

Chronic Exposure to Two Regimens of Waterpipe Smoke Elicits Lung Injury, Genotoxicity, and Mitochondrial Impairment with the Involvement of MAPKs Activation in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
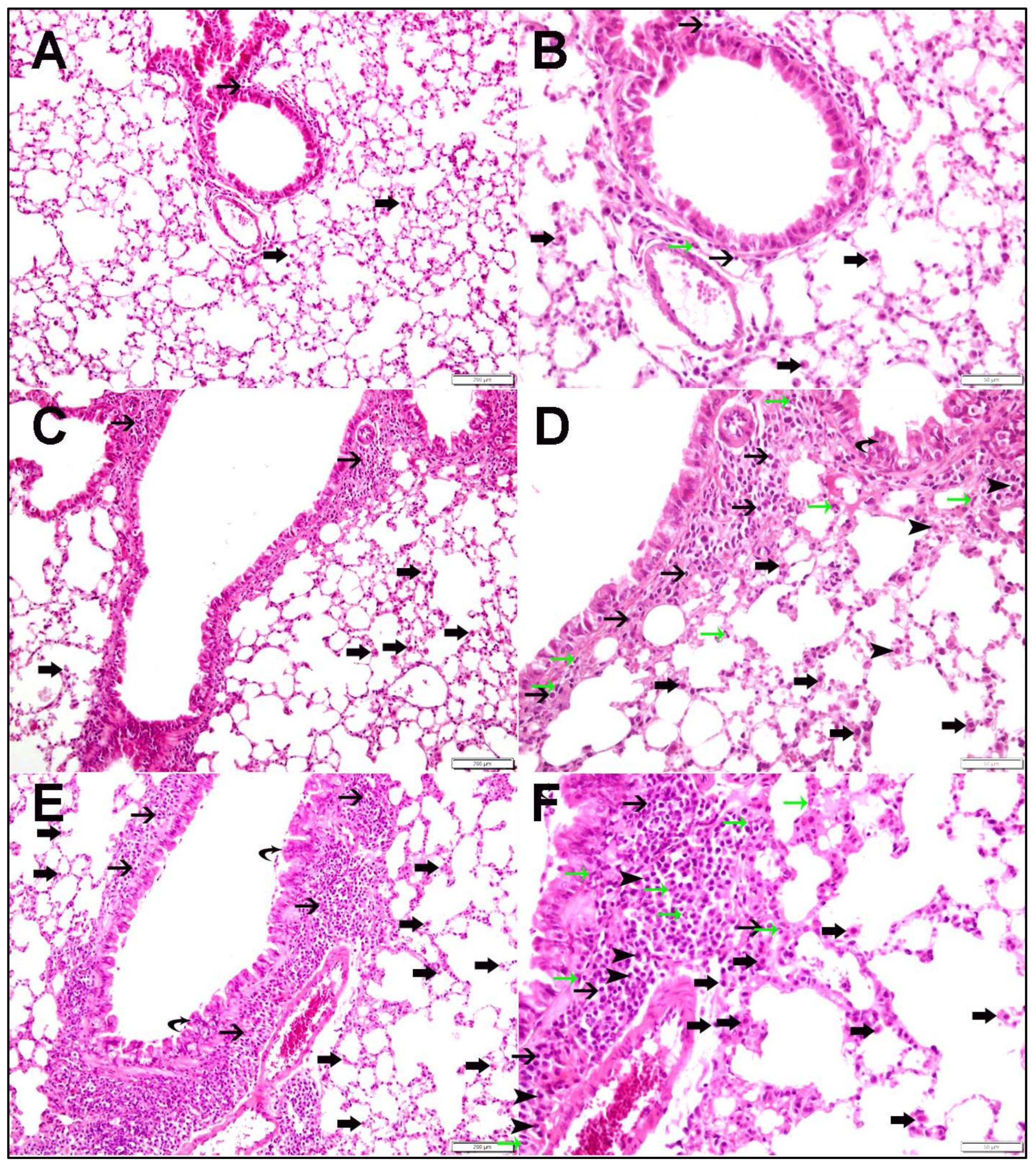
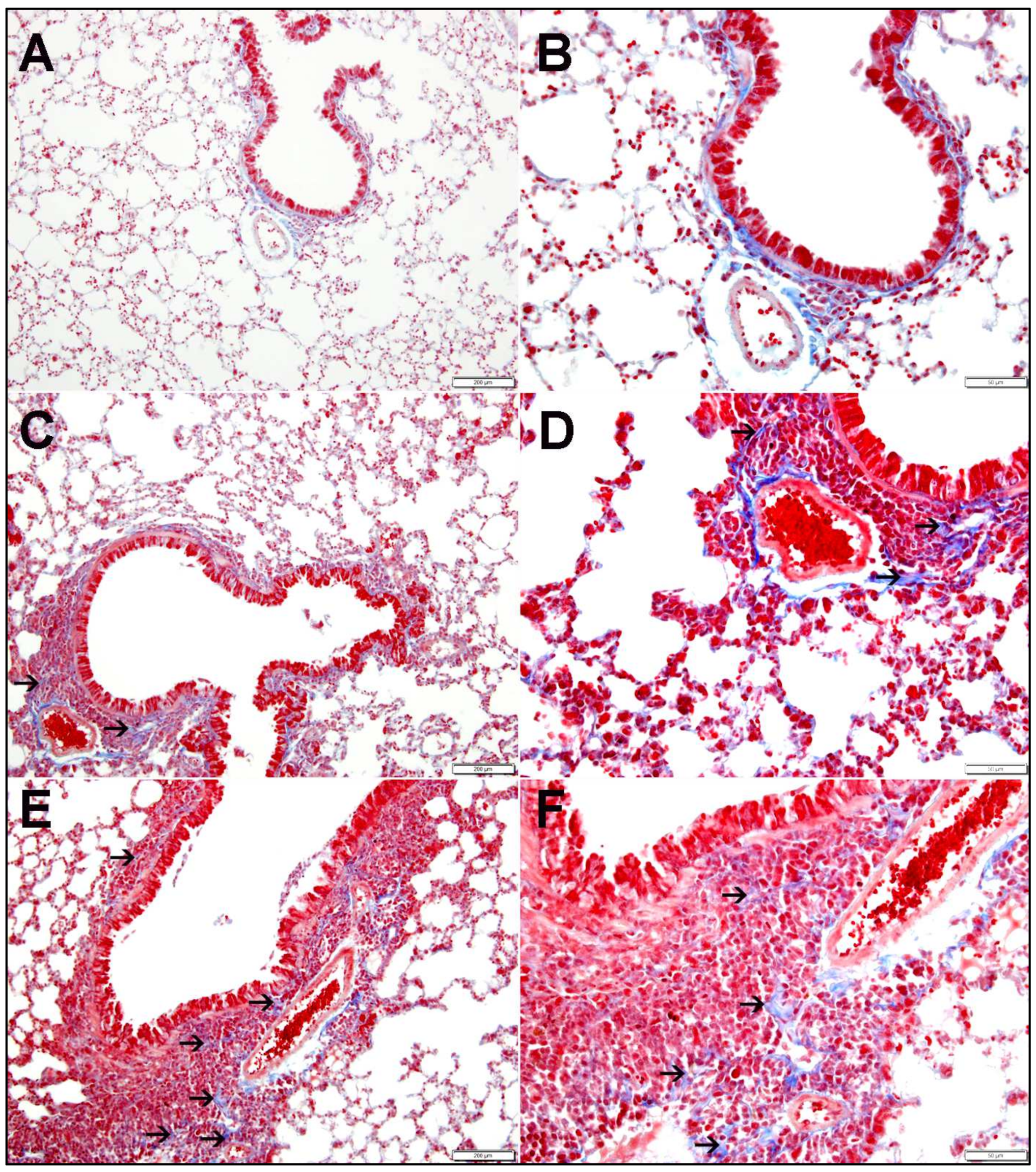
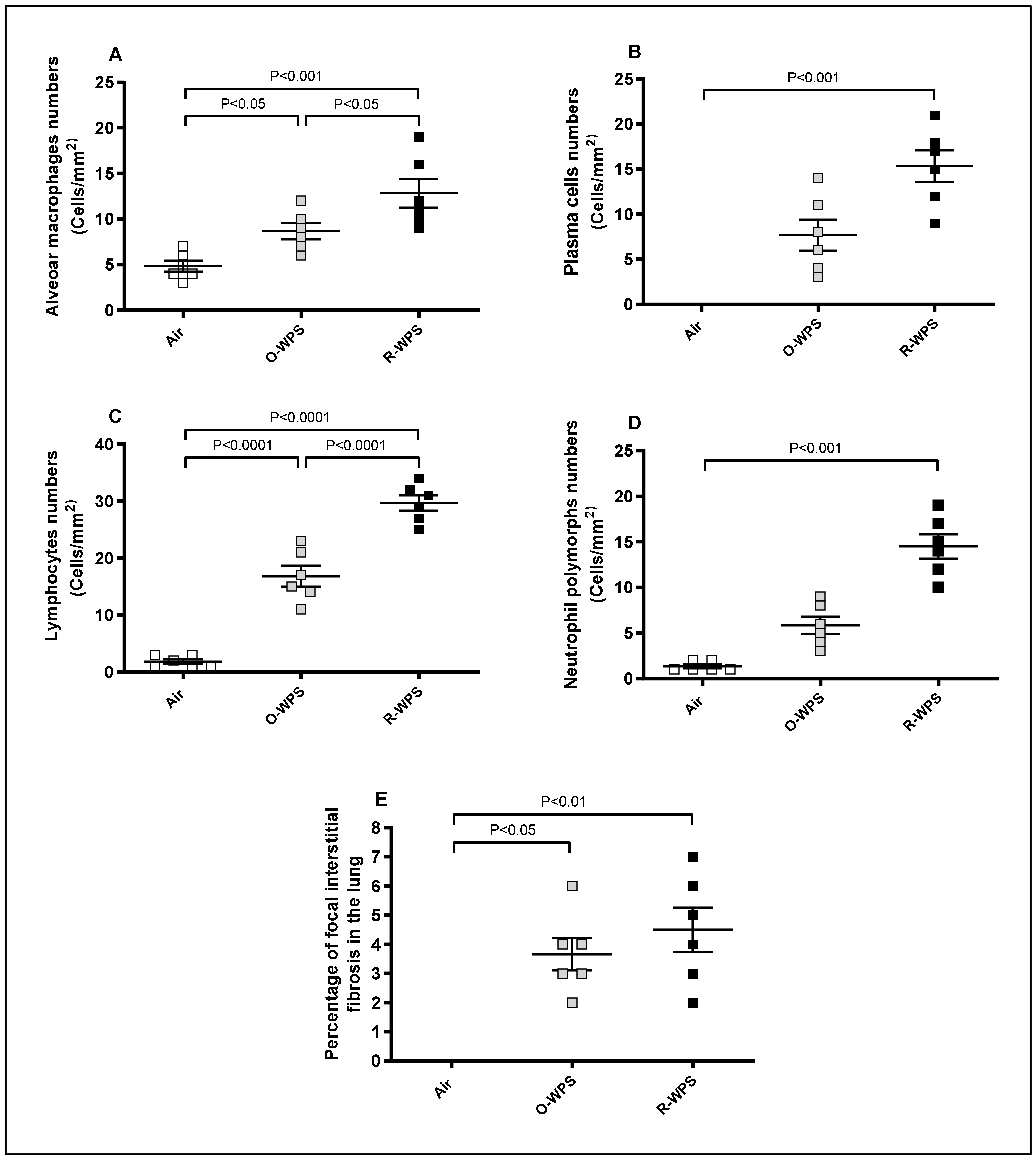
2.1. Lung Histopathology
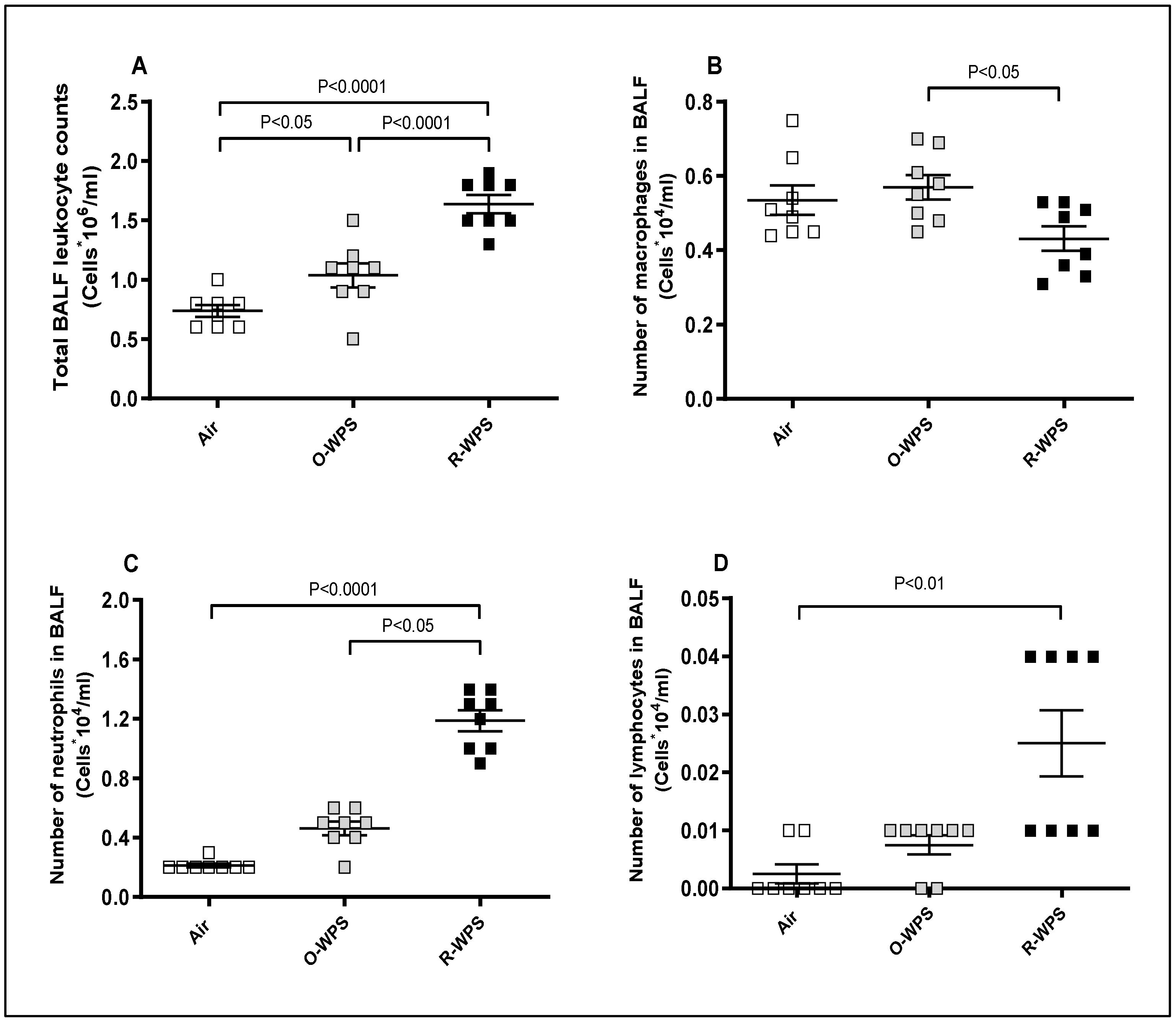
2.2. Cell Counts in the BALF
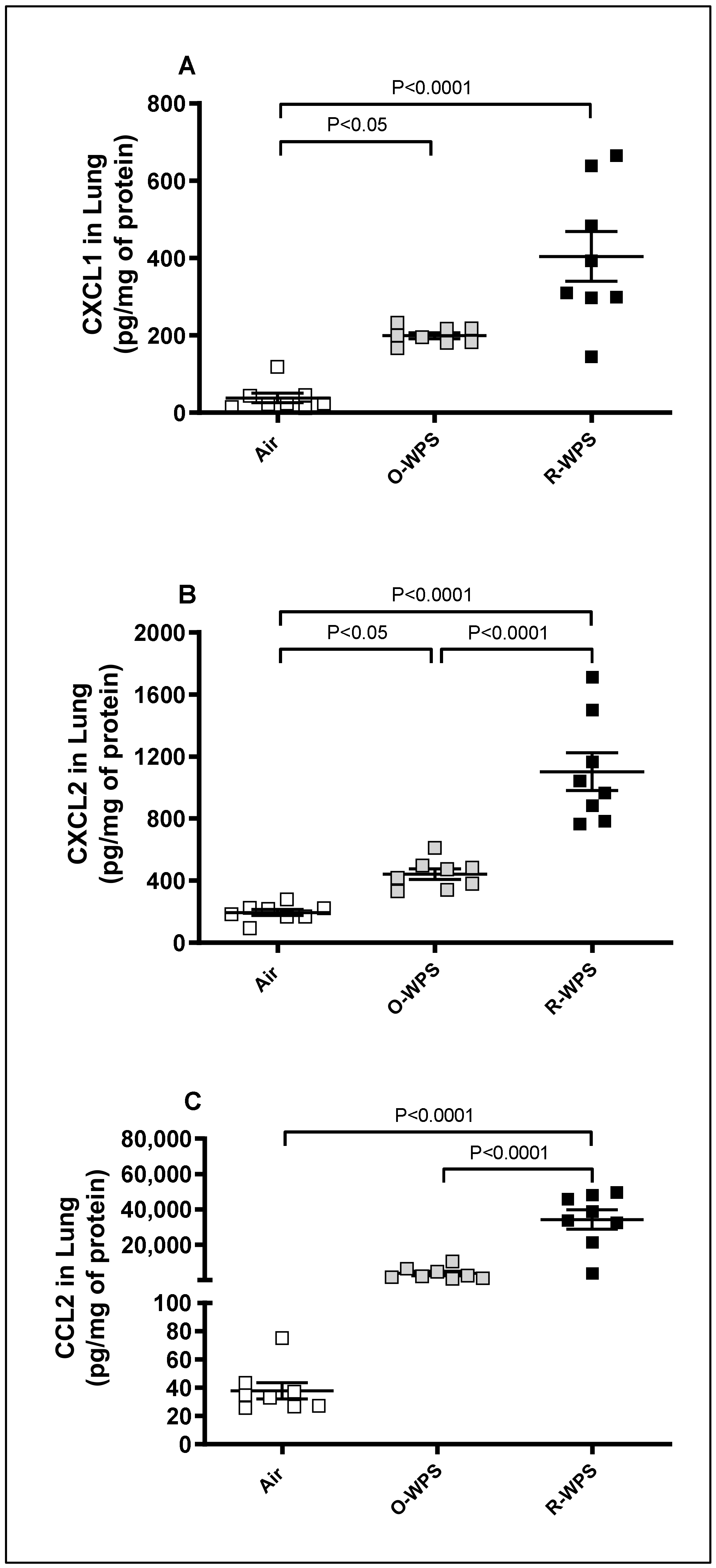
2.3. Concentrations of CXCL1, CXCL2 and CCL2 in Lung Tissue Homogenates
2.4. Concentrations of CXCL1, CXCL2 and CCL2 in BALF
2.5. Concentrations of CXCL1, CXCL2 and CCL2 in Plasma
2.6. Levels of MMP-9, MPO, and LDH in Lung Tissue Homogenates
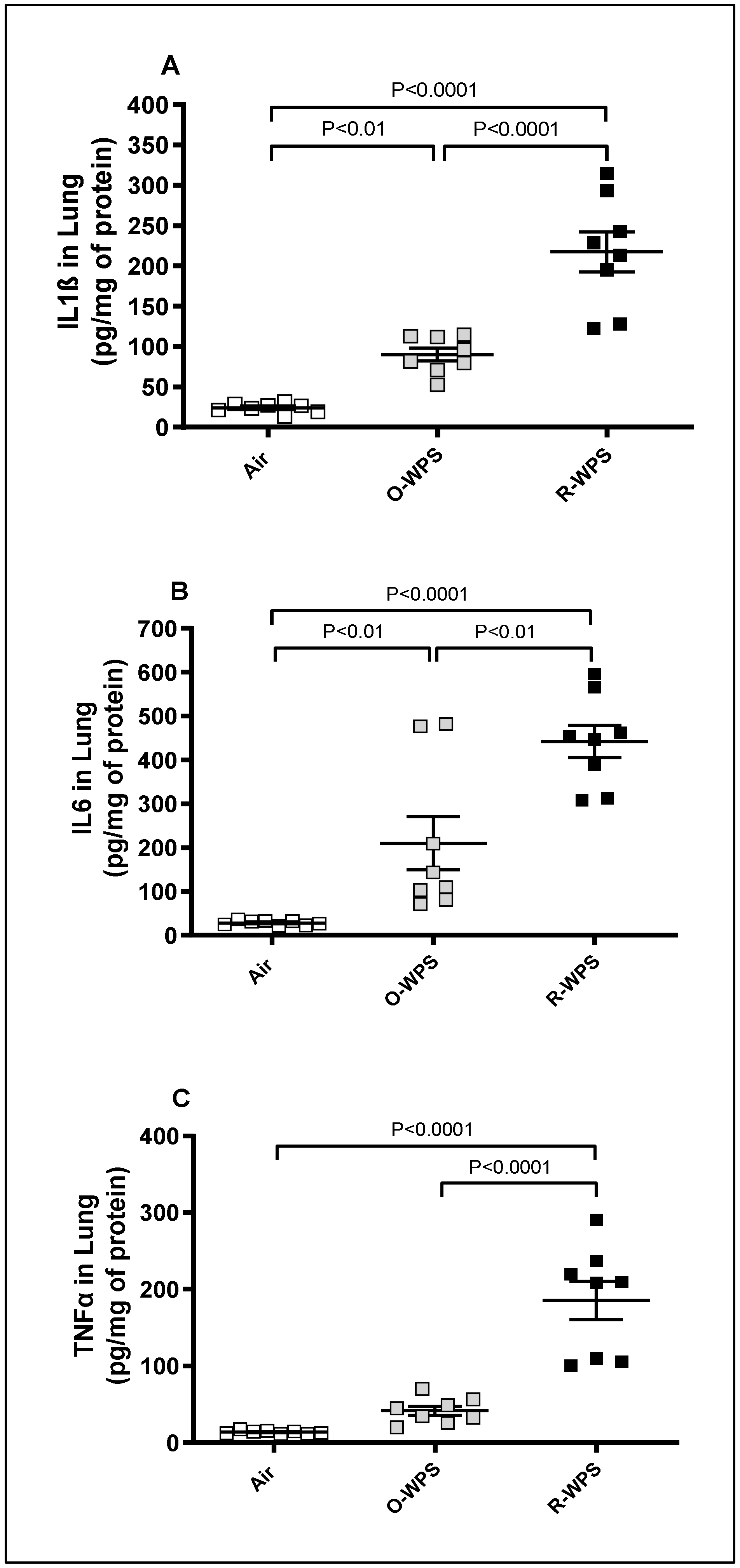
2.7. Concentrations of IL1β, IL6 and TNFα in Lung Tissue Homogenates
2.8. Concentrations of IL1β, IL6 and TNFα in BALF
2.9. Concentrations of IL1β, IL6 and TNFα in Plasma
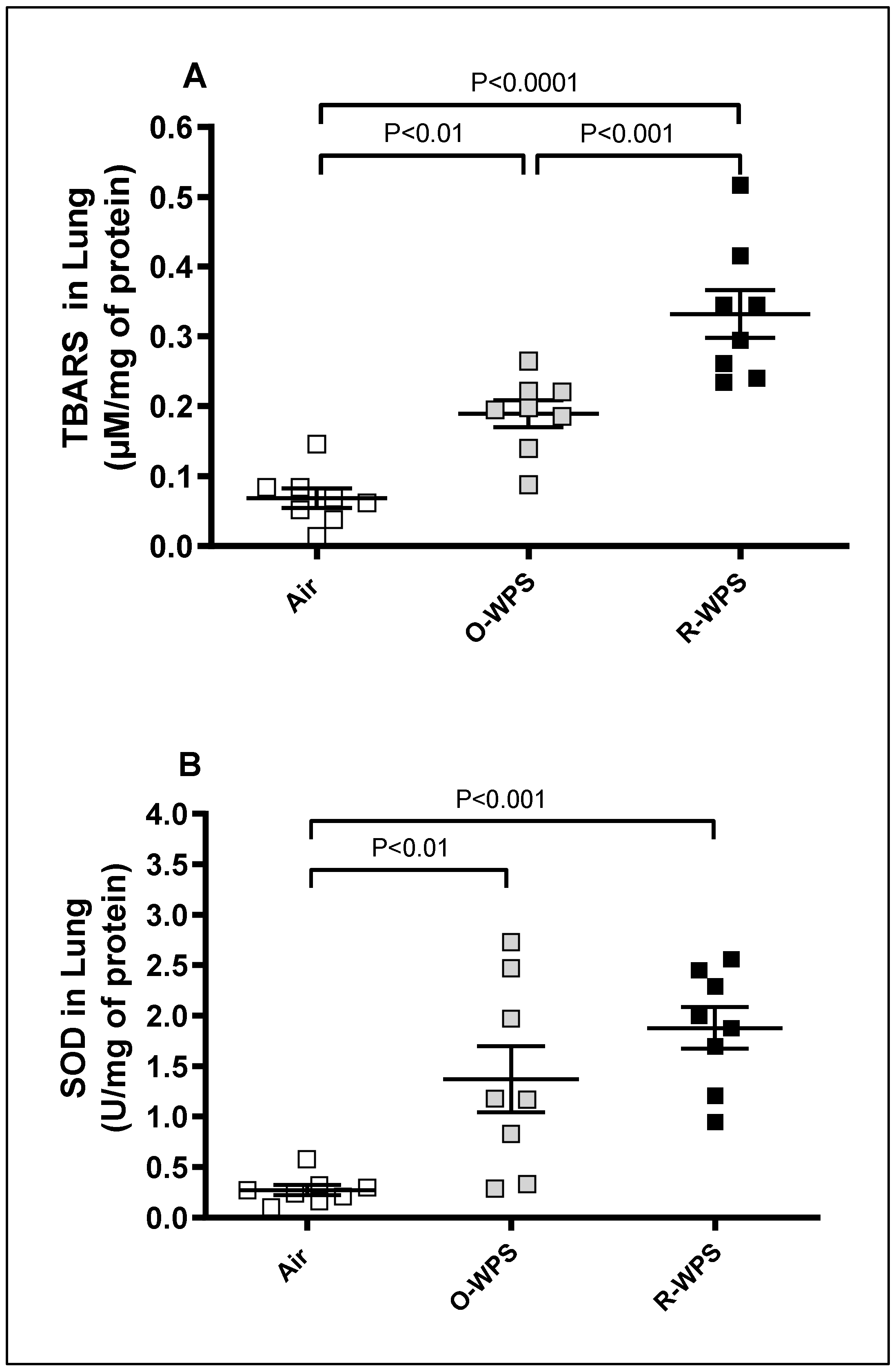
2.10. Levels of TBARS and SOD in Lung Tissue Homogenates
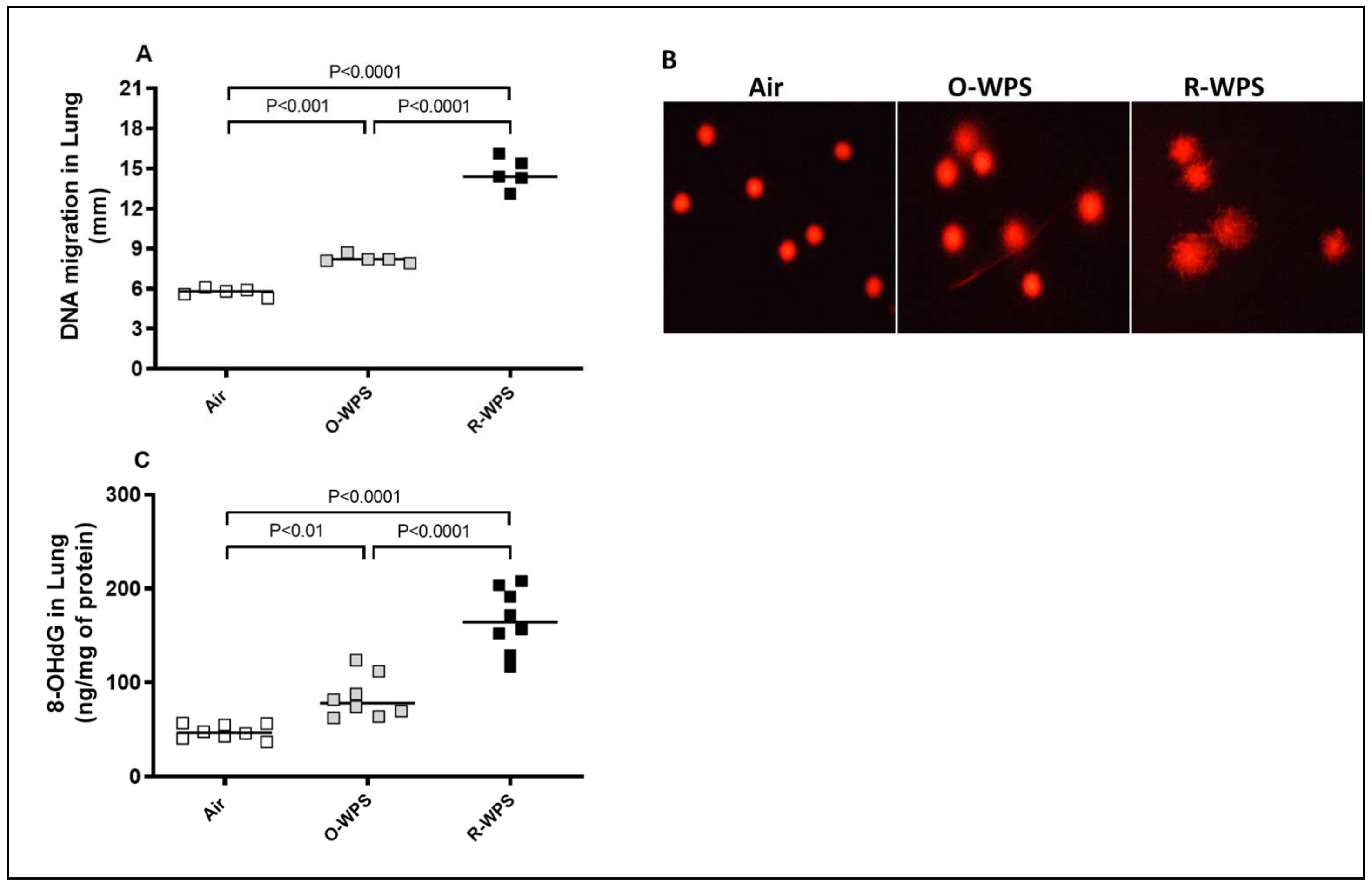
2.11. DNA Migration and Concentrations of 8-OHdG in Lung Tissue Homogenates
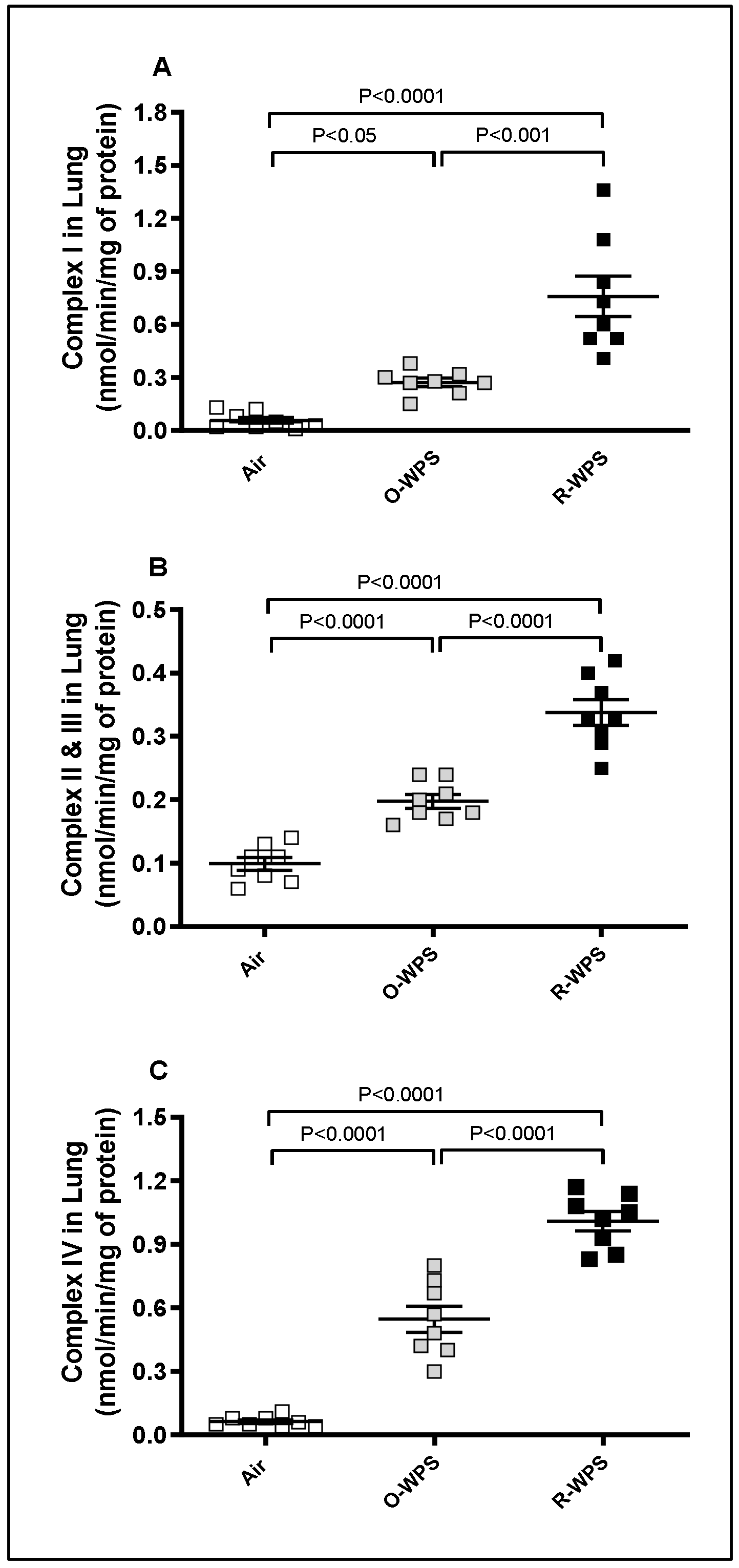
2.12. Mitochondrial Complexes I (A), II and III (B), IV (C) Activities in Lung Tissue Homogenates
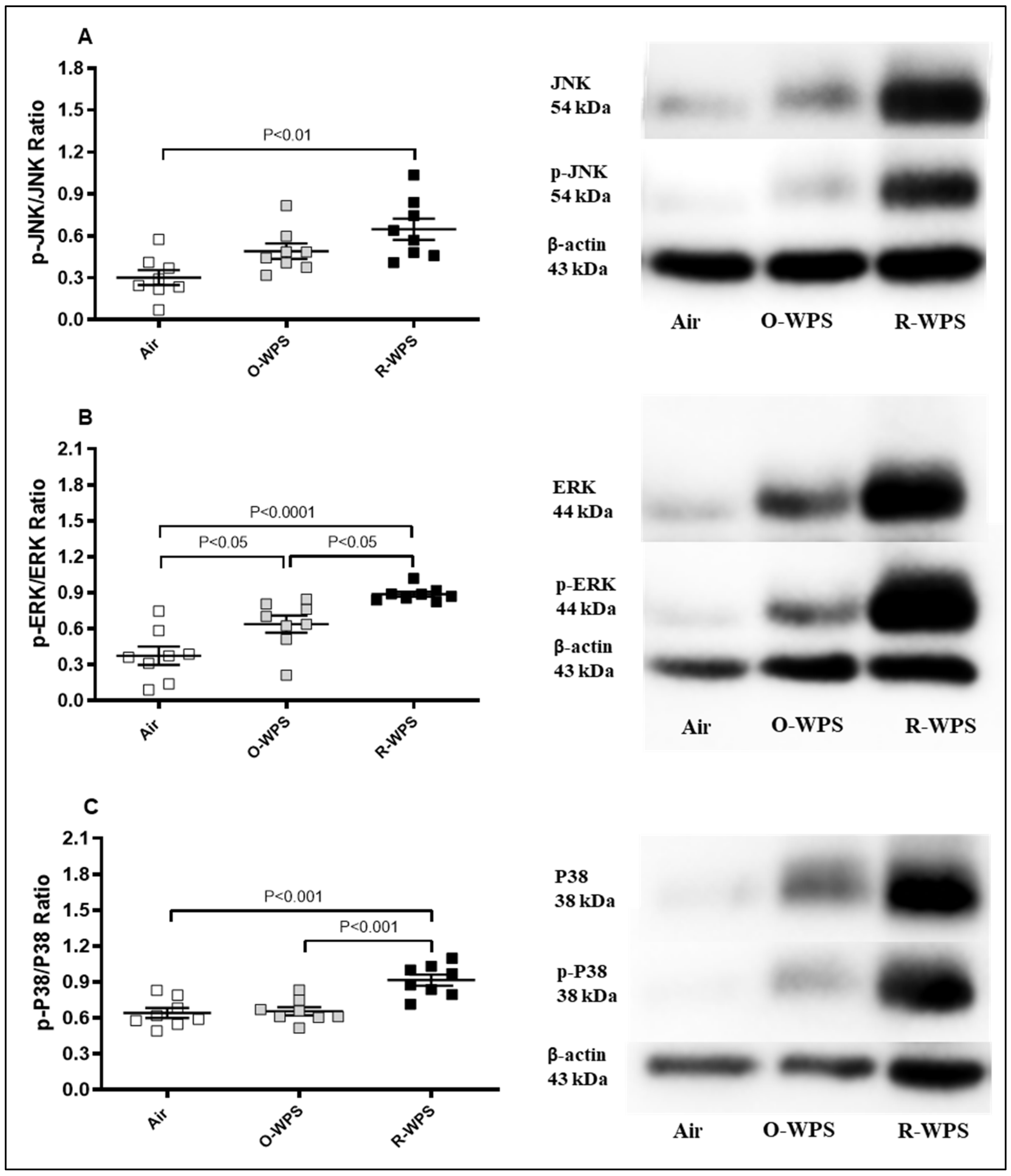
2.13. Expression of p-JNK/JNK, p-ERK/ERK and p-p38/p38 Ratios in Lung Tissue Homogenates
3. Discussion
4. Materials and Methods
4.1. Animals and WPS Exposure
4.2. Lung Histopathology
4.3. Blood Collection and Biochemical Analysis
4.4. Collection and Analysis of BAL Fluid
4.5. Measurement of Chemokines (CXCL1, CXCL2 and CCL2), Matrix Metalloproteinase-9 (MMP-9), Myeloperoxidase (MPO), Lactate Dehydrogenase (LDH), Interleukin-1β (IL1β), IL6, Tumor Necrosis Factor α (TNFα) and 8-Hydroxy-2-Deoxyguanosine (8-OHdG) in Lung Homogenates
4.6. DNA Damage Assessment in the Lung by COMET Assay
4.7. Measurement of Thiobarbituric Acid Reactive Substances (TBARS) Concentration and Superoxide Dismutase (SOD) Activity in Lung Homogenates
4.8. Isolation of Lung Mitochondria and Assessment of Mitochondrial Electron Transport Complexes
4.9. Quantification of Mitogen-Activated Protein Kinases (MAPKs) (Phospho-JNK/JNK, Phospho-ERK/ERK and Phospho-p38/P38) Ratios Expression in Lung Homogenates
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almomen, S.; Aldossari, M.; Khaleel, Y.; Altamimi, M.; Alharbi, O.; Alsuwaydani, A.; Almutairi, M.; Alyousef, S.; Hafiz, R.; Alshomer, F.; et al. Effect of glycerol concentration on levels of toxicants emissions from water-pipe tobacco smoking (WTS). BMC Public Health 2023, 23, 1858. [Google Scholar] [CrossRef] [PubMed]
- Chaouachi, K. A critique of the WHO TobReg’s “Advisory Note” report entitled: “Waterpipe tobacco smoking: Health effects, research needs and recommended actions by regulators”. J. Negat. Results Biomed. 2006, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Martinasek, M.P.; McDermott, R.J.; Martini, L. Waterpipe (hookah) tobacco smoking among youth. Curr. Probl. Pediatr. Adolesc. Health Care 2011, 41, 34–57. [Google Scholar] [CrossRef]
