
TH301 Emerges as a Novel Anti-Oncogenic Agent for Human Pancreatic Cancer Cells: The Dispensable Roles of p53, CRY2 and BMAL1 in TH301-Induced CDKN1A/p21CIP1/WAF1 Upregulation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
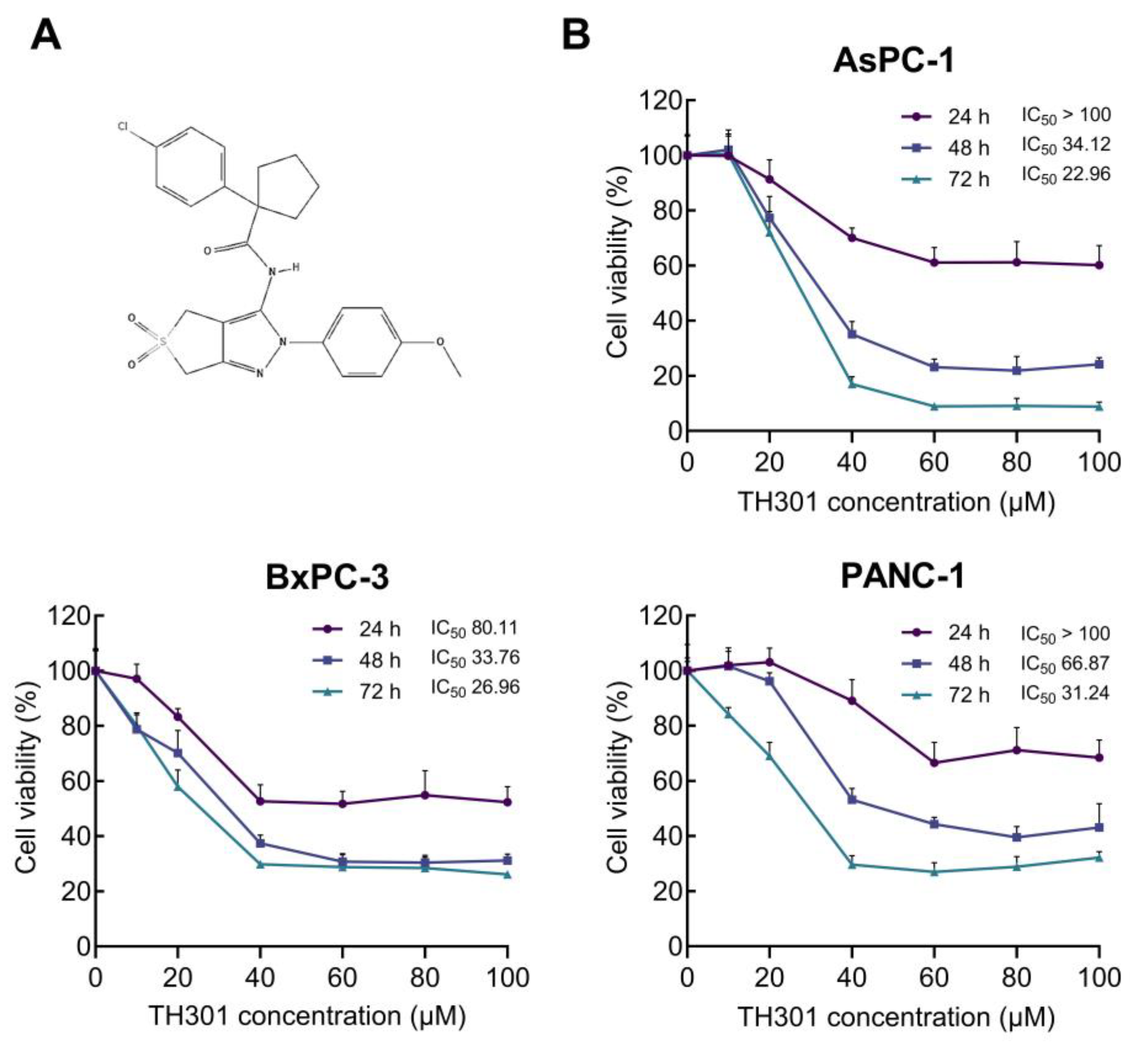
2.1. TH301-Induced Reduction of Cell Viability and Growth of Pancreatic Cancer Cells in a Mutational Signature-Dependent Fashion
