
Association of Novel Pathogenic Variant (p. Ile366Asn) in PLA2G6 Gene with Infantile Neuroaxonal Dystrophy


Abstract
1. Introduction
2. Results
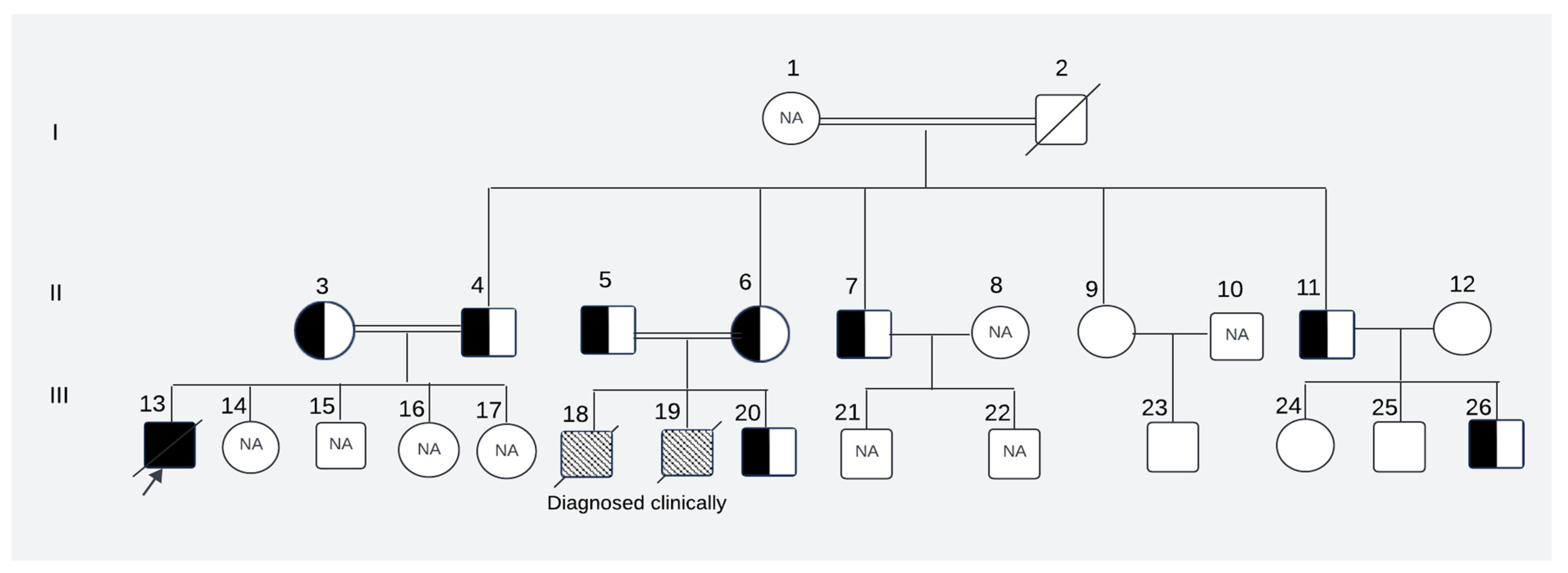
2.1. Clinical Presentation
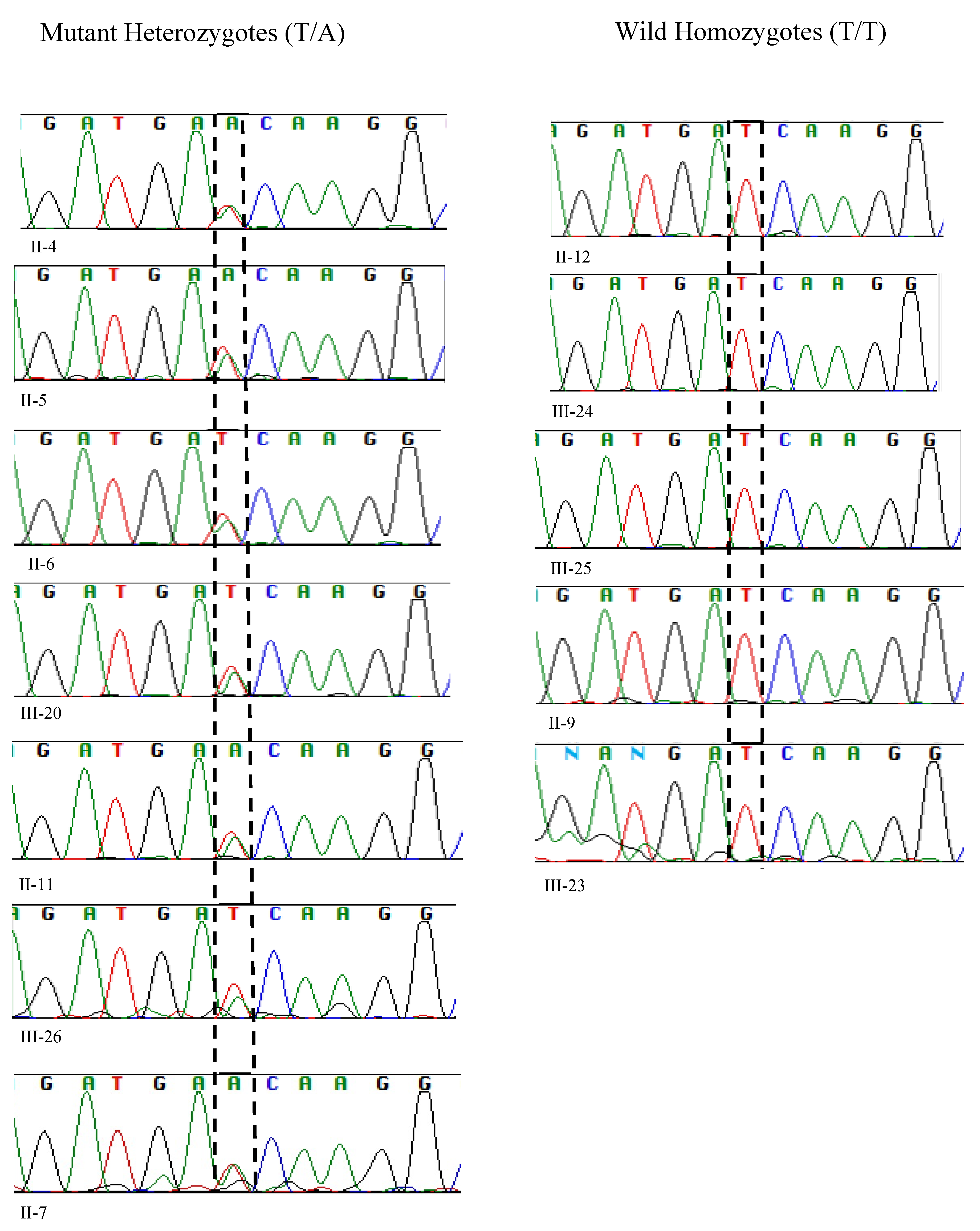
2.2. Genetic Analysis
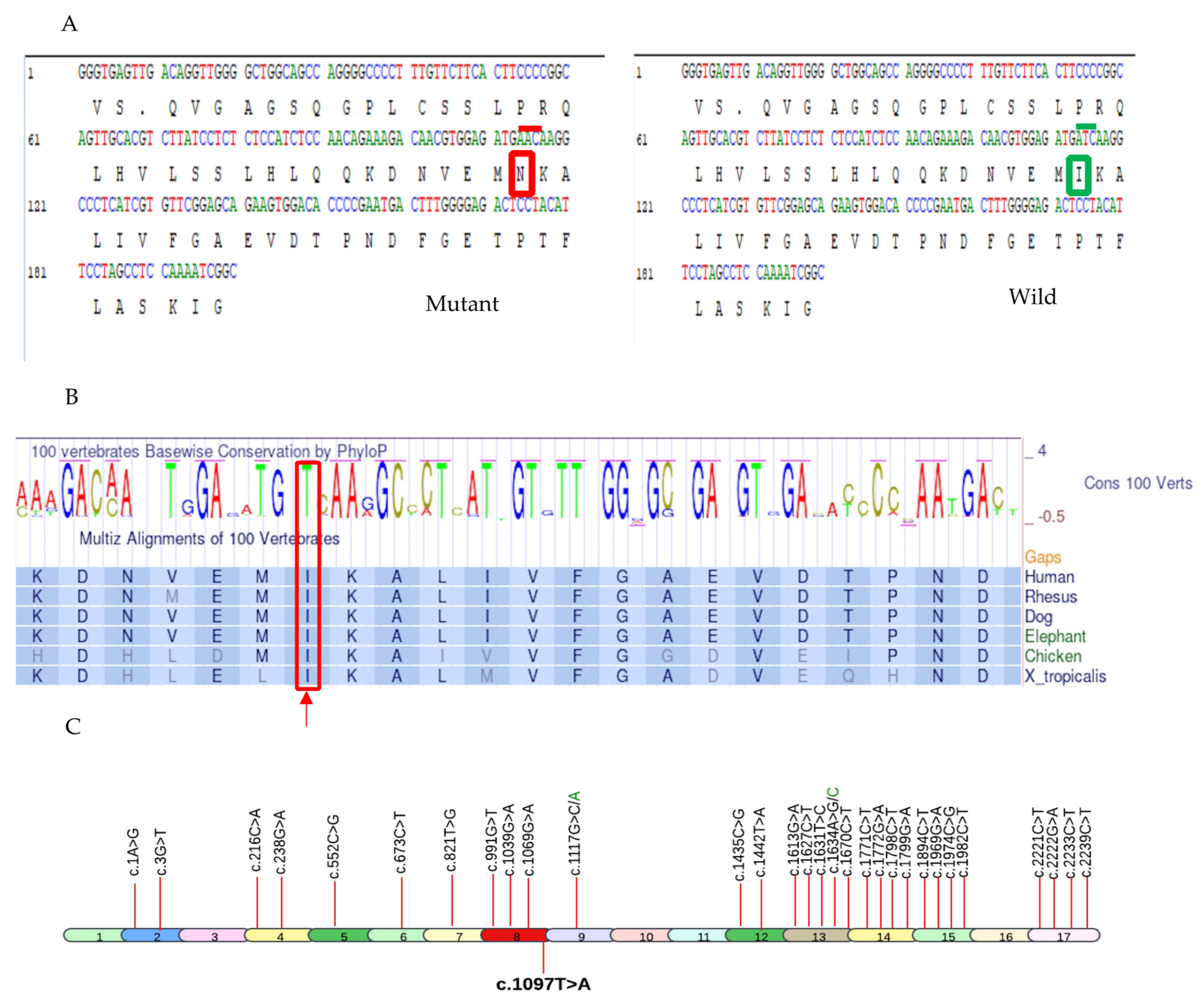
2.3. Bioinformatic Analyses
3. Discussion
4. Materials and Methods
4.1. Case Description
4.2. Clinical Data and Specimen Collection
4.3. Laboratory Testing
4.4. Bioinformatic Tools
4.4.1. Genomic Position and Protein Domain
4.4.2. Pathogenic Variant Annotation
4.4.3. Structural Modeling of Pathogenic Variant and Prediction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, P.; Gao, Z.; Jiang, Y.; Wang, J.; Zhang, F.; Wang, S.; Yang, Y.; Xiong, H.; Zhang, Y.; Bao, X.; et al. Follow-up study of 25 Chinese children with PLA 2G6-associated neurodegeneration. Eur. J. Neurol. 2013, 20, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Altuame, F.D.; Foskett, G.; Atwal, P.S.; Endemann, S.; Midei, M.; Milner, P.; Salih, M.A.; Hamad, M.; Al-Muhaizea, M.; Hashem, M.; et al. The natural history of infantile neuroaxonal dystrophy. Orphanet. J. Rare Dis. 2020, 15, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Atwal, P.S.; Midei, M.; Adams, D.; Fay, A.; Heerinckx, F.; Milner, P. The infantile neuroaxonal dystrophy rating scale (INAD-RS). Orphanet. J. Rare Dis. 2020, 15, 195. [Google Scholar] [CrossRef] [PubMed]
- Toth-Bencsik, R.; Balicza, P.; Varga, E.T.; Lengyel, A.; Rudas, G.; Gal, A.; Molnar, M.J. New insights of phospholipase A2 associated neurodegeneration phenotype based on the long-term follow-up of a large Hungarian family. Front. Genet. 2021, 12, 628904. [Google Scholar] [CrossRef]
