cC1qR further reinforces the suggestion that cC1q does not contribute to lupus development through the regulation of T cell proliferation and cytokine production, though it may potentially do so via other indirect pathways.
In summary, the results obtained in these studies seem to suggest that C1q deficiency is intimately linked to the overproduction of the potent pro-inflammatory cytokines IL-6 and TNF-α. Previous studies have demonstrated that IL-6 regulates and promotes CD4+ T cell proliferation, thereby creating an exaggerated immune response and thus leading to autoimmunity and the development of SLE. Further investigation into the roles of C1q and gC1qR on the surface of CD4+ T cells could not only provide new insight into the roles of these molecules in the immune activation leading to SLE, but may also help in the design of new therapeutic treatment options in a manner similar to treatments of angioedema with recombinant C1.
4. Materials and Methods
4.1. Chemicals and General Reagents
The following reagents and chemicals were purchased or obtained from the sources indicated: RPMI 1640, 100× Penicillin/Streptomycin, and trypsin/EDTA (Invitrogen, Carlsbad, CA, USA); heat-inactivated FBS (Hyclone, Logan, UT, USA); Dulbecco’s PBS (D-PBS) without calcium and magnesium (Mediatech Inc, Manassas, VA, USA); alkaline phosphatase (AP)-conjugated Streptavidin (Invitrogen, Carlsbad, CA, USA); p-nitrophenyl phosphate (pNPP) (Pierce, Rockford, IL, USA); and BSA (Thermo Fisher, Waltham, MA, USA).
4.2. Proteins and Antibodies
Rabbit antibodies to C1q and recombinant human gC1qR/p33, as well as human cC1qR/calreticulin, were made and purified in our laboratory and have been described in previous publications [
32,
33]. Purified human C1q was purchased from Quidel (San Diego, CA, USA). The receptors of the globular heads and collagen stalks of C1q, gC1qR, and cC1qR were purified from human cell lines, as described in previous publications [
34]. Cytokine antibodies, such as rat anti-IL-6, anti-IL-10, anti-IFN-γ, or anti-TNF-α, were obtained from Thermo Fisher (Waltham, MA, USA).
4.3. Cultured Cells
The Molt-4 cell line was originally developed from cells taken from a 19-year-old male patient with T cell lymphoblastic acute cell leukemia in relapse (ATCC CRL-1582). Molt-4 CD4+ T cells can be utilized as proxies for SLE CD4+ T cells in mechanistic and molecular signaling experiments such as these. They provide an advantage in experimental convenience due to their indefinite proliferation and easier accessibility than primary SLE patient samples. Furthermore, we have successfully used these cells previously to show that the incubation of these cells with C1q induces an anti-proliferative response [
22]. The cells were incubated in a 37 °C incubator with 5% CO
2 and grown in RPMI 1640 medium with 10% FBS and 1% Penicillin/Streptomycin antibiotic solution (Pen-Strep).
The cell suspension was incubated in 10 mL of media in a T25 Falcon flask. After 48 h intervals, 100 μL of cells were taken out, to which 10 μL of Trypan blue dye was added, and the cells were evaluated for viability by counting under a microscope using a hematocytometer.
4.4. Enzyme-Linked Immunosorbent Assays (ELISAs)
Both indirect and direct whole-cell ELISAs were conducted in clear, 96-well, flat-bottomed, tissue culture-treated Falcon plates. For the indirect ELISAs, the plates were first coated with poly-L-lysine, and 105 Molt-4 cells per well suspended in 100 μL of PBS were then added. The plates were centrifuged for 5 min at 1500 rpm and incubated with 100 μL of 0.5% glutaraldehyde in cold PBS for 30 min. The wells were washed with PBS and the unreacted sites blocked with 0.1% BSA in PBS. After washing, an indirect ELISA was conducted with primary Rabbit anti-C1q, anti-gC1qR, or anti-cC1qR, followed by reaction with AP-Donkey-anti-Rabbit. Then, 100 μL of diluted pNPP was added for color development, and the relative absorbance of the wells was measured at 405 nm using an ELISA plate reader.
For the direct ELISAs, 106 Molt-4 cells were first cultured (37 °C for 36 h) in 1 mL of RPMI 1640 medium with 10% FBS and 1% Pen-Strep in duplicate wells of a 24-well, flat-bottomed, tissue culture-treated plate in the presence or absence of 10 μg/mL of either C1q, gC1qR, or cC1qR. After incubation, the samples were centrifuged for 5 min at 1100 rpm, and 100 μL of supernatant was collected. The wells of a 96-well plate were then coated with the supernatant, and after incubation (1 h, 37 °C), the wells were blocked with 300 μL of TBS-BSA. Then, biotinylated rat anti-IL-6, anti-IL-10, anti-IFN-γ, or anti-TNF-α were added and incubated for 60 min at 37 °C, followed by additional incubation with AP-conjugated Streptavidin. Finally, the color development was visualized by the addition of 100 μL of diluted pNPP, and the relative absorbance of the wells was measured at 405 nm using a plate reader.
4.5. Cell Proliferation Assays
The Molt-4 cells used in the cell proliferation assays were cultured as described above at 37 °C with 5% CO2 in RPMI 1640 medium, 10% FBS, and 1% Pen-Strep. The cells were plated in clear 24-well flat-bottomed Falcon plates, with each well containing 1 mL of 1 × 106 cells/mL. The cells were then treated with increasing concentrations (0 μg/mL, 5 μg/mL, 10 μg/mL, and 20 μg/mL) of purified C1q, gC1qR, or cC1qR.
The assay was run for a total of 96 h, allowing the cells to grow in the treated media at 37 °C with 5% carbon dioxide (CO2). At 24 h intervals, data were collected from an undisturbed column of wells with two samples of each treatment, and the cells were photographed. Then, 100 μL of each sample was taken out, and the cell viability was assessed by the addition of 10 μL of Trypan blue dye as described above.
4.6. Statistical Analysis
Statistical analyses were calculated using GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA, USA) and Excel (Microsoft, Redmond, Washington, NA, USA). Nonparametric ANOVA tests were applied to compare the control and experimental groups throughout. Differences were considered significant for p < 0.05 (n = separate experiments performed in duplicates).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}