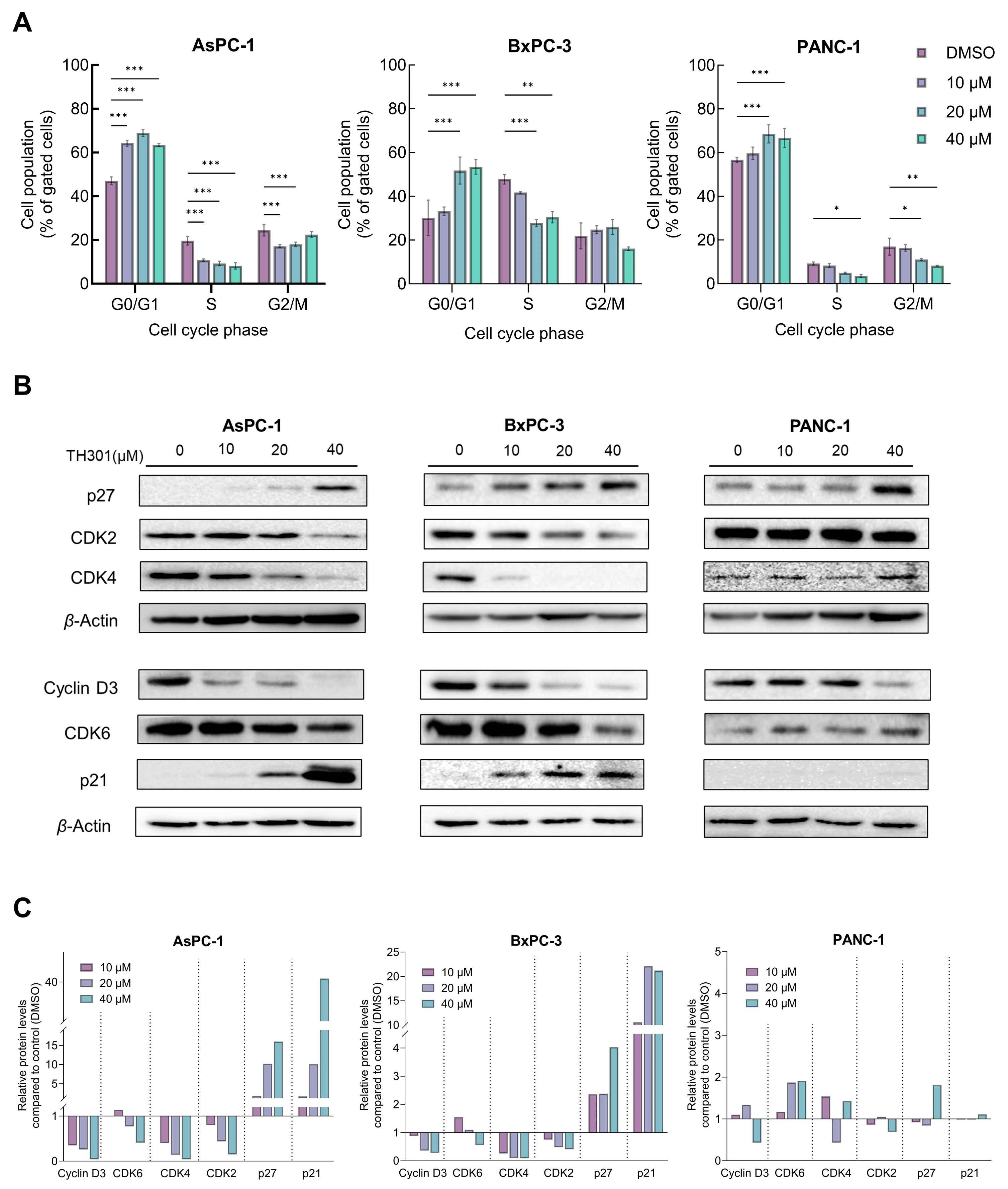
2.2. TH301 Causes Cell Cycle Arrest at the G1-Phase and Alters Protein Expression Profiles of Critical Regulators in Pancreatic Cancer Cells: Mutational Load-Dependent Responses
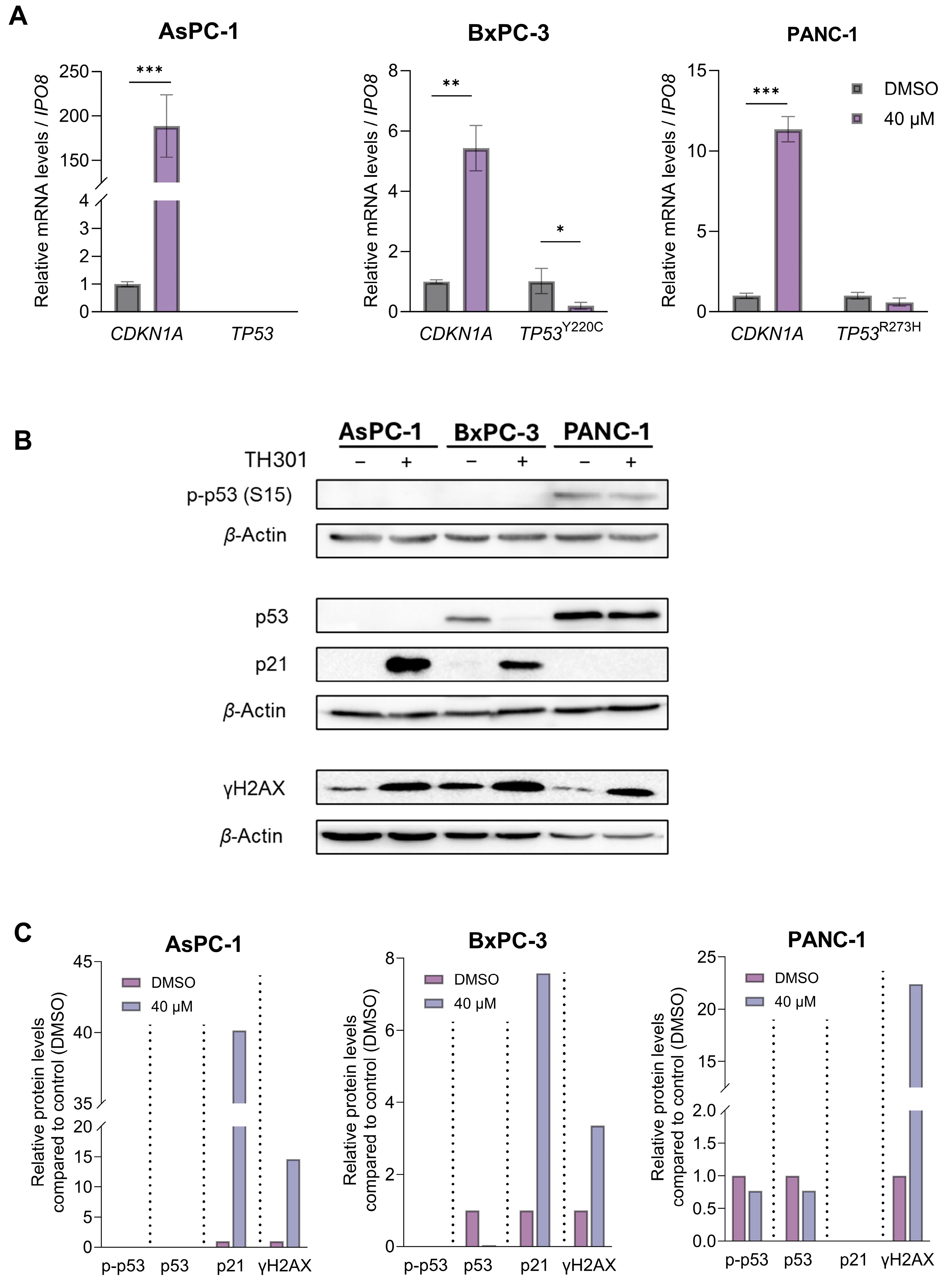
2.3. TH301 Causes a Strong p53-Independent Induction of the CDKN1A/p21 Cell Cycle Inhibitor in Pancreatic Cancer Cell Environments, Following Mutational Load-Specific Patterns
