
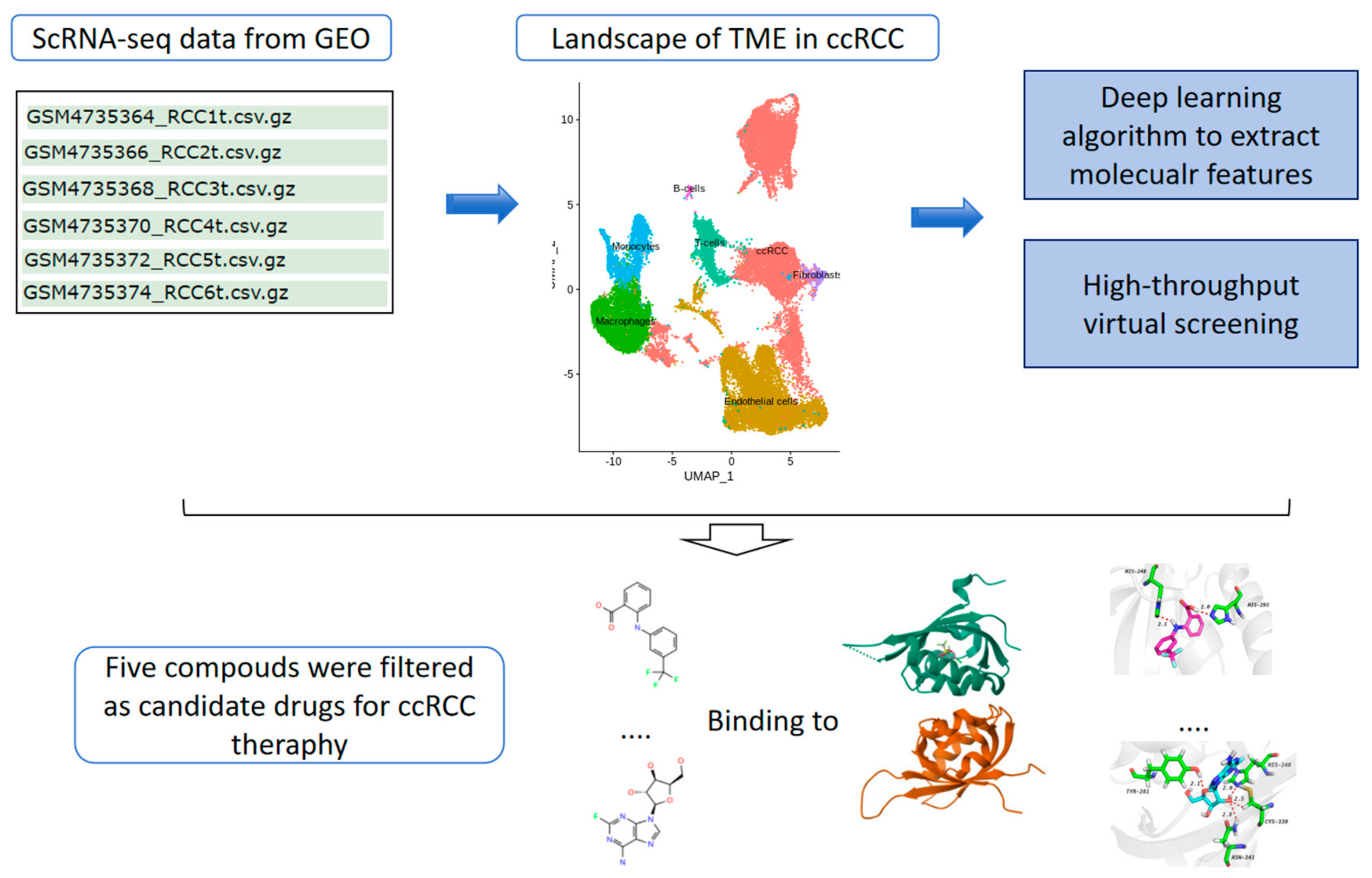
Boosting Clear Cell Renal Carcinoma-Specific Drug Discovery Using a Deep Learning Algorithm and Single-Cell Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
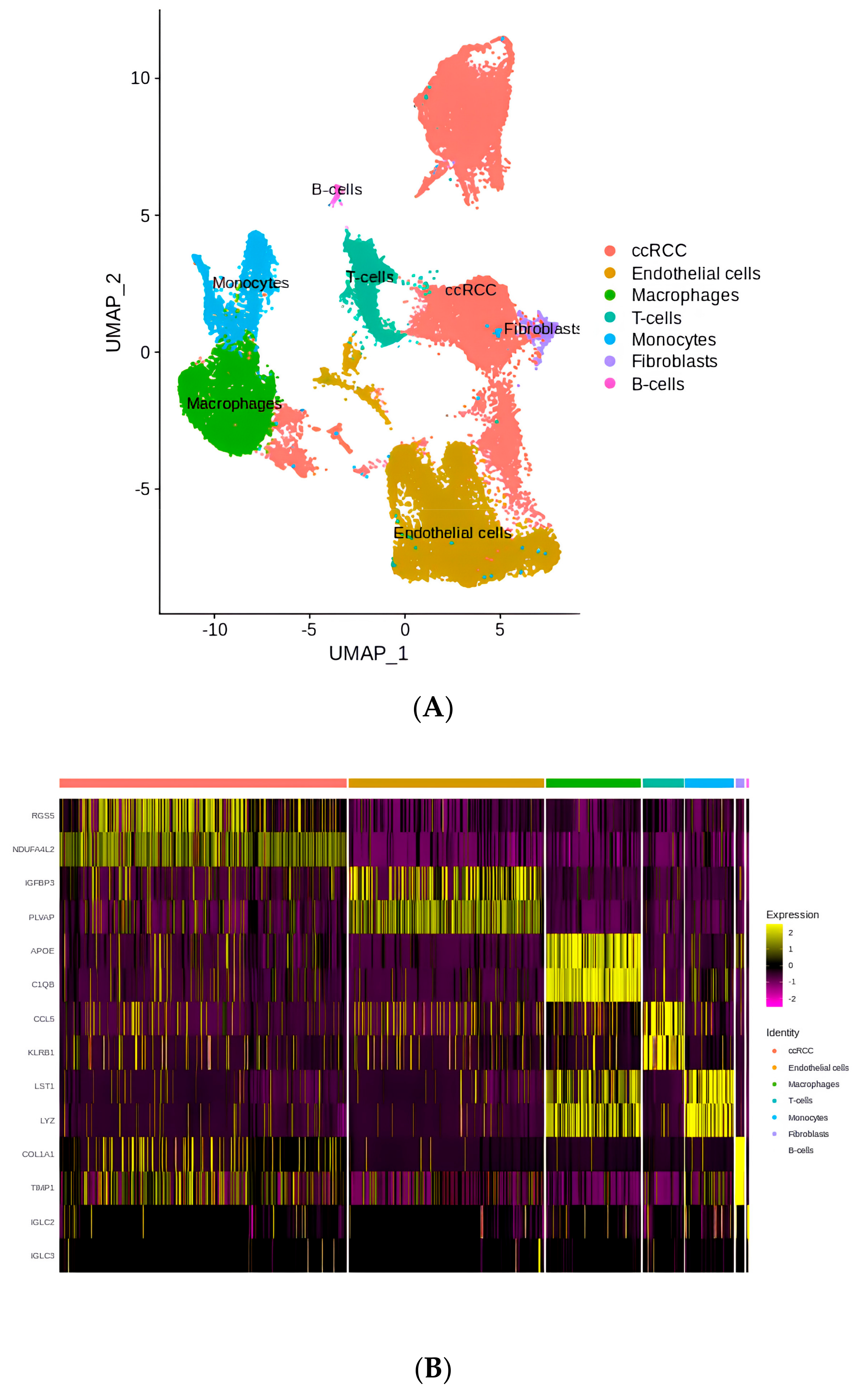
2.1. The Landscape of Cell Type Clustering in ccRCC According to an scRNA-seq Analysis
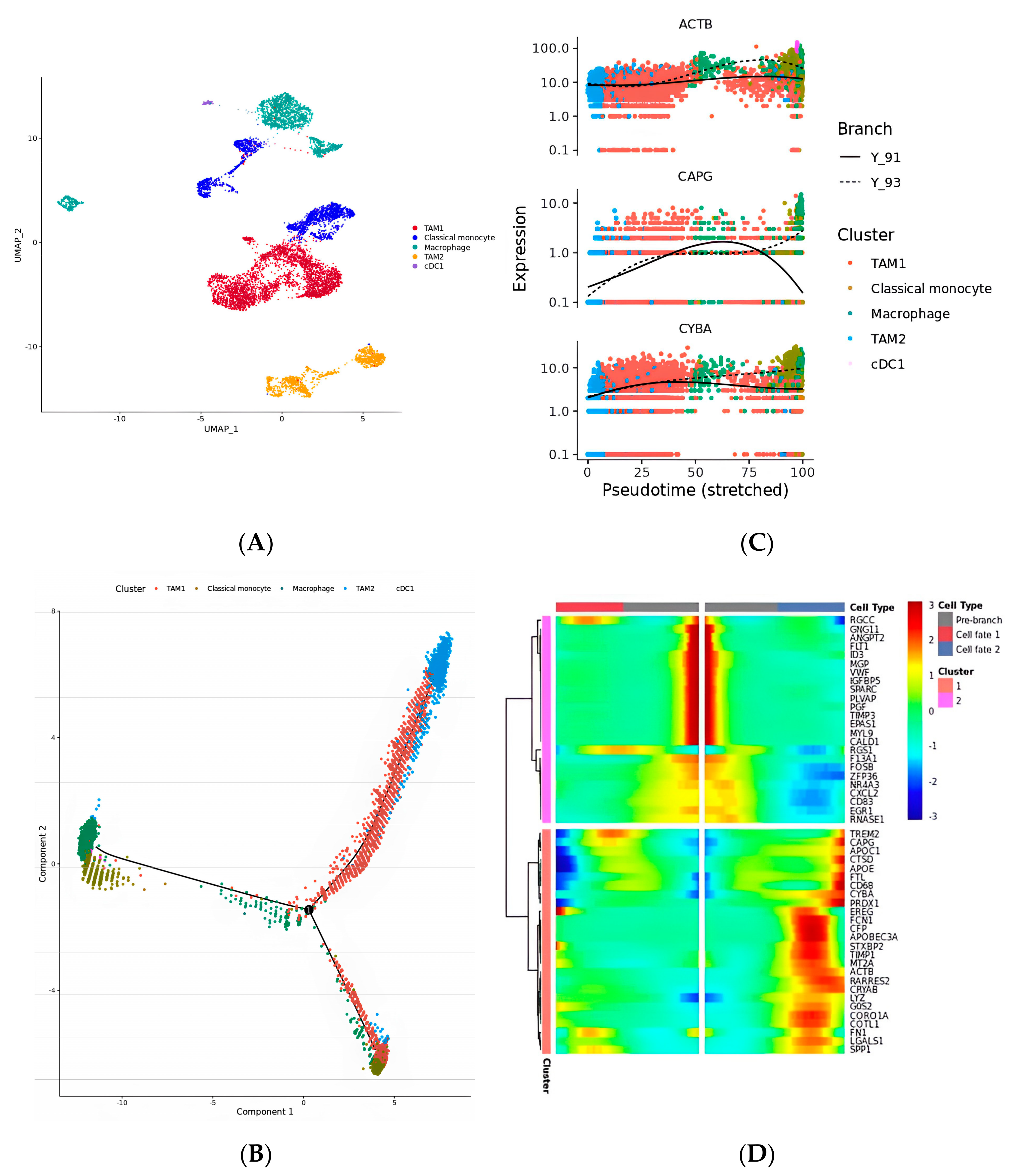
2.2. The Four Kinds of Subsets Identified in Macrophage Cells
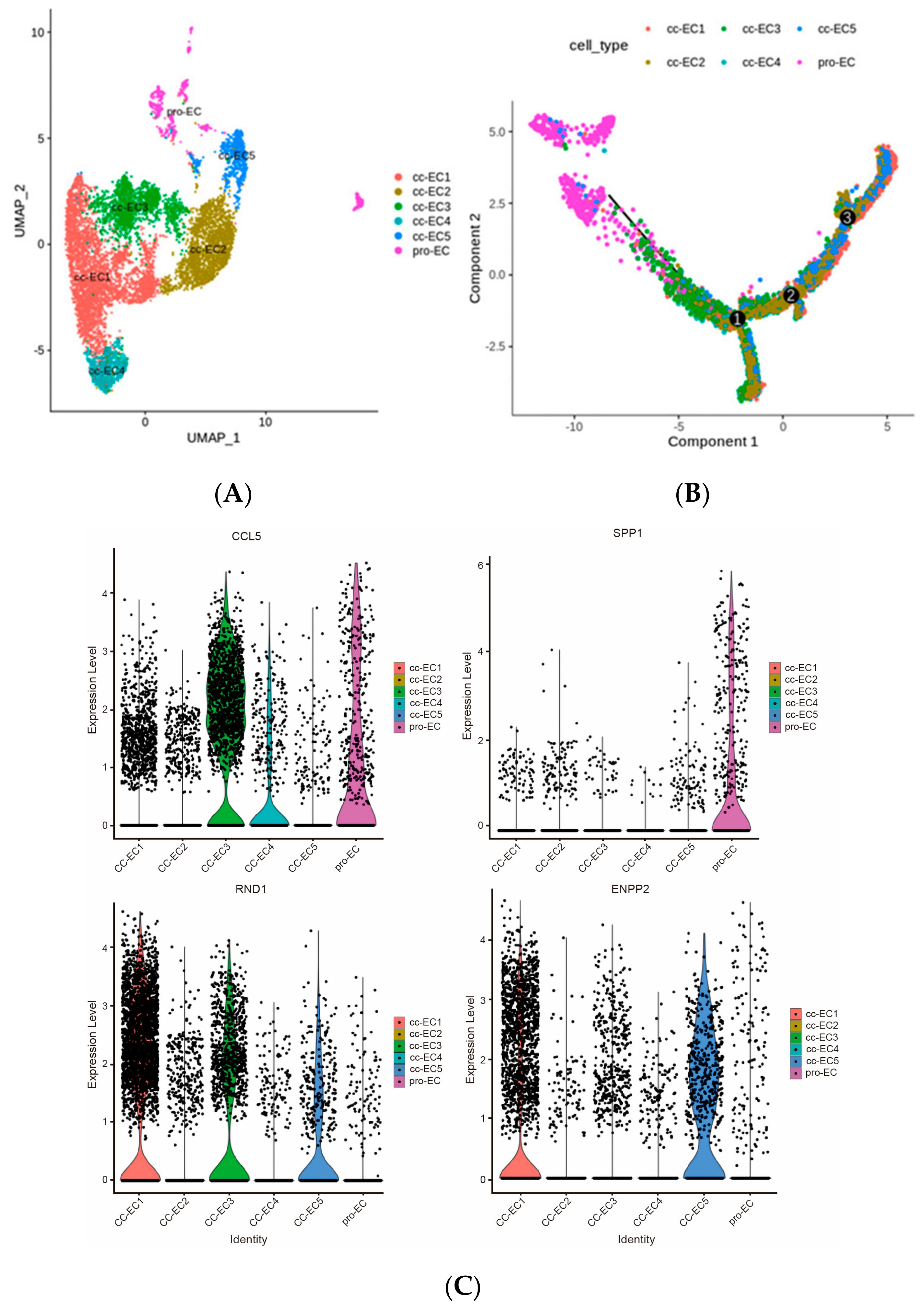
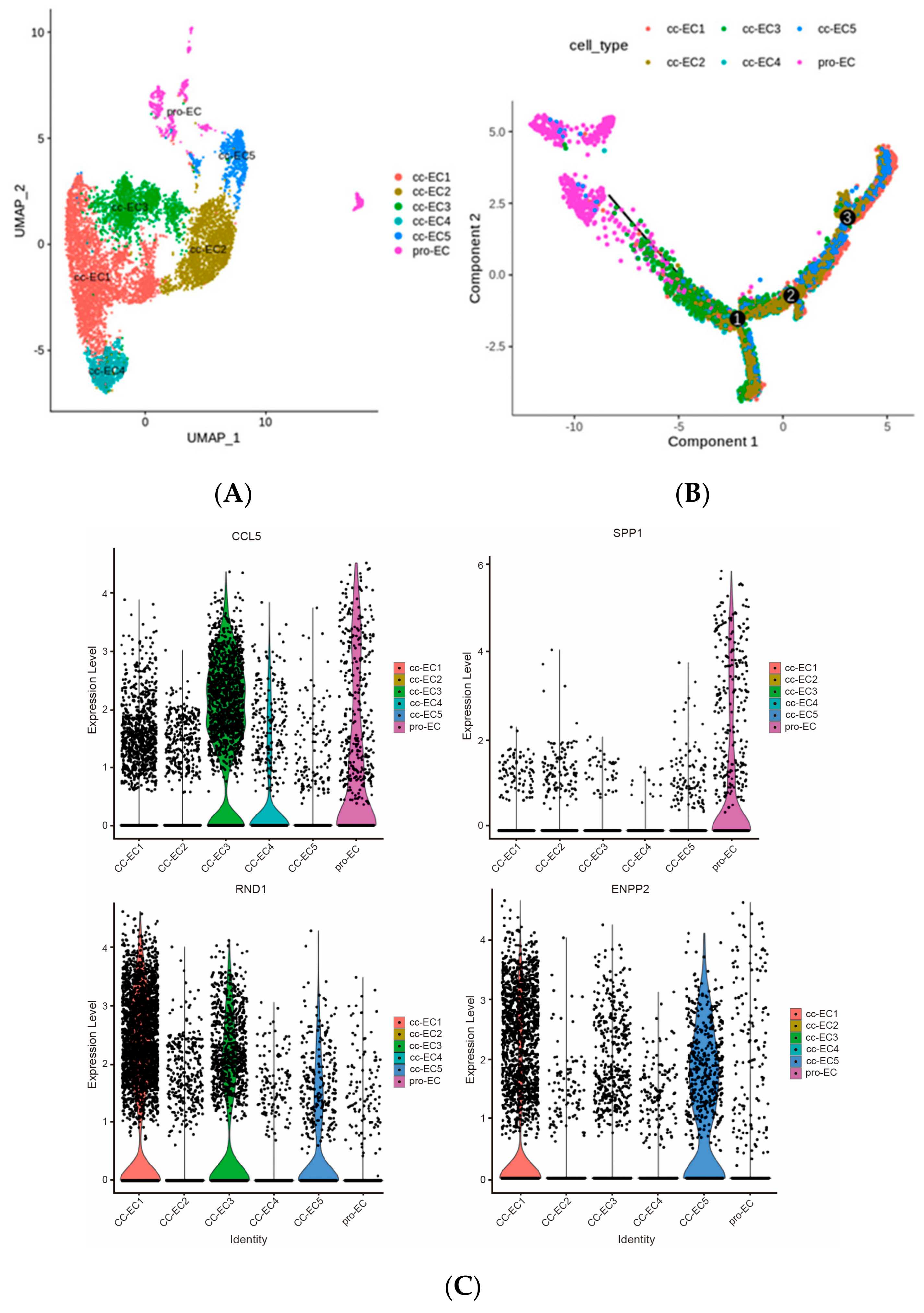
2.3. Six Subtypes of Endothelial Cells in ccRCC
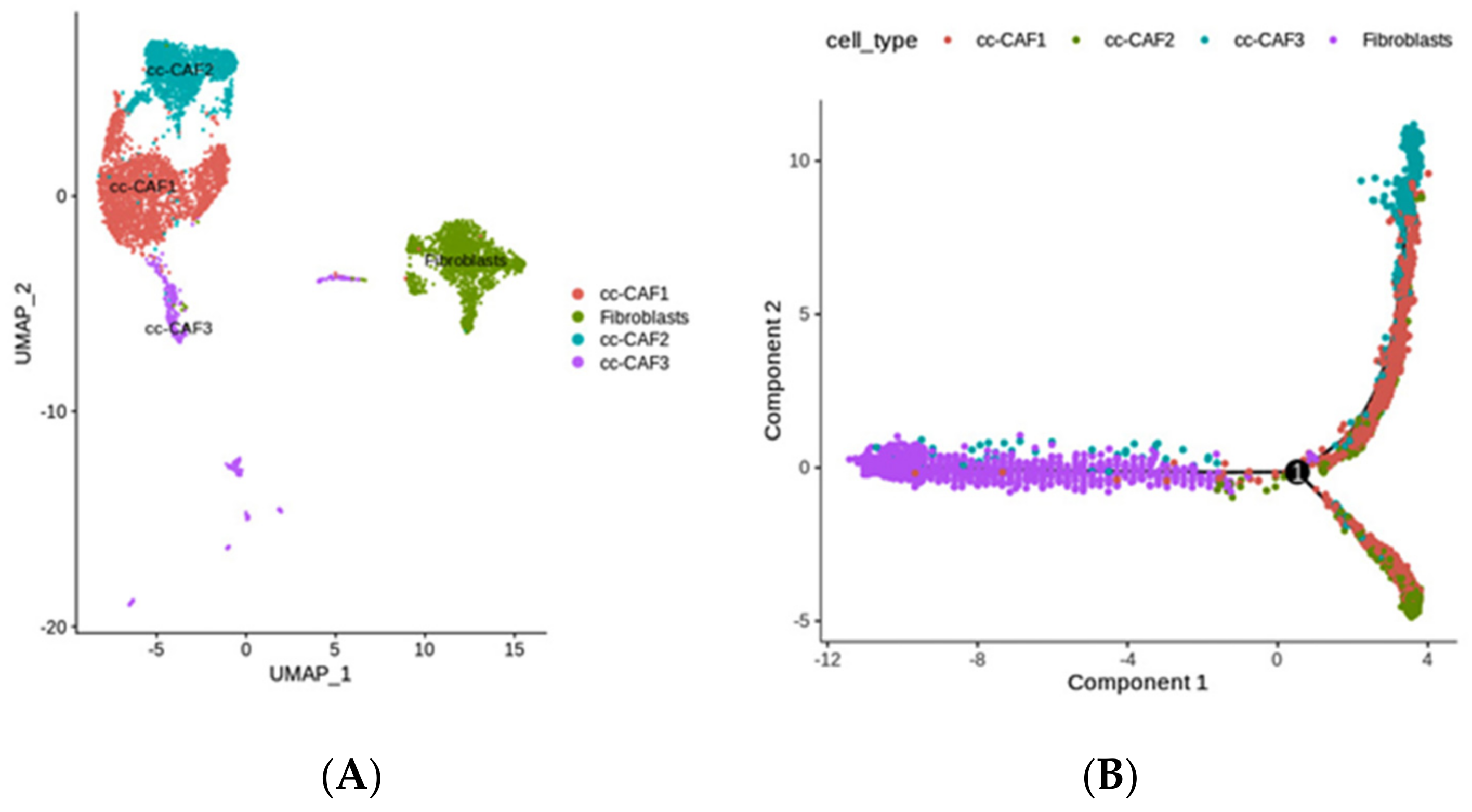
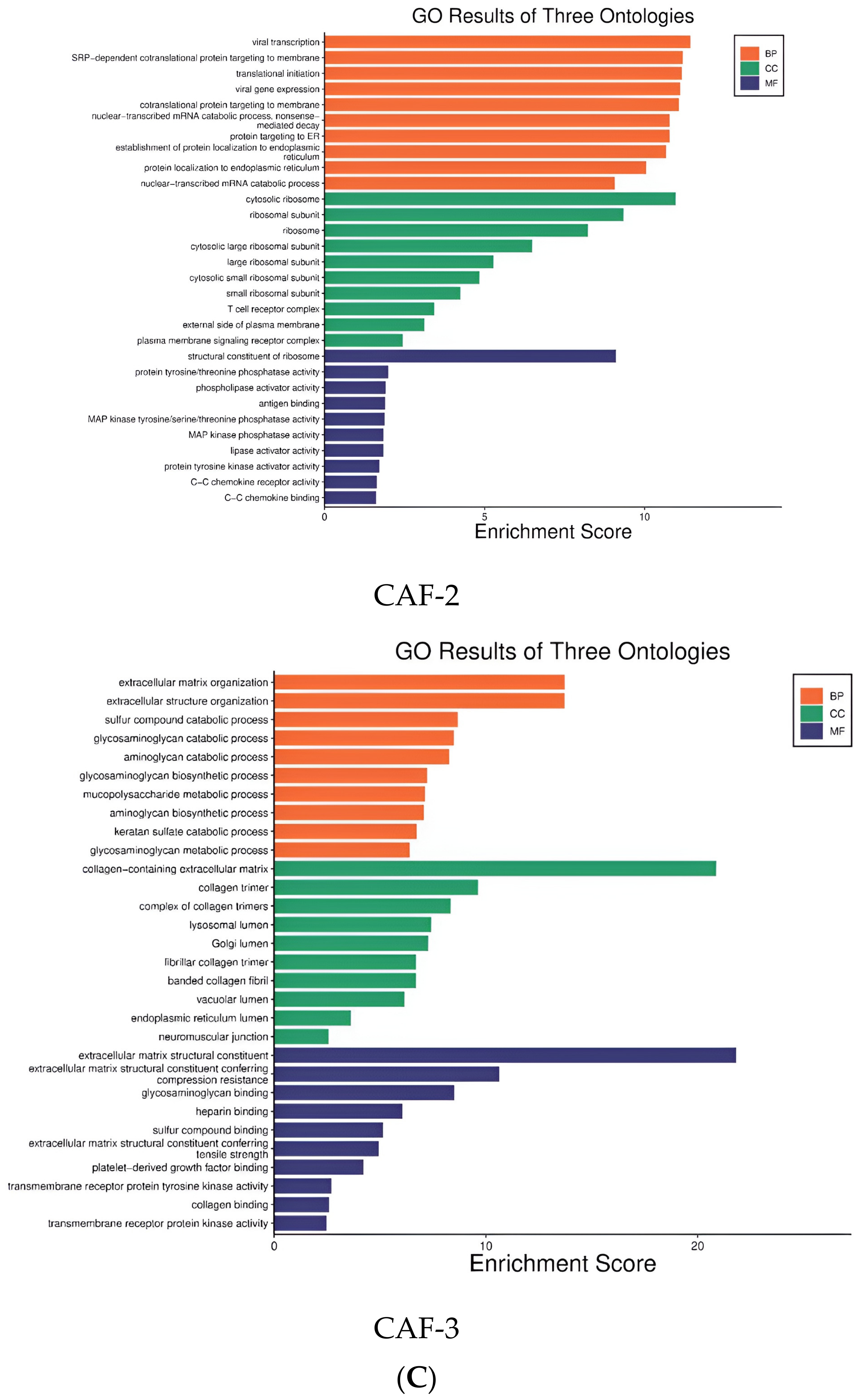
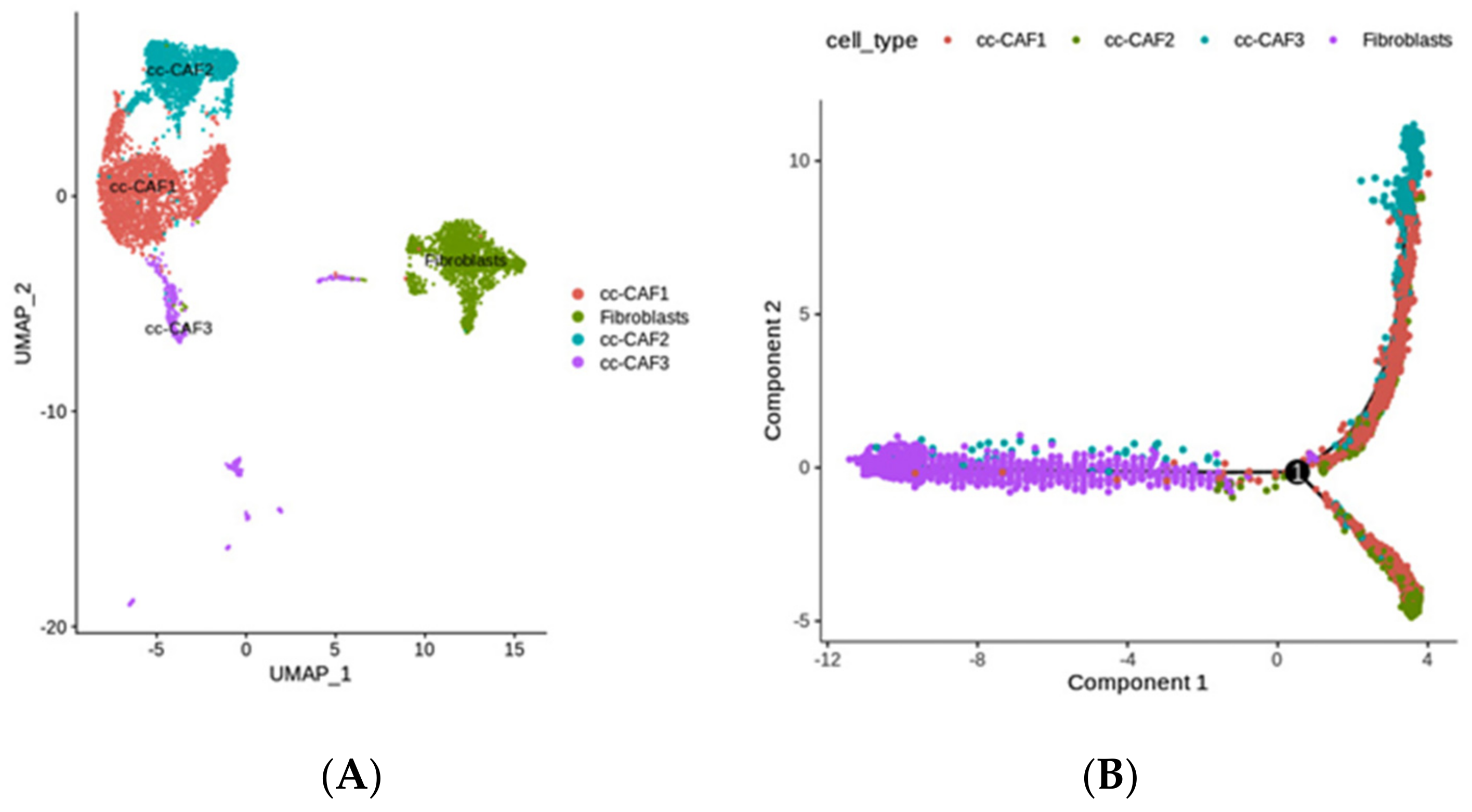
2.4. The Diversity of Fibroblasts (Including Multiple CAFs) in ccRCC, Revealed by scRNA-seq
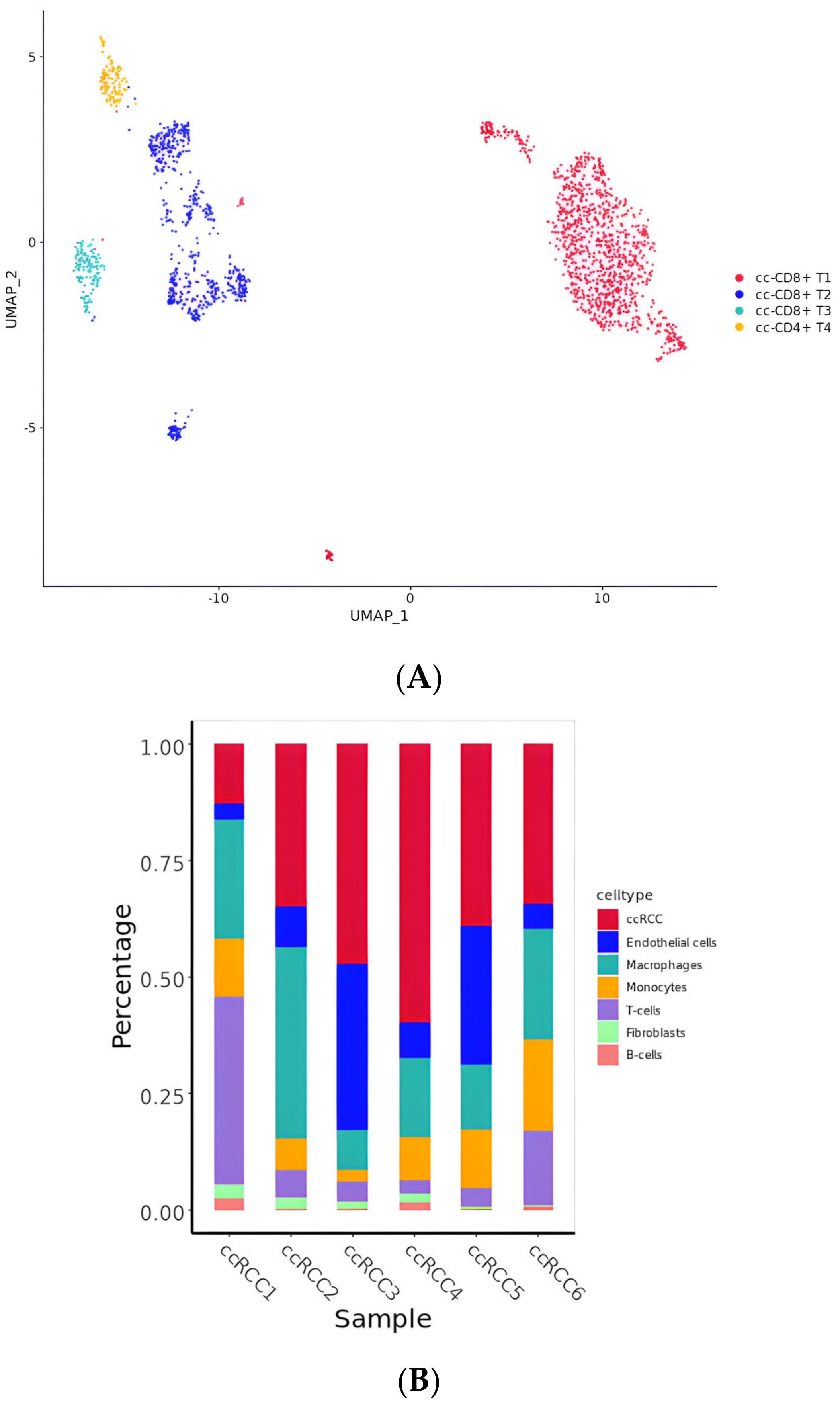
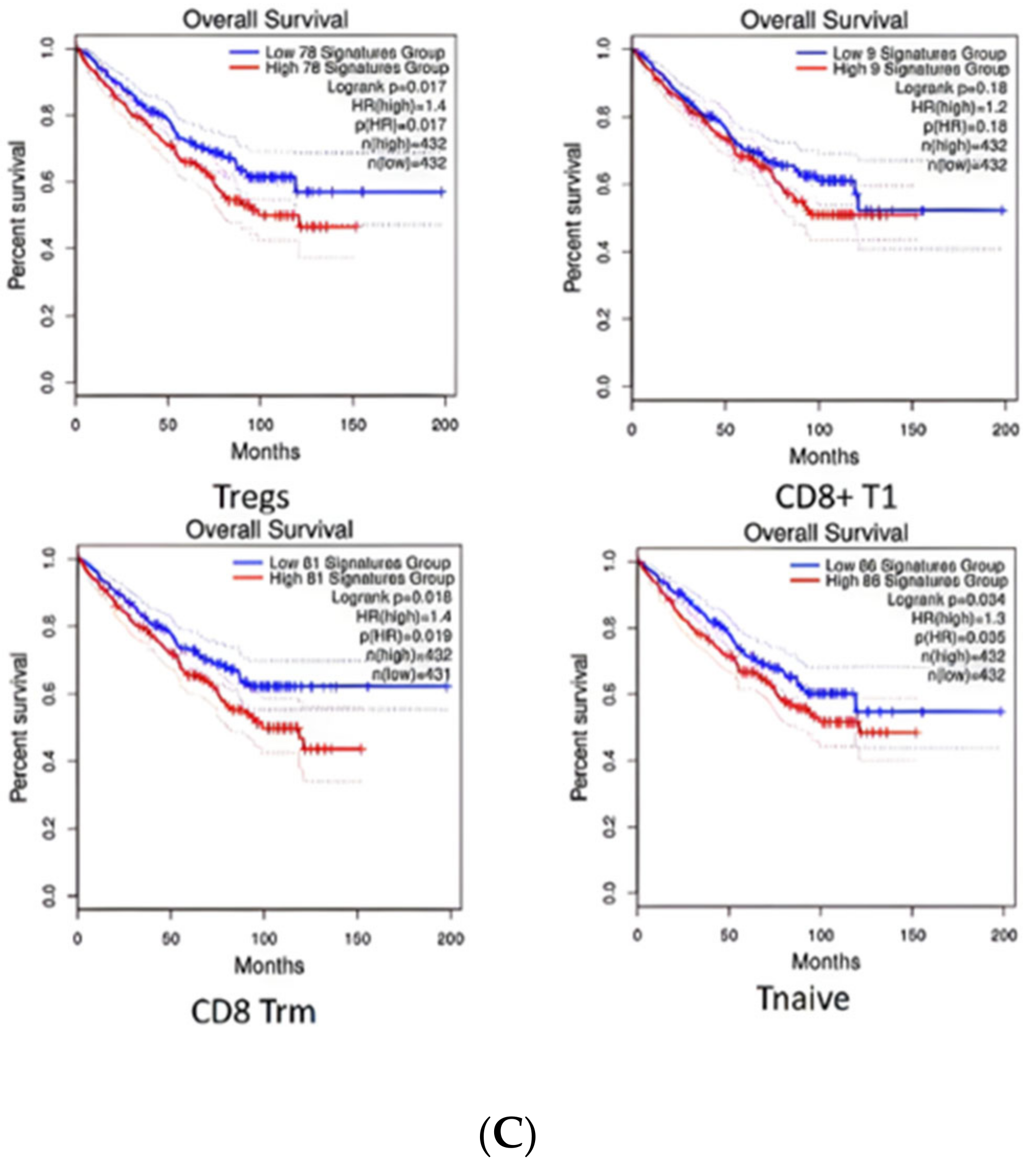
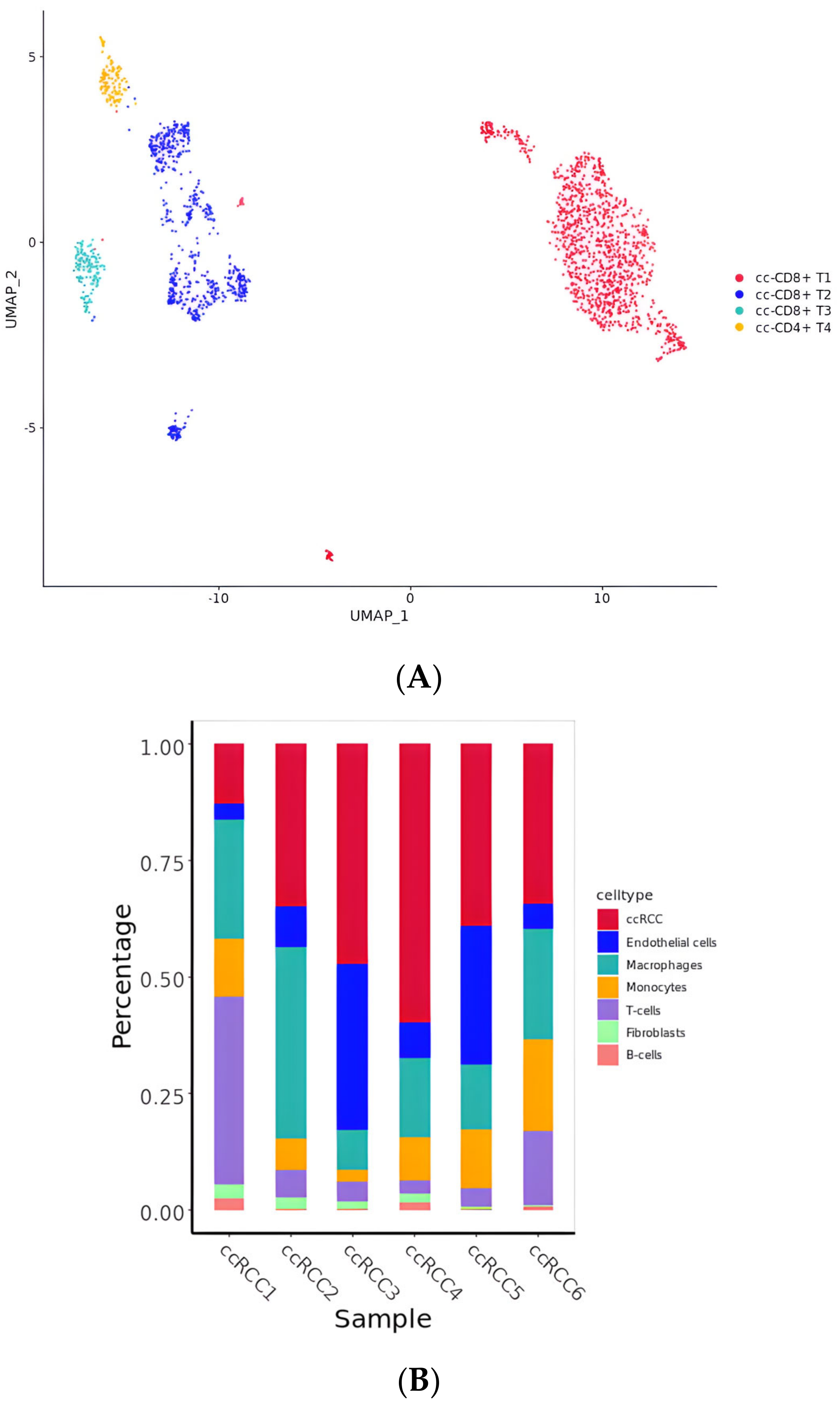
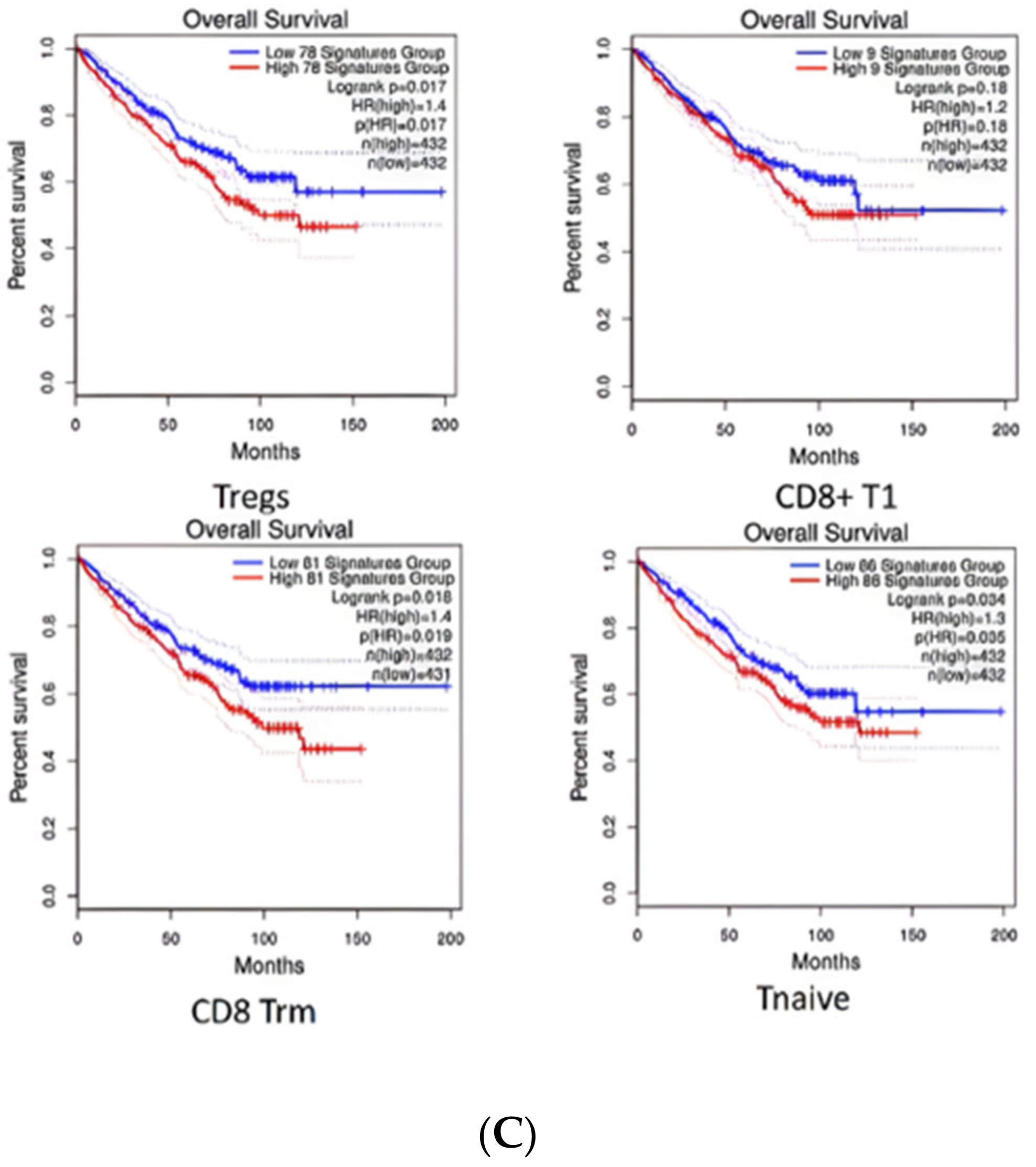
2.5. Diversity of T Cell Characteristics in the ccRCC Immune Microenvironment
2.6. TFs and Their Related Significantly Active Compounds
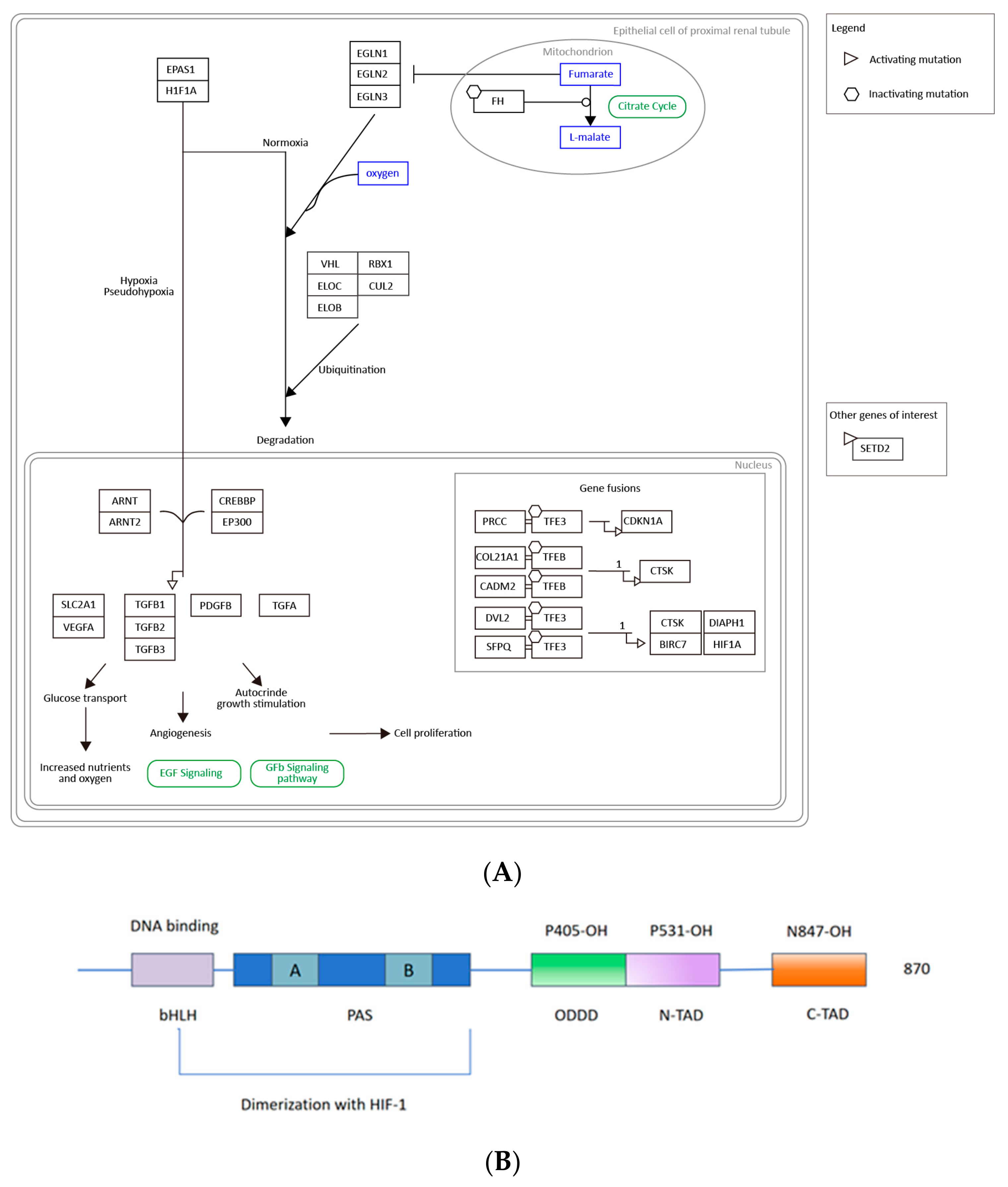
2.7. EPAS1/HIF-2α Is Highly Associated with ccRCC and May Be a Clinical Biomarker and Drug Target