- Hayashi, D.; Dennis, E.A. Molecular basis of unique specificity and regulation of group VIA calcium-independent phospholipase A2 (PNPLA9) and its role in neurodegenerative diseases. Pharmacol. Therapeut. 2023, 245, 108395. [Google Scholar] [CrossRef]
- Hartley, T.; Lemire, G.; Kernohan, K.D.; Howley, H.E.; Adams, D.R.; Boycott, K.M. New diagnostic approaches for undiagnosed rare genetic diseases. Ann. Rev. Genom. Hum. Genet. 2020, 21, 351–372. [Google Scholar] [CrossRef]
- Deng, X.; Yuan, L.; Jankovic, J.; Deng, H. Phenotypic spectrum of neurodegeneration associated with pathogenic variants in the PLA2G6 gene (PLAN). Neurology 2008, 70, 1623–1629. [Google Scholar] [CrossRef]
- Zou, Y.; Luo, H.; Yuan, H.; Xie, K.; Yang, Y.; Huang, S.; Yang, B.; Liu, Y. PLA2G6 pathogenic variant underlies infantile neuroaxonal dystrophy. Am. J. Hum. Genet. 2006, 79, 942–948. [Google Scholar] [CrossRef]
- Wan, Y.; Jiang, Y.; Xie, Z.; Ling, C.; Du, K.; Li, R.; Yuan, Y.; Wang, Z.; Sun, W.; Jin, H. Novel PLA2G6 pathogenic variants in Chinese patients with pla2g6-associated neurodegeneration. Front. Neurol. 2022, 13, 922528. [Google Scholar] [CrossRef]
- Ahmed, S.; Jafri, H.; Rashid, Y.; Ehsan, Y.; Bashir, S.; Ahmed, M. Cascade screening for beta-thalassemia in Pakistan: Development, feasibility and acceptability of a decision support intervention for relatives. Eur. J. Hum. Genet. 2022, 30, 73–80. [Google Scholar] [CrossRef]