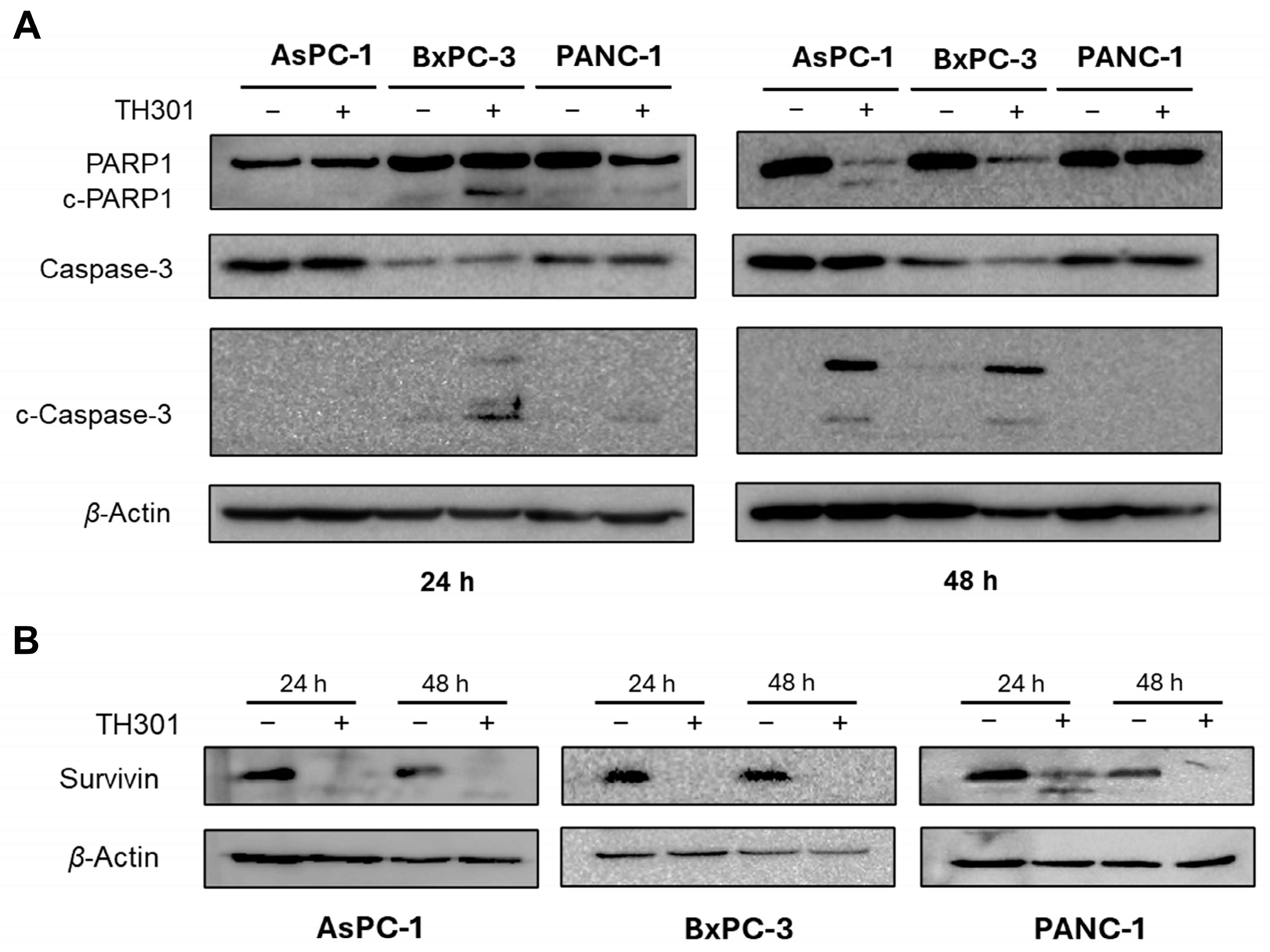
2.4. TH301 Induces Caspase Repertoire-Mediated Apoptosis in Pancreatic Cancer Cell Settings: Mutational Signature-Dependent Responses
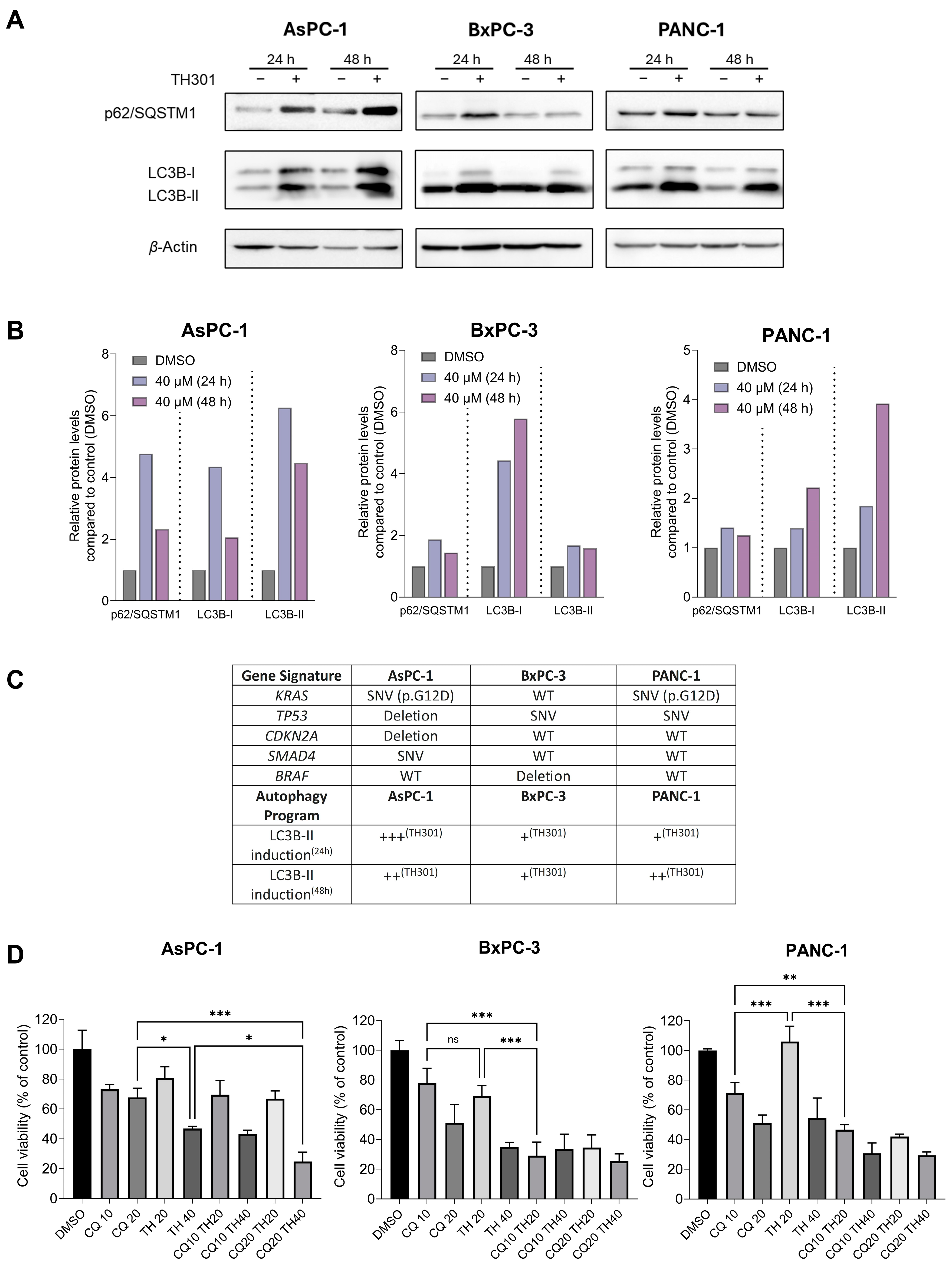
2.5. TH301 Upregulates LC3B-II-Depenent Autophagy in Pancreatic Cancer Cells, Following a Mutational Signature-Independent Pattern