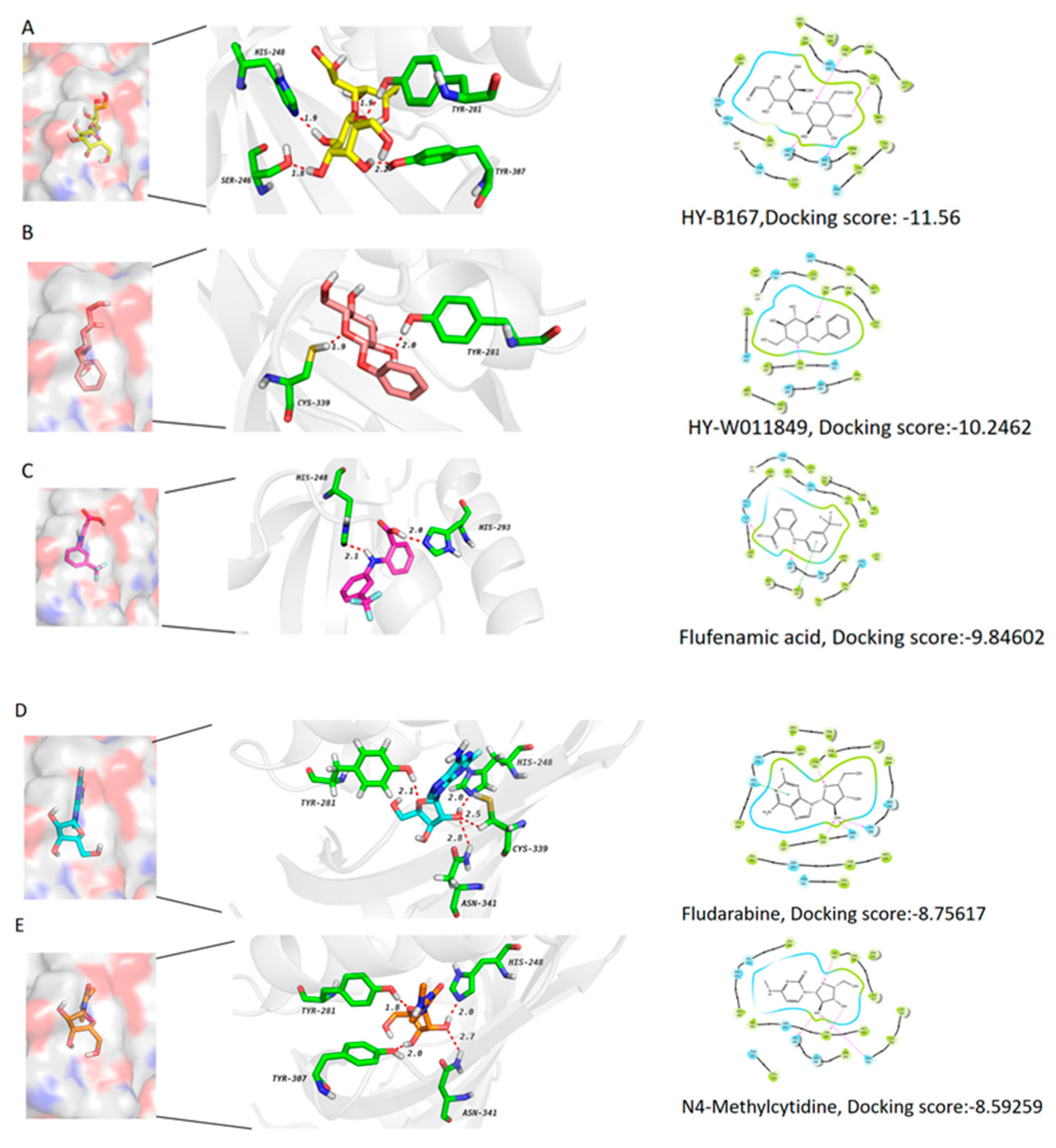
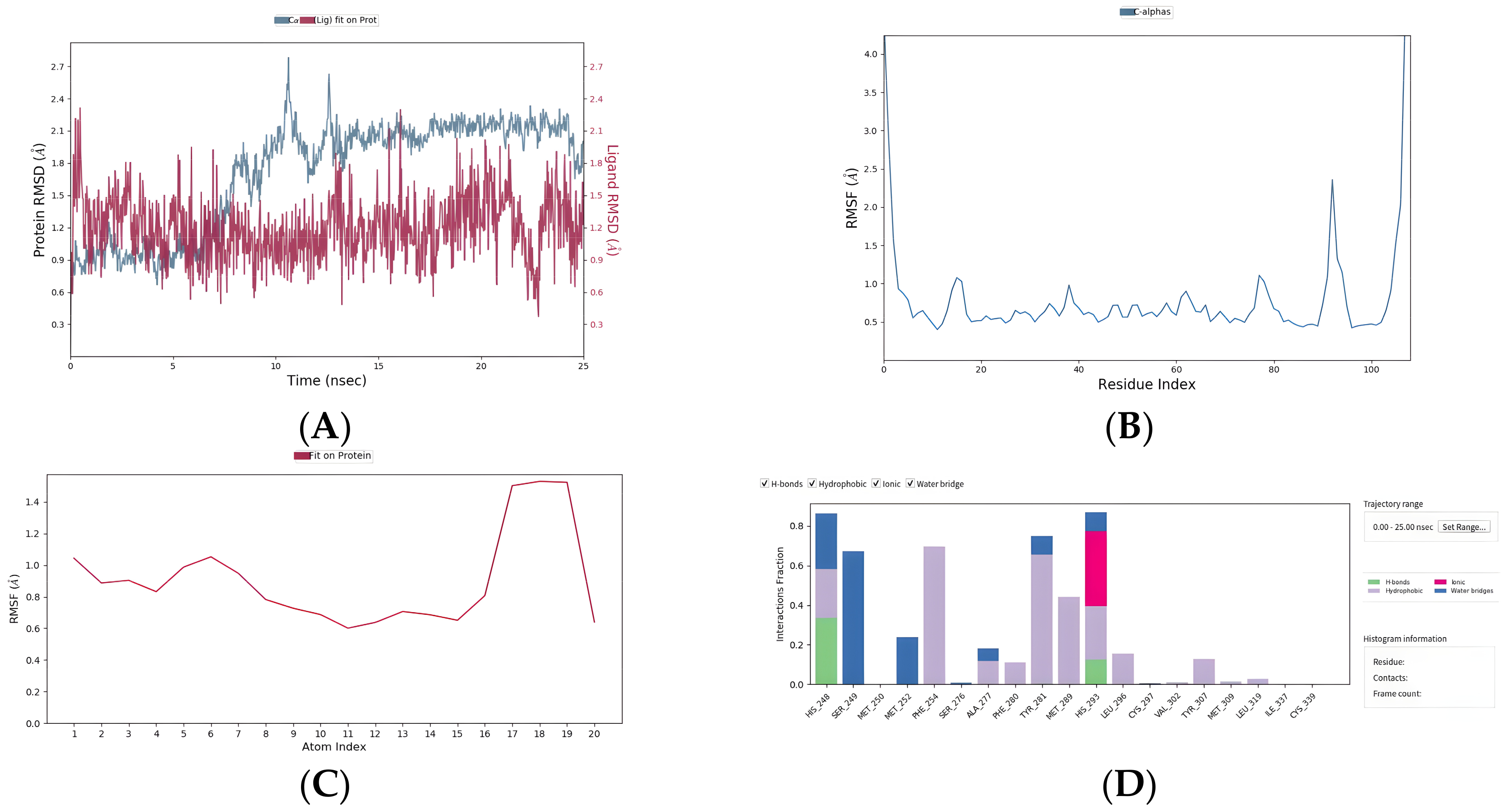
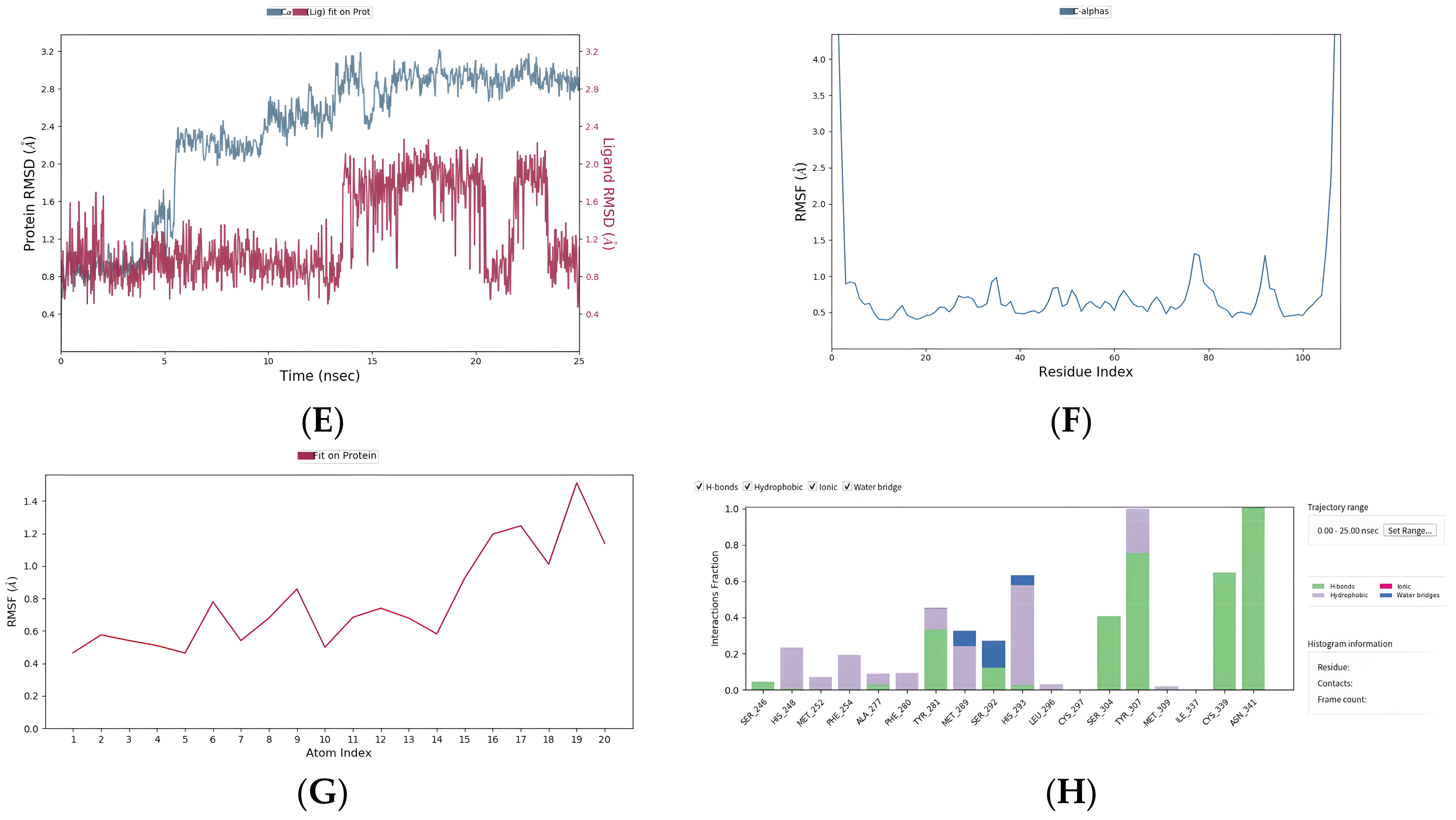
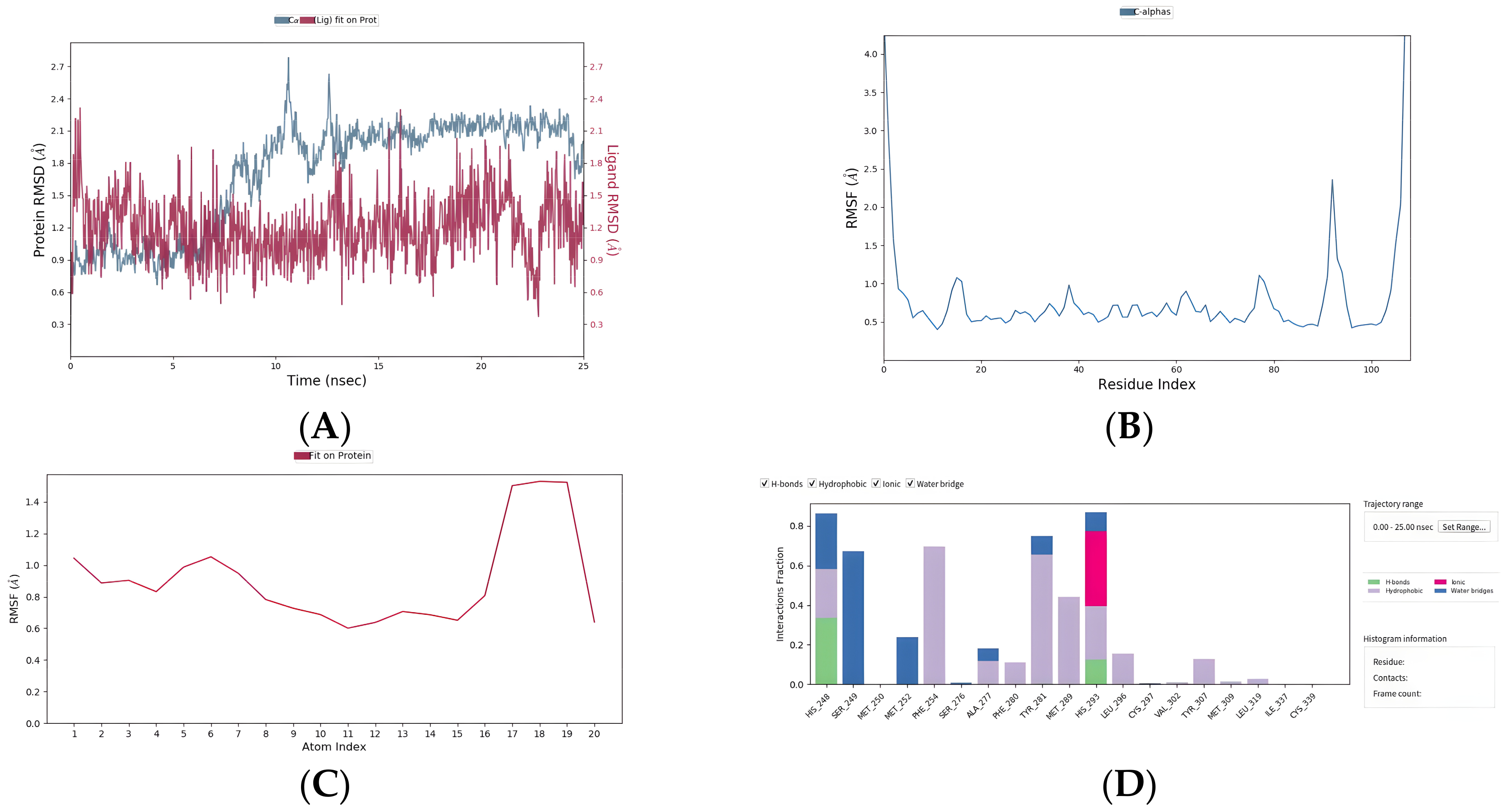
2.8. Five Compounds Were Identified to Target the TF EPAS1
3. Materials and Methods
3.1. Dataset Preparation
3.2. Molecular Feature Extraction Model and Virtual Screening Software
3.3. Cell Clustering Analysis, Visualization, and Annotation
3.4. Functional Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Pham, C.G.; Hsieh, J.J. A clear picture of renal cell carcinoma. Nat. Genet. 2013, 45, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.-H.; Lu, W.; Liang, L.; Chen, G.; Lan, H.-H.; Liang, X.-Y.; Zhu, X. Prognosis of clear cell renal cell carcinoma (ccRCC) based on a six-lncRNA-based risk score: An investigation based on RNA-sequencing data. J. Transl. Med. 2019, 17, 281. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Mitchell, T.J.; Vieira Braga, F.A.; Tran, M.G.B.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.-M.; Loudon, K.W.; et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Lässer, C.; Shelke, G.V.; Yeri, A.; Kim, D.