- Cheema, H.; Bertoli-Avella, A.M.; Skrahina, V.; Anjum, M.N.; Waheed, N.; Saeed, A.; Beetz, C.; Perez-Lopez, J.; Rocha, M.E.; Alawbathani, S.; et al. Genomic testing in 1019 individuals from 349 Pakistani families results in high diagnostic yield and clinical utility. NPJ Genom. Med. 2020, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.P.; Tang, B.S.; Guo, J.F. PLA2G6-associated neurodegeneration (PLAN): Review of clinical phenotypes and genotypes. Front. Neurol. 2018, 9, 1100. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Luo, H.; Yuan, H.; Xie, K.; Yang, Y.; Huang, S.; Yang, B.; Liu, Y. Identification of a Novel Nonsense Pathogenic variant in PLA2G6 and Prenatal Diagnosis in a Chinese Family with Infantile Neuroaxonal Dystrophy. Front. Neurol. 2022, 13, 904027. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, X.; Nowatzke, W.; Ramanadham, S.; Turk, J. Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13. 1. J. Biol. Chem. 1999, 274, 9607–9616. [Google Scholar] [CrossRef]
- Balsinde, J.; Balboa, M.A. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell Signal. 2005, 17, 1052–1062. [Google Scholar] [CrossRef]
- Leslie, C.C. Thematic Review Series: Phospholipases: Central Role in Lipid Signaling and Disease: Cytosolic phospholipase A2: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef]
- Khan, S.A.; Ilies, M.A. The phospholipase A2 superfamily: Structure, isozymes, catalysis, physiologic and pathologic roles. Int. J. Mol. Sci. 2023, 24, 1353. [Google Scholar] [CrossRef]
- Deng, X.; Zheng, W.; Yang, Y.; Yang, Z.; Li, H.; Song, Z.; Wang, J.; Deng, H.; Yuan, L. Identification of PLA2G6 variants in a Chinese patient with Parkinson’s disease. Ageing Neur. Dis. 2023, 3, 9. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. Sequence and Structure-Based Analyses of Human Ankyrin Repeats. Molecules 2022, 27, 423. [Google Scholar] [CrossRef]