- Jukema, J.B.; Bagnasco, D.E.; Jukema, R.A. Waterpipe smoking: Not necessarily less hazardous than cigarette smoking: Possible consequences for (cardiovascular) disease. Neth. Heart J. Mon. J. Neth. Soc. Cardiol. Neth. Heart Found. 2014, 22, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Aljarrah, K.; Ababneh, Z.Q.; Al-Delaimy, W.K. Perceptions of hookah smoking harmfulness: Predictors and characteristics among current hookah users. Tob. Induc. Dis. 2009, 5, 16. [Google Scholar] [CrossRef]
- Maziak, W.; Taleb, Z.B.; Bahelah, R.; Islam, F.; Jaber, R.; Auf, R.; Salloum, R.G. The global epidemiology of waterpipe smoking. Tob. Control 2015, 24 (Suppl. S1), i3–i12. [Google Scholar] [CrossRef]
- Zheng, Z.; Xie, Z.; Li, D. Discussion of waterpipe tobacco smoking on reddit. Heliyon 2022, 8, e10635. [Google Scholar] [CrossRef]
- Rashidian, H.; Hadji, M.; Ansari-Moghaddam, A.; Bakhshi, M.; Nejatizadeh, A.; Marzban, M.; Rezaianzadeh, A.; Seyyedsalehi, M.S.; Moradi, A.; Gholipour, M.; et al. Association between waterpipe smoking and lung cancer: A multicentre case-control study in Iran. Public Health 2024, 231, 166–172. [Google Scholar] [CrossRef]
- Jawad, M.; Bakir, A.; Ali, M.; Grant, A. Impact of Waterpipe Tobacco Pack Health Warnings on Waterpipe Smoking Attitudes: A Qualitative Analysis among Regular Users in London. BioMed Res. Int. 2015, 2015, 745865. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Maziak, W.; Eissenberg, T.; Ward, K.D.; Thurston, G.; King, B.A.; Sutfin, E.L.; Cobb, C.O.; Griffiths, M.; Goldstein, L.B.; et al. Water Pipe (Hookah) Smoking and Cardiovascular Disease Risk: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e917–e936. [Google Scholar] [CrossRef]
- Jacob, P., 3rd; Abu Raddaha, A.H.; Dempsey, D.; Havel, C.; Peng, M.; Yu, L.; Benowitz, N.L. Comparison of nicotine and carcinogen exposure with water pipe and cigarette smoking. Cancer Epidemiol. Biomark. Prev. 2013, 22, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.D.; Cobb, C.O.; Kilgalen, B.; Austin, J.; Weaver, M.F.; Shihadeh, A.; Eissenberg, T. Acute effects of waterpipe tobacco smoking: A double-blind, placebo-control study. Drug Alcohol Depend. 2011, 116, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Khabour, O.F.; Alzoubi, K.H.; Bani-Ahmad, M.; Dodin, A.; Eissenberg, T.; Shihadeh, A. Acute exposure to waterpipe tobacco smoke induces changes in the oxidative and inflammatory markers in mouse lung. Inhal. Toxicol. 2012, 24, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Magun, B.E.; Wood, L.J. Lung inflammation caused by inhaled toxicants: A review. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 1391–1401. [Google Scholar] [CrossRef]
- Salameh, P.; Waked, M.; Khoury, F.; Akiki, Z.; Nasser, Z.; Abou Abbass, L.; Dramaix, M. Waterpipe smoking and dependence are associated with chronic bronchitis: A case-control study in Lebanon. East. Mediterr. Health J. 2012, 18, 996–1004. [Google Scholar] [CrossRef]