2.6. TH301 Potentiates the Cytopathic Effects of Oxaliplatin by Reducing Pancreatic Cancer Cell Viability
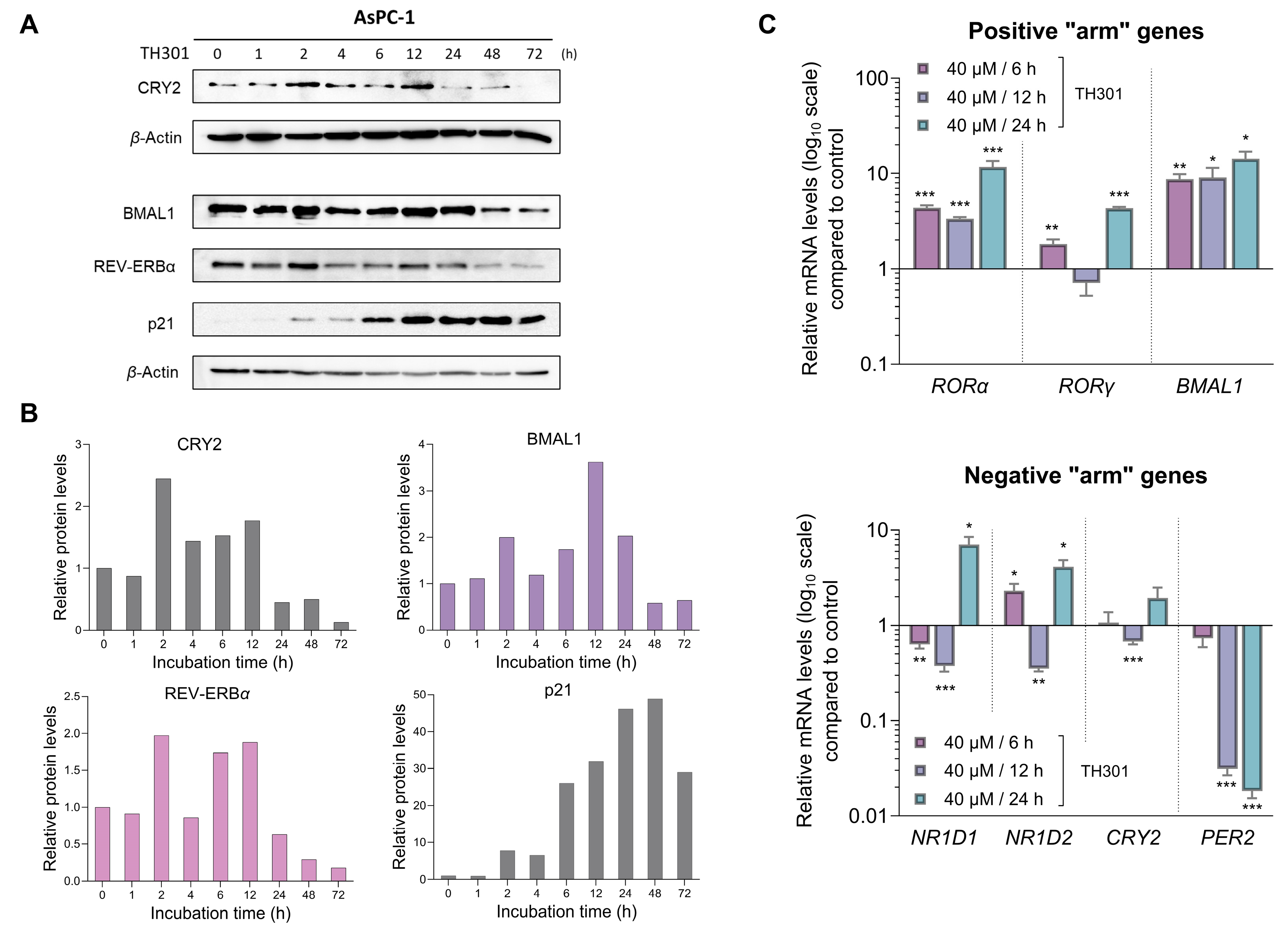
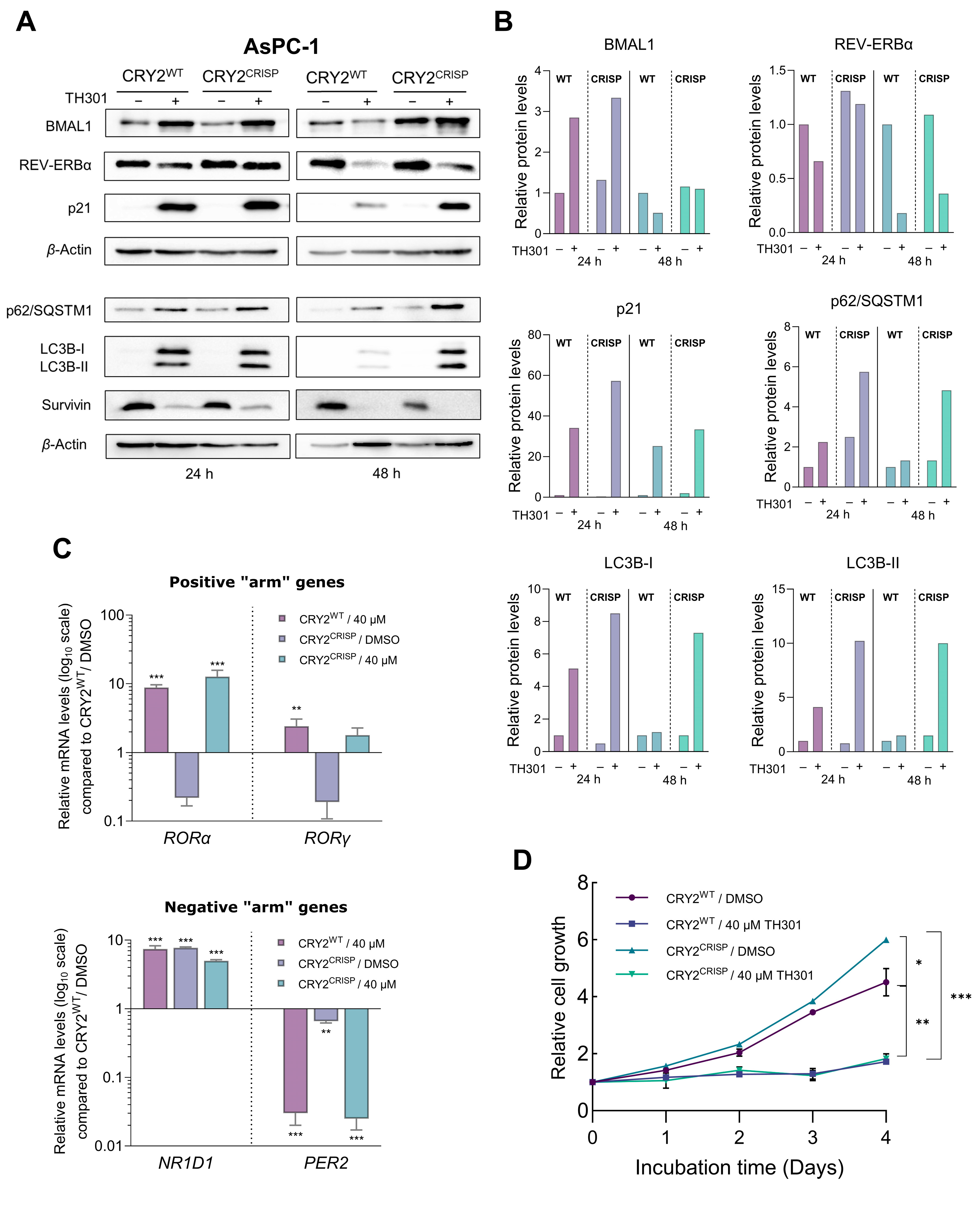
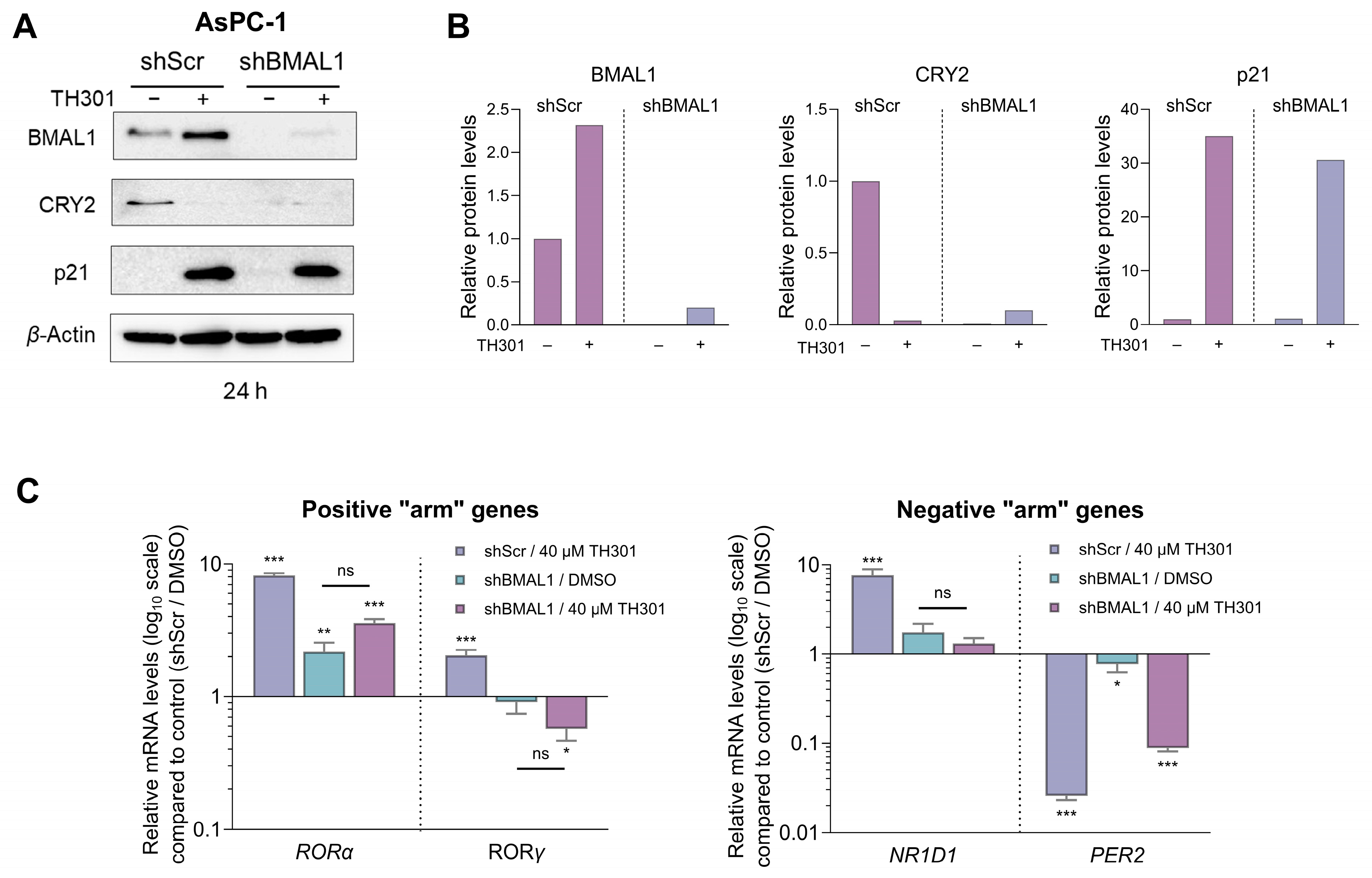
2.7. CRY2 and BMAL1 Are Not Required for the TH301-Driven Induction of p21 Cell Cycle Inhibitor in Pancreatic Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Inhibitors
4.3. MTT Assay
4.4. Cell Cycle Analysis
4.5. RNA Extraction
4.6. RT-qPCR
4.7. Western Blotting
4.8. Plasmids
4.9. Evaluation of BMAL1 Knock-Down Efficiency
4.10. Lentivirus Production
4.11. Lentivirus Transduction
4.12. Genomic DNA Extraction and PCR Amplification
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- National Cancer Institute. Surveillance, Epidemiology and End Results Program. Available online: www.seer.cancer.gov (accessed on 16 December 2024).