-K.; Crescitelli, R.; Raimondo, S.; Sjöstrand, M.; Gho, Y.S.; Van Keuren Jensen, K.; Lötvall, J. Two distinct extracellular RNA signatures released by a single cell type identified by microarray and next-generation sequencing. RNA Biol. 2017, 14, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Gross-Goupil, M. Overcoming resistance to tyrosine kinase inhibitors in renal cell carcinoma. Cancer Treat. Rev. 2012, 38, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Bukowski, R.M. Targeted therapy for metastatic renal cell carcinoma: A home run or a work in progress? Oncology 2008, 22, 388–396. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Z.; Bao, L.; Zhou, L.; Hou, Y.; Liu, L.; Xiong, M.; Zhang, Y.; Wang, B.; Tao, Z.; et al. Single-Cell Transcriptome Analysis Reveals Intratumoral Heterogeneity in ccRCC, which Results in Different Clinical Outcomes. Mol. Ther. 2020, 28, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Gao, L.; Hao, J.; Zou, X.; Hu, X. An immunotherapy response prediction model derived from proliferative CD4+ T cells and antigen-presenting monocytes in ccRCC. Front. Immunol. 2022, 13, 972227. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Dudani, S.; Graham, J.; Wells, J.C.; Bakouny, Z.; Pal, S.K.; Dizman, N.; Donskov, F.; Porta, C.; de Velasco, G.; Hansen, A.; et al. First-line (1L) immuno-oncology (IO) combination therapies in metastatic renal cell carcinoma (mRCC): Preliminary results from the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC). J. Clin. Oncol. 2019, 37, 584. [Google Scholar] [CrossRef]
- Borcherding, N.; Vishwakarma, A.; Voigt, A.P.; Bellizzi, A.; Kaplan, J.; Nepple, K.; Salem, A.K.; Jenkins, R.W.; Zakharia, Y.; Zhang, W. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun. Biol. 2021, 4, 122. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594.e12. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, O.; Borau, P.G.; Alonso-Gordoa, T.; Molina-Cerrillo, J.; Grande, E. Targeting HIF-2 α in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit. Rev. Oncol. 2017, 111, 117–123. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2–VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- Chang, K.; Chen, Y.; Zhang, X.; Zhang, W.; Xu, N.; Zeng, B.; Wang, Y.; Feng, T.; Dai, B.; Xu, F.; et al. DPP9 Stabilizes NRF2 to Suppress Ferroptosis and Induce Sorafenib Resistance in Clear Cell Renal Cell Carcinoma. Cancer Res. 2023, 83, 3940–3955. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Cella, D.; Motzer, R.J.; Suarez, C.; I Blum, S.; Ejzykowicz, F.; Hamilton, M.; Wallace, J.F.; Simsek, B.; Zhang, J.; Ivanescu, C.; et al. Patient-reported outcomes with first-line nivolumab plus cabozantinib versus sunitinib in patients with advanced renal cell carcinoma treated in CheckMate 9ER: An open-label, randomised, phase 3 trial. Lancet Oncol. 2022, 23, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human transcription factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Chen, X.; Zhao, L. HIV-1/HBV Coinfection Accurate Multitarget Prediction Using a Graph Neural Network-Based Ensemble Predicting Model. Int. J. Mol. Sci. 2023, 24, 7139. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by endothelial cells—Partnering up with the immune system? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Alchahin, A.M.; Mei, S.; Tsea, I.; Hirz, T.; Kfoury, Y.; Dahl, D.; Wu, C.-L.; Subtelny, A.O.; Wu, S.; Scadden, D.T.; et al. A transcriptional metastatic signature predicts survival in clear cell renal cell carcinoma. Nat. Commun. 2022, 13, 5747. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-L.; Chang, F.-C.; Schrimpf, C.; Chen, Y.-T.; Wu, C.-F.; Wu, V.-C.; Chiang, W.-C.; Kuhnert, F.; Kuo, C.J.; Chen, Y.-M.; et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am. J. Pathol. 