- She, J.; Yang, P.; Wang, Y.; Qin, X.; Fan, J.; Wang, Y.; Gao, G.; Luo, G.; Ma, K.; Li, B.; et al. Chinese water-pipe smoking and the risk of COPD. Chest 2014, 146, 924–931. [Google Scholar] [CrossRef]
- Nemmar, A.; Raza, H.; Yuvaraju, P.; Beegam, S.; John, A.; Yasin, J.; Hameed, R.S.; Adeghate, E.; Ali, B.H. Nose-only water-pipe smoking effects on airway resistance, inflammation, and oxidative stress in mice. J. Appl. Physiol. 2013, 115, 1316–1323. [Google Scholar] [CrossRef]
- Nemmar, A.; Al-Salam, S.; Yuvaraju, P.; Beegam, S.; Yasin, J.; Ali, B.H. Chronic Exposure to Water-Pipe Smoke Induces Alveolar Enlargement, DNA Damage and Impairment of Lung Function. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 38, 982–992. [Google Scholar] [CrossRef]
- Inoue-Choi, M.; Christensen, C.H.; Rostron, B.L.; Cosgrove, C.M.; Reyes-Guzman, C.; Apelberg, B.; Freedman, N.D. Dose-Response Association of Low-Intensity and Nondaily Smoking With Mortality in the United States. JAMA Netw. Open 2020, 3, e206436. [Google Scholar] [CrossRef]
- Hamadi, N.; Beegam, S.; Zaaba, N.E.; Elzaki, O.; Ali, B.H.; Nemmar, A. Comparative Study on the Chronic Vascular Responses Induced by Regular Versus Occasional Waterpipe Smoke Inhalation in Mice. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2022, 56, 13–27. [Google Scholar]
- Hamadi, N.; Al-Salam, S.; Beegam, S.; Zaaba, N.E.; Elzaki, O.; Nemmar, A. Impact of prolonged exposure to occasional and regular waterpipe smoke on cardiac injury, oxidative stress and mitochondrial dysfunction in male mice. Front. Physiol. 2024, 15, 1286366. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Beegam, S.; Zaaba, N.E.; Elzaki, O.; Pathan, A.; Ali, B.H. Waterpipe smoke inhalation induces lung injury and aortic endothelial dysfunction in mice. Physiol. Res. 2023, 72, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Nakhaee, M.R.; Zolfaghari, M.R.; Joukar, S.; Nakhaee, N.; Masoumi-Ardakani, Y.; Iranpour, M.; Nazari, M. Swimming Exercise Training Attenuates the Lung Inflammatory Response and Injury Induced by Exposing to Waterpipe Tobacco Smoke. Addict. Health 2020, 12, 109–117. [Google Scholar] [PubMed]
- Khabour, O.F.; Alzoubi, K.H.; Al-Sawalha, N.; Ahmad, M.B.; Shihadeh, A.; Eissenberg, T. The effect of chronic exposure to waterpipe tobacco smoke on airway inflammation in mice. Life Sci. 2018, 200, 110–114. [Google Scholar] [CrossRef]
- Nemmar, A.; Al-Salam, S.; Beegam, S.; Zaaba, N.E.; Ali, B.H. Effect of smoking cessation on chronic waterpipe smoke inhalation-induced airway hyperresponsiveness, inflammation, and oxidative stress. Am. J. Physiology. Lung Cell. Mol. Physiol. 2021, 320, L791–L802. [Google Scholar] [CrossRef]
- Braber, S.; Henricks, P.A.; Nijkamp, F.P.; Kraneveld, A.D.; Folkerts, G. Inflammatory changes in the airways of mice caused by cigarette smoke exposure are only partially reversed after smoking cessation. Respir. Res. 2010, 11, 99. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Ellwanger, A.; Solon, M.; Cambier, C.J.; Pinkerton, K.E.; Koth, L.L. Alveolar macrophage recruitment and activation by chronic second hand smoke exposure in mice. Copd 2009, 6, 86–94. [Google Scholar] [CrossRef]