- Gourgou-Bourgade, S.; Bascoul-Mollevi, C.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Boige, V.; et al. Impact of FOLFIRINOX Compared with Gemcitabine on Quality of Life in Patients with Metastatic Pancreatic Cancer: Results from the PRODIGE 4/ACCORD 11 Randomized Trial. J. Clin. Oncol. 2013, 31, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Korhan, P.; Verkerk, R.; Critchley, W.R. Scientific Rationale for Integrative and Personalised Strategies for Pancreatic Ductal Adenocarcinoma Management. Integr. Mol. Med. 2017, 4, 1–32. [Google Scholar] [CrossRef]
- Giovannetti, E.; van der Borden, C.L.; Frampton, A.E.; Ali, A.; Firuzi, O.; Peters, G.J. Never Let It Go: Stopping Key Mechanisms Underlying Metastasis to Fight Pancreatic Cancer. Semin. Cancer Biol. 2017, 44, 43–59. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Gurreri, E.; Genovese, G.; Perelli, L.; Agostini, A.; Piro, G.; Carbone, C.; Tortora, G. KRAS-Dependency in Pancreatic Ductal Adenocarcinoma: Mechanisms of Escaping in Resistance to KRAS Inhibitors and Perspectives of Therapy. Int. J. Mol. Sci. 2023, 24, 9313. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The Role of Stroma in Pancreatic Cancer: Diagnostic and Therapeutic Implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Li, C.; Simeone, D.M. Human Pancreatic Cancer Stem Cells: Implications for How We Treat Pancreatic Cancer. Transl. Oncol. 2008, 1, 14–18. [Google Scholar] [CrossRef]
- Battaglin, F.; Chan, P.; Pan, Y.; Soni, S.; Qu, M.; Spiller, E.R.; Castanon, S.; Roussos Torres, E.T.; Mumenthaler, S.M.; Kay, S.A.; et al. Clocking Cancer: The Circadian Clock as a Target in Cancer Therapy. Oncogene 2021, 40, 3187–3200. [Google Scholar] [CrossRef] [PubMed]
- Roenneberg, T.; Merrow, M. Circadian Clocks—The Fall and Rise of Physiology. Nat. Rev. Mol. Cell Biol. 2005, 6, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control Mechanism of the Circadian Clock for Timing of Cell Division in Vivo. Science 2003, 302, 255–259. [Google Scholar] [CrossRef]
- Ma, D.; Li, S.; Molusky, M.M.; Lin, J.D. Circadian Autophagy Rhythm: A Link between Clock and Metabolism? Trends Endocrinol. Metab. 2012, 23, 319–325. [Google Scholar] [CrossRef]
- Wende, A.R.; Young, M.E.; Chatham, J.; Zhang, J.; Rajasekaran, N.S.; Darley-Usmar, V.M. Redox Biology and the Interface between Bioenergetics, Autophagy and Circadian Control of Metabolism. Free Radic. Biol. Med. 2016, 100, 94–107. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Kang, T.H.; Reardon, J.T.; Lee, J.H.; Ozturk, N. Circadian Clock Control of the Cellular Response to DNA Damage. FEBS Lett. 2010, 584, 2618–2625. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yang, Y.; Selby, C.P.; Liu, Z.; Sancar, A. Molecular Mechanism of the Repressive Phase of the Mammalian Circadian Clock. Proc. Natl. Acad. Sci. USA 2020, 118, e2021174118. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional Architecture of the Mammalian Circadian Clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Roenneberg, T.; Merrow, M. The Network of Time: Understanding the Molecular Circadian System. Curr. Biol. 2003, 13, R198–R207. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.; Sassone-Corsi, P. Riding Tandem: Circadian Clocks and the Cell Cycle. Cell 2007, 129, 461–464. [Google Scholar] [CrossRef]
- Gréchez-Cassiau, A.; Rayet, B.; Guillaumond, F.; Teboul, M.; Delaunay, F. The Circadian Clock Component BMAL1 Is a Critical Regulator of P21 WAF1/CIP1 Expression and Hepatocyte Proliferation. J. Biol. Chem. 2008, 283, 4535–4542. [Google Scholar] [CrossRef]
- Zhu, X.; Maier, G.; Panda, S. Learning from Circadian Rhythm to Transform Cancer Prevention, Prognosis, and Survivorship Care. Trends Cancer 2024, 10, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Kondratov, R.V. Clock at the Core of Cancer Development. Biology 2021, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.B.; Nukaya, M.; Berres, M.E.; Rubinstein, C.D.; Wu, G.; Hogenesch, J.B.; Bradfield, C.A.; Ronnekleiv-Kelly, S.M. The Circadian Clock Is Disrupted in Pancreatic Cancer. PLoS Genet. 2023, 19, e1010770. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Adnan, D.; Abdel-Reheem, M.K.; Anafi, R.C.; Leary, D.D.; Bishehsari, F. Circadian Transcriptome of Pancreatic Adenocarcinoma Unravels Chronotherapeutic Targets. JCI Insight 2024, 9, e177697. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Lee, J.W.; St. John, P.C.; Sawa, M.; Iwaisako, K.; Noguchi, T.; Pongsawakul, P.Y.; Sonntag, T.; Welsh, D.K.; Brenner, D.A.; et al. Identification of Small Molecule Activators of Cryptochrome. Science 2012, 337, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef]
- Chun, S.K.; Jang, J.; Chung, S.; Yun, H.; Kim, N.J.; Jung, J.W.; Son, G.H.; Suh, Y.G.; Kim, K. Identification and Validation of Cryptochrome Inhibitors That Modulate the Molecular Circadian Clock. ACS Chem. Biol. 2014, 9, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.K.; Chung, S.; Kim, H.D.; Lee, J.H.; Jang, J.; Kim, J.; Kim, D.; Son, G.H.; Oh, Y.J.; Suh, Y.G.; et al. A Synthetic Cryptochrome Inhibitor Induces Anti-Proliferative Effects and Increases Chemosensitivity in Human Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2015, 467, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Li, S.; Yang, Y.; Shi, X.; Zhao, B.; Lv, L.; Xin, Z.; Kang, J.; Ren, P.; Wu, H. Cryptochrome 2 Acetylation Attenuates Its Antiproliferative Effect in Breast Cancer. Cell Death Dis. 2023, 14, 250. [Google Scholar] [CrossRef]
- Huber, A.L.; Papp, S.J.; Chan, A.B.; Henriksson, E.; Jordan, S.D.; Kriebs, A.; Nguyen, M.; Wallace, M.; Li, Z.; Metallo, C.M.; et al. CRY2 and FBXL3 Cooperatively Degrade C-MYC. Mol. Cell 2016, 64, 774–789. [Google Scholar] [CrossRef]
- Fang, L.; Yang, Z.; Zhou, J.; Tung, J.Y.; Hsiao, C.D.; Wang, L.; Deng, Y.; Wang, P.; Wang, J.; Lee, M.H. Circadian Clock Gene CRY2 Degradation Is Involved in Chemoresistance of Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1476–1487. [Google Scholar] [CrossRef]
- Hoffman, A.E.; Zheng, T.; Ba, Y.; Stevens, R.G.; Yi, C.-H.; Leaderer, D.; Zhu, Y. Phenotypic Effects of the Circadian Gene Cryptochrome 2 on Cancer-Related Pathways. BMC Cancer 2010, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Son, Y.L.; Aikawa, Y.; Makino, E.; Nagai, Y.; Srivastava, A.; Oshima, T.; Sugiyama, A.; Hara, A.; Abe, K.; et al. Isoform-Selective Regulation of Mammalian Cryptochromes. Nat. Chem. Biol. 2020, 16, 676–685. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 4653191. 2024. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/4653191 (accessed on 16 December 2024).