- Kumar, A.; Balbach, J. Folding and stability of ankyrin repeats control biological protein function. Biomolecules 2021, 11, 840. [Google Scholar] [CrossRef]
- Malley, K.R.; Koroleva, O.; Miller, I.; Sanishvili, R.; Jenkins, C.M.; Gross, R.W.; Korolev, S. The structure of iPLA2β reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 2018, 9, 765. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Azcona, L.J.; Paisán-Ruiz, C. Pla2g6 deficiency in zebrafish leads to dopaminergic cell death, axonal degeneration, increased β-synuclein expression, and defects in brain functions and pathways. Mol. Neurobiol. 2018, 55, 6734–6754. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cocheme, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef]
- Larsson, P.K.; Claesson, H.E.; Kennedy, B.P. Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J. Biol. Chem. 1998, 273, 207–214. [Google Scholar] [CrossRef]
- UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2023, 51, 2699. [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; ClinVar, D.M. Public archive of relationships among sequence variation and human phenotype. Nuclei Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Genome Aggregation Database Consortium; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2022, 43, 1012–1030. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Röner, S.; Kircher, M. CADD v1. 7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024, 52, D1143–D1154. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Tordai, H.; Torres, O.; Csepi, M.; Padányi, R.; Lukács, G.L.; Hegedűs, T. Analysis of AlphaMissense data in different protein groups and structural context. Sci. Data 2024, 11, 495. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB. org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Silico Tools | Threshold | Score | Predicted Effect |
|---|---|---|---|
| SIFT | ≤0.05 | 0.00 | Pathogenic |
| CADD | >15 | 27.4 | Pathogenic |
| PolyPhen-2 | >0.5 | 0.92 | Pathogenic |
| REVEL | ≥0.5 | 0.58 | Pathogenic |
| Alpha missense | >0.5 | 0.68 | Pathogenic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheema, A.N.; Shi, R.; Kamboh, M.I. Association of Novel Pathogenic Variant (p. Ile366Asn) in PLA2G6 Gene with Infantile Neuroaxonal Dystrophy. Int. J. Mol. Sci. 2025, 26, 352. https://doi.org/10.3390/ijms26010352
Cheema AN, Shi R, Kamboh MI. Association of Novel Pathogenic Variant (p. Ile366Asn) in PLA2G6 Gene with Infantile Neuroaxonal Dystrophy. International Journal of Molecular Sciences. 2025; 26(1):352. https://doi.org/10.3390/ijms26010352
Chicago/Turabian StyleCheema, Asma Naseer, Ruyu Shi, and M. Ilyas Kamboh. 2025. "Association of Novel Pathogenic Variant (p. Ile366Asn) in PLA2G6 Gene with Infantile Neuroaxonal Dystrophy" International Journal of Molecular Sciences 26, no. 1: 352. https://doi.org/10.3390/ijms26010352
APA StyleCheema, A. N., Shi, R., & Kamboh, M. I. (2025). Association of Novel Pathogenic Variant (p. Ile366Asn) in PLA2G6 Gene with Infantile Neuroaxonal Dystrophy. International Journal of Molecular Sciences, 26(1), 352. https://doi.org/10.3390/ijms26010352