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Ten Hacken, N.H.; Krysko, D.V.; Vandenabeele, P.; de Vries, M.; van Oosterhout, A.J.; et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L377–L386. [Google Scholar] [CrossRef]
- Jia, J.; Conlon, T.M.; Ballester Lopez, C.; Seimetz, M.; Bednorz, M.; Zhou-Suckow, Z.; Weissmann, N.; Eickelberg, O.; Mall, M.A.; Yildirim, A. Cigarette smoke causes acute airway disease and exacerbates chronic obstructive lung disease in neonatal mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L602–L610. [Google Scholar] [CrossRef]
- Moazed, F.; Burnham, E.L.; Vandivier, R.W.; O’Kane, C.M.; Shyamsundar, M.; Hamid, U.; Abbott, J.; Thickett, D.R.; Matthay, M.A.; McAuley, D.F.; et al. Cigarette smokers have exaggerated alveolar barrier disruption in response to lipopolysaccharide inhalation. Thorax 2016, 71, 1130–1136. [Google Scholar] [CrossRef]
- Tanigawa, T.; Araki, S.; Nakata, A.; Kitamura, F.; Yasumoto, M.; Sakurai, S.; Kiuchi, T. Increase in memory (CD4+CD29+ and CD4+CD45RO+) T and naive (CD4+CD45RA+) T-cell subpopulations in smokers. Arch. Environ. Health 1998, 53, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Betsuyaku, T.; Hamamura, I.; Hata, J.; Takahashi, H.; Mitsuhashi, H.; Adair-Kirk, T.L.; Senior, R.M.; Nishimura, M. Bronchiolar chemokine expression is different after single versus repeated cigarette smoke exposure. Respir. Res. 2008, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Allais, L.; Verschuere, S.; Maes, T.; De Smet, R.; Devriese, S.; Gonzales, G.B.; Peeters, H.; Van Crombruggen, K.; Bachert, C.; De Vos, M.; et al. Translational research into the effects of cigarette smoke on inflammatory mediators and epithelial TRPV1 in Crohn’s disease. PLoS ONE 2020, 15, e0236657. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Marudamuthu, A.S.; Tsukasaki, Y.; Ikebe, M.; Fu, J.; Shetty, S. p53- and PAI-1-mediated induction of C-X-C chemokines and CXCR2: Importance in pulmonary inflammation due to cigarette smoke exposure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L496–L506. [Google Scholar] [CrossRef]
- Bratcher, P.E.; Weathington, N.M.; Nick, H.J.; Jackson, P.L.; Snelgrove, R.J.; Gaggar, A. MMP-9 cleaves SP-D and abrogates its innate immune functions in vitro. PLoS ONE 2012, 7, e41881. [Google Scholar] [CrossRef]
- Ishii, T.; Abboud, R.T.; Wallace, A.M.; English, J.C.; Coxson, H.O.; Finley, R.J.; Shumansky, K.; Paré, P.D.; Sandford, A.J. Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. Eur. Respir. J. 2014, 43, 82–91. [Google Scholar] [CrossRef]
- Zhou, L.; Le, Y.; Tian, J.; Yang, X.; Jin, R.; Gai, X.; Sun, Y. Cigarette smoke-induced RANKL expression enhances MMP-9 production by alveolar macrophages. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 81–91. [Google Scholar] [CrossRef]
- Haegens, A.; Vernooy, J.H.; Heeringa, P.; Mossman, B.T.; Wouters, E.F. Myeloperoxidase modulates lung epithelial responses to pro-inflammatory agents. Eur. Respir. J. 2008, 31, 252–260. [Google Scholar] [CrossRef]