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Edward Mercer, W.; Kinzler, K.W.; Vogelstein, B. WAF1, a Potential Mediator of P53 Tumor Suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase Chromatin Domains Involved in DNA Double-Strand Breaks In Vivo. J. Cell Biol. 1999, 146, 905–915. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The Histone Guardian of the Genome. DNA Repair 2004, 3, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Arif, T.; Shteinfer-Kuzmine, A. Apoptotic Proteins with Non-Apoptotic Activity: Expression and Function in Cancer. Apoptosis 2023, 28, 730–753. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Greene, M.I. Survivin as a Therapeutic Target for the Treatment of Human Cancer. Cancers 2024, 16, 1705. [Google Scholar] [CrossRef]
- Brown, M.; Zhang, W.; Yan, D.; Kenath, R.; Le, L.; Wang, H.; Delitto, D.; Ostrov, D.; Robertson, K.; Liu, C.; et al. The Role of Survivin in the Progression of Pancreatic Ductal Adenocarcinoma (PDAC) and a Novel Survivin-Targeted Therapeutic for PDAC. PLoS ONE 2020, 15, e0226917. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qian, D.; Wang, Y.; Meng, L.; Chen, D.; Ji, X.; Feng, W. Survivin Expression and Serum Levels in Pancreatic Cancer. World J. Surg. Oncol. 2015, 13, 189. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouysségur, J. Hypoxia-Induced Autophagy: Cell Death or Cell Survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stomme, J.M.; Dell’Antonio, G.; et al. Pancreatic Cancers Require Autophagy for Tumor Growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an Autophagy Inhibitor, Potentiates the Radiosensitivity of Glioma Initiating Cells by Inhibiting Autophagy and Activating Apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef]
- Fu, Z.; Cheng, X.; Kuang, J.; Feng, H.; Chen, L.; Liang, J.; Shen, X.; Yuen, S.; Peng, C.; Shen, B.; et al. CQ Sensitizes Human Pancreatic Cancer Cells to Gemcitabine through the Lysosomal Apoptotic Pathway via Reactive Oxygen Species. Mol. Oncol. 2018, 12, 529–544. [Google Scholar] [CrossRef] [PubMed]
- DepMap Portal. Available online: https://depmap.org/portal (accessed on 16 December 2024).
- Miki, T.; Matsumoto, T.; Zhao, Z.; Lee, C.C. P53 Regulates Period2 Expression and the Circadian Clock. Nat. Commun. 2013, 4, 2444. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Vila-Caballer, M.; Liu, J.; Schiffhauer, S.; Finkielstein, C.V. Association of the Circadian Factor Period 2 to P53 Influences P53’s Function in DNA-Damage Signaling. Mol. Biol. Cell 2015, 26, 359–372. [Google Scholar] [CrossRef]
- Molinari, M. Cell Cycle Checkpoints and Their Inactivation in Human Cancer. Cell Prolif. 2000, 33, 261–274. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Wei, W. Cell Cycle on the Crossroad of Tumorigenesis and Cancer Therapy. Trends Cell Biol. 2022, 32, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Horoszewicz, J.S.; Leong, S.S.; Shimano, T.; Penetrante, R.; Sanders, W.H.; Berjian, R.; Douglass, H.O.; Martin, E.W.; Chu, T.N. Human pancreatic adenocarcinoma: In vitro and in vivo morphology of a new tumor line established from ascites. In Vitro 1982, 18, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Nowak, N.J.; Loor, R.; Ochi, H.; Sandberg, A.A.; Lopez, C.; Pickren, J.W.; Berjian, R.; Douglass, H.O.; Chu, T.M. Characterization of a New Primary Human Pancreatic Tumor Line. Cancer Investig. 1986, 4, 15–23. [Google Scholar] [CrossRef]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (PANC-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge’, S.J. The P21 Cdk-Interacting Protein Cipl Is a Potent Inhibitor of Gl Cyclin-Dependent Kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK Inhibitors: Positive and Negative Regulators of G 1-Phase Progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kaida, D. Upregulation of P27 Cyclin-Dependent Kinase Inhibitor and a C-Terminus Truncated Form of P27 Contributes to G1 Phase Arrest. Sci. Rep. 2016, 6, 27829. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific Inhibition of Cyclin-Dependent Kinase 4/6 by PD 0332991 and Associated Antitumor Activity in Human Tumor Xenografts. Mol. Cancer Ther. 2004, 3, 1427–1464. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Bujanda, L.; Billadeau, D.D.; Zhang, J.S. Embryonic Stem Cell Factors and Pancreatic Cancer. World J. Gastroenterol. 2014, 20, 2247–2254. [Google Scholar] [CrossRef]
- Butera, G.; Brandi, J.; Cavallini, C.; Scarpa, A.; Lawlor, R.T.; Scupoli, M.T.; Marengo, E.; Cecconi, D.; Manfredi, M.; Donadelli, M. The Mutant P53-Driven Secretome Has Oncogenic Functions in Pancreatic Ductal Adenocarcinoma Cells. Biomolecules 2020, 10, 884. [Google Scholar] [CrossRef]