- Khabour, O.F.; Alzoubi, K.H.; Abu Thiab, T.M.; Al-Husein, B.A.; Eissenberg, T.; Shihadeh, A.L. Changes in the expression and protein level of matrix metalloproteinases after exposure to waterpipe tobacco smoke. Inhal. Toxicol. 2015, 27, 689–693. [Google Scholar] [CrossRef]
- Nemmar, A.; Al-Salam, S.; Beegam, S.; Yuvaraju, P.; Ali, B.H. Comparative Study on Pulmonary Toxicity in Mice Induced by Exposure to Unflavoured and Apple- and Strawberry-Flavoured Tobacco Waterpipe Smoke. Oxidative Med. Cell. Longev. 2020, 2020, 6450450. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.J.; Lutey, B.A.; Suzuki, Y.; Toennies, H.M.; Kelley, D.G.; Kobayashi, D.K.; Ijem, W.G.; Deslee, G.; Moore, C.H.; Jacobs, M.E.; et al. The role of matrix metalloproteinase-9 in cigarette smoke-induced emphysema. Am. J. Respir. Crit. Care Med. 2011, 183, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.D.; Baumlin, N.; Dennis, J.S.; Yoshida, M.; Kis, A.; Aguiar, C.; Schmid, A.; Mendes, E.; Salathe, M. Losartan reduces cigarette smoke-induced airway inflammation and mucus hypersecretion. ERJ Open Res. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Liu, X.; Zhang, G.; Ming, Z.; Wang, T. Isoliquiritigenin Inhibits Cigarette Smoke-Induced COPD by Attenuating Inflammation and Oxidative Stress via the Regulation of the Nrf2 and NF-κB Signaling Pathways. Front. Pharmacol. 2018, 9, 1001. [Google Scholar] [CrossRef] [PubMed]
- Nath, D.; Shivasekar, M.; Vinodhini, V.M. Smoking Induces the Circulating Levels of Matrix Metalloproteinase-9 and Its Association with Cardiovascular Risk in Young Smokers. Medeni. Med. J. 2022, 37, 306–312. [Google Scholar] [CrossRef]
- Klein, R.; Nagy, O.; Tóthová, C.; Chovanová, F. Clinical and Diagnostic Significance of Lactate Dehydrogenase and Its Isoenzymes in Animals. Vet. Med. Int. 2020, 2020, 5346483. [Google Scholar] [CrossRef]
- Nemmar, A.; Al Hemeiri, A.; Al Hammadi, N.; Yuvaraju, P.; Beegam, S.; Yasin, J.; Elwasila, M.; Ali, B.H.; Adeghate, E. Early pulmonary events of nose-only water pipe (shisha) smoking exposure in mice. Physiol. Rep. 2015, 3, e12258. [Google Scholar] [CrossRef]
- Anbarasi, K.; Sabitha, K.E.; Devi, C.S. Lactate dehydrogenase isoenzyme patterns upon chronic exposure to cigarette smoke: Protective effect of bacoside A. Environ. Toxicol. Pharmacol. 2005, 20, 345–350. [Google Scholar] [CrossRef]
- Ranawat, P.; Kaur, N.; Koul, A. Modulation of cigarette smoke induced alterations by aqueous Ocimum sanctum leaf extract in pulmonary tissue of rodents. Sci. Rep. 2023, 13, 15806. [Google Scholar] [CrossRef]
- Zhou, L.; Jian, T.; Wan, Y.; Huang, R.; Fang, H.; Wang, Y.; Liang, C.; Ding, X.; Chen, J. Luteolin Alleviates Oxidative Stress in Chronic Obstructive Pulmonary Disease Induced by Cigarette Smoke via Modulation of the TRPV1 and CYP2A13/NRF2 Signaling Pathways. Int. J. Mol. Sci. 2023, 25, 369. [Google Scholar] [CrossRef]
- Nemmar, A.; Al-Salam, S.; Yuvaraju, P.; Beegam, S.; Ali, B.H. Exercise Training Mitigates Water Pipe Smoke Exposure-Induced Pulmonary Impairment via Inhibiting NF-κB and Activating Nrf2 Signalling Pathways. Oxidative Med. Cell. Longev. 2018, 2018, 7459612. [Google Scholar] [CrossRef] [PubMed]
- Belchamber, K.; Hall, D.A.; Hourani, S.M. Smoking enhances the proinflammatory effects of nucleotides on cytokine release from human lung. PLoS ONE 2014, 9, e99711. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Rahman, I. Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: Implications for COPD and lung cancer. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1245–L1258. [Google Scholar] [CrossRef] [PubMed]
- Frangie, C.; Daher, J. Role of myeloperoxidase in inflammation and atherosclerosis (Review). Biomed. Rep. 2022, 16, 53. [Google Scholar] [CrossRef]
- Park, H.Y.; Man, S.F.; Tashkin, D.; Wise, R.A.; Connett, J.E.; Anthonisen, N.A.; Sin, D.D. The relation of serum myeloperoxidase to disease progression and mortality in patients with chronic obstructive pulmonary disease (COPD). PLoS ONE 2013, 8, e61315. [Google Scholar] [CrossRef]
- Agustí, A.; Hogg, J.C. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative Stress in Chronic Obstructive Pulmonary Disease. Antioxidants 2022, 11, 965. [Google Scholar] [CrossRef]
- Hakim, F.; Hellou, E.; Goldbart, A.; Katz, R.; Bentur, Y.; Bentur, L. The acute effects of water-pipe smoking on the cardiorespiratory system. Chest 2011, 139, 775–781. [Google Scholar] [CrossRef]
- Khan, N.A.; Sundar, I.K.; Rahman, I. Strain- and sex-dependent pulmonary toxicity of waterpipe smoke in mouse. Physiol. Rep. 2018, 6, e13579. [Google Scholar] [CrossRef]
- Badran, M.; Laher, I. Waterpipe (shisha, hookah) smoking, oxidative stress and hidden disease potential. Redox Biol. 2020, 34, 101455. [Google Scholar] [CrossRef]
- Alsaad, A.M.; Al-Arifi, M.N.; Maayah, Z.H.; Attafi, I.M.; Alanazi, F.E.; Belali, O.M.; Alhoshani, A.; Asiri, Y.A.; Korashy, H.M. Genotoxic impact of long-term cigarette and waterpipe smoking on DNA damage and oxidative stress in healthy subjects. Toxicol. Mech. Methods 2019, 29, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.G.; Zilhão, R.; Thorsteinsdóttir, S.; Carlos, A.R. Linking Oxidative Stress and DNA Damage to Changes in the Expression of Extracellular Matrix Components. Front. Genet. 2021, 12, 673002. [Google Scholar] [CrossRef] [PubMed]
- Grillo, R.; Khemiss, M.; da Silva, Y.S. Cytotoxic and Genotoxic Effects of Waterpipe on Oral Health Status: Systematic review and meta-analysis. Sultan Qaboos Univ. Med. J. 2023, 23, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Al-Salam, S.; Beegam, S.; Zaaba, N.E.; Elzaki, O.; Ali, B.H. Waterpipe smoke inhalation potentiates cardiac oxidative stress, inflammation, mitochondrial dysfunction, apoptosis and autophagy in experimental hypertension. Biomed. Pharmacother. Biomed. Pharmacother. 2023, 158, 114144. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Goerlitz, D.S.; Dumitrescu, R.G.; Han, D.; Seillier-Moiseiwitsch, F.; Spernak, S.M.; Orden, R.A.; Chen, J.; Goldman, R.; Shields, P.G. Associations between cigarette smoking and mitochondrial DNA abnormalities in buccal cells. Carcinogenesis 2008, 29, 1170–1177. [Google Scholar] [CrossRef]