- Pan, M.; Jiang, C.; Zhang, Z.; Achacoso, N.; Alexeeff, S.; Solorzano, A.V.; Tse, P.; Chung, E.; Sundaresan, T.; Suga, J.M.; et al. TP53 Gain-of-Function and Non-Gain-of-Function Mutations Are Associated With Differential Prognosis in Advanced Pancreatic Ductal Adenocarcinoma. JCO Precis. Oncol. 2023, 7, 2200570. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When Mutants Gain New Powers: News from the Mutant P53 Field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant P53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Tyner, A.L. Transcriptional Regulation of the P21 (WAF1/CIP1) Gene. Exp. Cell Res. 1999, 246, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O.; Gascan, H. Functional Interaction of STAT3 Transcription Factor with the Cell Cycle Inhibitor P21(WAF1/CIP1/SDI1). J. Biol. Chem. 2000, 275, 18794–18800. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras Requires Autophagy to Maintain Oxidative Metabolism and Tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological Activation of REV-ERBs Is Lethal in Cancer and Oncogene-Induced Senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Smith, M.; Milazzo, G.; Tao, L.; Fekry, B.; Zhu, B.; Mohammad, M.A.; Di Giacomo, S.; Borkar, R.; Reddy, K.R.K.; Capasso, M.; et al. Restoration of the Molecular Clock Is Tumor Suppressive in Neuroblastoma. Nat. Commun. 2021, 12, 4006. [Google Scholar] [CrossRef] [PubMed]
- De Mei, C.; Ercolani, L.; Parodi, C.; Veronesi, M.; Vecchio, C.L.; Bottegoni, G.; Torrente, E.; Scarpelli, R.; Marotta, R.; Ruffili, R.; et al. Dual Inhibition of REV-ERBβ and Autophagy as a Novel Pharmacological Approach to Induce Cytotoxicity in Cancer Cells. Oncogene 2015, 34, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Solt, L.A.; Kojetin, D.J.; Burris, T.P. Regulation of P53 Stability and Apoptosis by a ROR Agonist. PLoS ONE 2012, 7, e34921. [Google Scholar] [CrossRef]
- Kojetin, D.; Wang, Y.; Kamenecka, T.M.; Burris, T.P. Identification of SR8278, a Synthetic Antagonist of the Nuclear Heme Receptor REV-ERB. ACS Chem. Biol. 2011, 6, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Aikawa, Y.; Sugiyama, A.; Nagai, Y.; Hara, A.; Oshima, T.; Amaike, K.; Kay, S.A.; Itami, K.; Hirota, T. An Isoform-Selective Modulator of Cryptochrome 1 Regulates Circadian Rhythms in Mammals. Cell Chem. Biol. 2020, 27, 1192–1198.e5. [Google Scholar] [CrossRef]
- Chan, A.B.; Lamia, K.A. Cancer, Hear My Battle CRY. J. Pineal Res. 2020, 69, e12658. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; McNair, C.M.; McCann, J.J.; Alshalalfa, M.; Shostak, A.; Severson, T.M.; Zhu, Y.; Bergman, A.; Gordon, N.; Mandigo, A.C.; et al. The Circadian Cryptochrome, CRY1, Is a pro-Tumorigenic Factor That Rhythmically Modulates DNA Repair. Nat. Commun. 2021, 12, 401. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhao, S.; Jiang, X.; Zhang, E.; Hu, G.; Hu, B.; Zheng, P.; Xiao, J.; Lu, Z.; Lu, Y.; et al. The Circadian Clock Gene Bmal1 Acts as a Potential Anti-Oncogene in Pancreatic Cancer by Activating the P53 Tumor Suppressor Pathway. Cancer Lett. 2016, 371, 314–325. [Google Scholar] [CrossRef]
- Stephenson, E.M.; Usselmann, L.E.J.; Tergaonkar, V.; Virshup, D.M.; Dallmann, R. Cancer Clocks in Tumourigenesis: The P53 Pathway and Beyond. Endocr. Relat. Cancer 2021, 28, R95–R110. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, N.; Lee, J.H.; Gaddameedhi, S.; Sancar, A. Loss of Cryptochrome Reduces Cancer Risk in P53 Mutant Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.L.; Wang, H.; Hu, A.; Chauhan, J.; Jung, K.Y.; Gharavi, R.B.; Prochownik, E.V.; Fletcher, S. Pharmacophore Identification of C-Myc Inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 2013, 23, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-Molecule Inhibitors of the Myc Oncoprotein. Biochim. Biophys. Acta 2014, 1849, 525. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chen, C.; Okayama, H. High-Efficiency Transformation of Mammalian Cells by Plasmid DNA. Mol. Cell Biol. 1987, 7, 2745–2752. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farmakis, D.; Stravopodis, D.J.; Prombona, A. TH301 Emerges as a Novel Anti-Oncogenic Agent for Human Pancreatic Cancer Cells: The Dispensable Roles of p53, CRY2 and BMAL1 in TH301-Induced CDKN1A/p21CIP1/WAF1 Upregulation. Int. J. Mol. Sci. 2025, 26, 178. https://doi.org/10.3390/ijms26010178
Farmakis D, Stravopodis DJ, Prombona A. TH301 Emerges as a Novel Anti-Oncogenic Agent for Human Pancreatic Cancer Cells: The Dispensable Roles of p53, CRY2 and BMAL1 in TH301-Induced CDKN1A/p21CIP1/WAF1 Upregulation. International Journal of Molecular Sciences. 2025; 26(1):178. https://doi.org/10.3390/ijms26010178
Chicago/Turabian StyleFarmakis, Danae, Dimitrios J. Stravopodis, and Anastasia Prombona. 2025. "TH301 Emerges as a Novel Anti-Oncogenic Agent for Human Pancreatic Cancer Cells: The Dispensable Roles of p53, CRY2 and BMAL1 in TH301-Induced CDKN1A/p21CIP1/WAF1 Upregulation" International Journal of Molecular Sciences 26, no. 1: 178. https://doi.org/10.3390/ijms26010178
APA StyleFarmakis, D., Stravopodis, D. J., & Prombona, A. (2025). TH301 Emerges as a Novel Anti-Oncogenic Agent for Human Pancreatic Cancer Cells: The Dispensable Roles of p53, CRY2 and BMAL1 in TH301-Induced CDKN1A/p21CIP1/WAF1 Upregulation. International Journal of Molecular Sciences, 26(1), 178. https://doi.org/10.3390/ijms26010178