- Ballweg, K.; Mutze, K.; Königshoff, M.; Eickelberg, O.; Meiners, S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am. J. Physiology. Lung Cell. Mol. Physiol. 2014, 307, L895–L907. [Google Scholar] [CrossRef]
- Wang, Z.; White, A.; Wang, X.; Ko, J.; Choudhary, G.; Lange, T.; Rounds, S.; Lu, Q. Mitochondrial Fission Mediated Cigarette Smoke-induced Pulmonary Endothelial Injury. Am. J. Respir. Cell Mol. Biol. 2020, 63, 637–651. [Google Scholar] [CrossRef]
- Cowan, K.J.; Storey, K.B. Mitogen-activated protein kinases: New signaling pathways functioning in cellular responses to environmental stress. J. Exp. Biol. 2003, 206, 1107–1115. [Google Scholar] [CrossRef]
- Chuang, S.M.; Wang, I.C.; Yang, J.L. Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis 2000, 21, 1423–1432. [Google Scholar] [CrossRef]
- Broom, O.J.; Widjaya, B.; Troelsen, J.; Olsen, J.; Nielsen, O.H. Mitogen activated protein kinases: A role in inflammatory bowel disease? Clin. Exp. Immunol. 2009, 158, 272–280. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, W.G.; Luo, Y.; Han, F.F.; Yao, X.H.; Yang, T.Y.; Zhang, Y.; Pi, W.F.; Guo, X.J. Cigarette smoke-induced skeletal muscle atrophy is associated with up-regulation of USP-19 via p38 and ERK MAPKs. J. Cell. Biochem. 2011, 112, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Zhou, H.; You, J.; Kaiser, V.A.; Khajuria, R.K.; Muhammad, S. Tobacco smoke condensate-induced senescence in endothelial cells was ameliorated by colchicine treatment via suppression of NF-κB and MAPKs P38 and ERK pathways activation. Cell Commun. Signal. CCS 2024, 22, 214. [Google Scholar] [CrossRef] [PubMed]
- Restivo, I.; Attanzio, A.; Giardina, I.C.; Di Gaudio, F.; Tesoriere, L.; Allegra, M. Cigarette Smoke Extract Induces p38 MAPK-Initiated, Fas-Mediated Eryptosis. Int. J. Mol. Sci. 2022, 23, 14730. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Yuvaraju, P.; Beegam, S.; John, A.; Raza, H.; Ali, B.H. Cardiovascular effects of nose-only water-pipe smoking exposure in mice. Am. J. Physiology. Heart Circ. Physiol. 2013, 305, H740–H746. [Google Scholar] [CrossRef]
- Nemmar, A.; Al-Salam, S.; Yuvaraju, P.; Beegam, S.; Yasin, J.; Ali, B.H. Chronic exposure to water-pipe smoke induces cardiovascular dysfunction in mice. Am. J. Physiology. Heart Circ. Physiol. 2017, 312, H329–H339. [Google Scholar] [CrossRef]
- Beegam, S.; Al-Salam, S.; Zaaba, N.E.; Elzaki, O.; Ali, B.H.; Nemmar, A. Effects of Waterpipe Smoke Exposure on Experimentally Induced Chronic Kidney Disease in Mice. Int. J. Mol. Sci. 2024, 25, 585. [Google Scholar] [CrossRef]
- Toukan, Y.; Hakim, F.; Bentur, Y.; Aharon-Peretz, J.; Elemy, A.; Gur, M.; Hanna, M.; Fisher, T.; Scherb, I.; Bentur, L. The Effect of a 30-Min Water-Pipe Smoking Session on Cognitive Measures and Cardio-Pulmonary Parameters. Nicotine Tob. Res. Off. J. Soc. Res. Nicotine Tob. 2020, 22, 1347–1353. [Google Scholar] [CrossRef]
- Zu, G.X.; Sun, K.Y.; Liu, X.J.; Tang, J.Q.; Huang, H.L.; Han, T. Banxia xiexin decoction prevents the development of gastric cancer. World J. Clin. Oncol. 2024, 15, 1293–1308. [Google Scholar] [CrossRef]
- Houchen, C.J.; Ghanem, S.; Kaartinen, V.; Bumann, E.E. TGF-β signaling in the cranial neural crest affects late-stage mandibular bone resorption and length. Front. Physiol. 2024, 15, 1435594. [Google Scholar] [CrossRef]
- Zaaba, N.E.; Al-Salam, S.; Beegam, S.; Elzaki, O.; Yasin, J.; Nemmar, A. Catalpol Attenuates Oxidative Stress and Inflammation via Mechanisms Involving Sirtuin-1 Activation and NF-κB Inhibition in Experimentally-Induced Chronic Kidney Disease. Nutrients 2023, 15, 237. [Google Scholar] [CrossRef]
- Hardy, R.D.; Coalson, J.J.; Peters, J.; Chaparro, A.; Techasaensiri, C.; Cantwell, A.M.; Kannan, T.R.; Baseman, J.B.; Dube, P.H. Analysis of pulmonary inflammation and function in the mouse and baboon after exposure to Mycoplasma pneumoniae CARDS toxin. PLoS ONE 2009, 4, e7562. [Google Scholar] [CrossRef]
- Bubeck, S.S.; Cantwell, A.M.; Dube, P.H. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect. Immun. 2007, 75, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Speit, G. The contribution of cytotoxicity to DNA-effects in the single cell gel test (comet assay). Toxicol. Lett. 1997, 90, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Speit, G. Genotoxic effects of chemicals in the single cell gel (SCG) test with human blood cells in relation to the induction of sister-chromatid exchanges (SCE). Mutat. Res. 1995, 346, 49–56. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamadi, N.; Al-Salam, S.; Beegam, S.; Zaaba, N.E.; Elzaki, O.; Nemmar, A. Chronic Exposure to Two Regimens of Waterpipe Smoke Elicits Lung Injury, Genotoxicity, and Mitochondrial Impairment with the Involvement of MAPKs Activation in Mice. Int. J. Mol. Sci. 2025, 26, 430. https://doi.org/10.3390/ijms26010430
Hamadi N, Al-Salam S, Beegam S, Zaaba NE, Elzaki O, Nemmar A. Chronic Exposure to Two Regimens of Waterpipe Smoke Elicits Lung Injury, Genotoxicity, and Mitochondrial Impairment with the Involvement of MAPKs Activation in Mice. International Journal of Molecular Sciences. 2025; 26(1):430. https://doi.org/10.3390/ijms26010430
Chicago/Turabian StyleHamadi, Naserddine, Suhail Al-Salam, Sumaya Beegam, Nur Elena Zaaba, Ozaz Elzaki, and Abderrahim Nemmar. 2025. "Chronic Exposure to Two Regimens of Waterpipe Smoke Elicits Lung Injury, Genotoxicity, and Mitochondrial Impairment with the Involvement of MAPKs Activation in Mice" International Journal of Molecular Sciences 26, no. 1: 430. https://doi.org/10.3390/ijms26010430
APA StyleHamadi, N., Al-Salam, S., Beegam, S., Zaaba, N. E., Elzaki, O., & Nemmar, A. (2025). Chronic Exposure to Two Regimens of Waterpipe Smoke Elicits Lung Injury, Genotoxicity, and Mitochondrial Impairment with the Involvement of MAPKs Activation in Mice. International Journal of Molecular Sciences, 26(1), 430. https://doi.org/10.3390/ijms26010430