2011, 178, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrød, B. The scavenger endothelial cell: A new player in homeostasis and immunity. Am. J. Physiol. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar] [CrossRef] [PubMed]
- Stamatiades, E.G.; Tremblay, M.-E.; Bohm, M.; Crozet, L.; Bisht, K.; Kao, D.; Coelho, C.; Fan, X.; Yewdell, W.T.; Davidson, A.; et al. Immune monitoring of trans-endothelial transport by kidney-resident macrophages. Cell 2016, 166, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Eichten, A.; Parveen, A.; Adler, C.; Huang, Y.; Wang, W.; Ding, Y.; Adler, A.; Nevins, T.; Ni, M.; et al. Single-cell transcriptome analyses reveal endothelial cell heterogeneity in tumors and changes following antiangiogenic treatment. Cancer Res. 2018, 78, 2370–2382. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Lan, T.; Wei, Y.; Wei, X. CCL5/CCR5 axis in human diseases and related treatments. Genes Dis. 2022, 9, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-J.; Liao, J.-M.; Zhu, F. Proliferating cell nuclear antigen clamp associated factor, a potential proto-oncogene with increased expression in malignant gastrointestinal tumors. World J. Gastrointest. Oncol. 2021, 13, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mishra, S.; Kumar, A.; Raut, A.A.; Sato, S.; Takaoka, A.; Kumar, H. Essential role of Rnd1 in innate immunity during viral and bacterial infections. Cell Death Dis. 2022, 13, 520. [Google Scholar] [CrossRef] [PubMed]
- Cholia, R.; Nayyar, H.; Kumar, R.; Mantha, A. Understanding the Multifaceted Role of Ectonucleotide Pyrophosphatase/Phosphodiesterase 2 (ENPP2) and its Altered Behaviour in Human Diseases. Curr. Mol. Med. 2015, 15, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, Z.; Lin, Q.; Bei, W.; Guo, J. Pathological and therapeutic roles of bioactive peptide trefoil factor 3 in diverse diseases: Recent progress and perspective. Cell Death Dis. 2022, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Malarkannan, S. NKG7 makes a better killer. Nat. Immunol. 2020, 21, 1139–1140. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.S.; Rivera, F.D.L.; Yan, J.; Corvino, D.; Das, I.; Zhang, P.; Kuns, R.; Chauhan, S.B.; Hou, J.; Li, X.-Y.; et al. The NK cell granule protein NKG7 regulates cytotoxic granule exocytosis and inflammation. Nat. Immunol. 2020, 21, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Galbo, P.M., Jr.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef]
- Sauls, R.S.; McCausland, C.; Taylor, B.N. Histology, T-Cell Lymphocyte. [Updated 2023 May 1]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK535433 (accessed on 10 January 2024).
- Cancer Genome Atlas Research Network; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef]
- McRitchie, B.R.; Akkaya, B. Exhaust the exhausters: Targeting regulatory T cells in the tumor microenvironment. Front. Immunol. 2022, 13, 940052. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; de Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Xia, G.; Kageyama, Y.; Hayashi, T.; Kawakami, S.; Yoshida, M.; Kihara, K. Regulation of vascular endothelial growth factor transcription by endothelial PAS domain protein 1 (EPAS1) and possible involvement of EPAS1 in the angiogenesis of renal cell carcinoma. Cancer 2001, 91, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.R.; Elks, P.M.; Marriott, H.M.; Eamsamarng, S.; Higgins, K.R.; Lewis, A.; Williams, L.; Parmar, S.; Shaw, G.; McGrath, E.E.; et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood 2014, 123, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Kristan, A.; Debeljak, N.; Kunej, T. Integration and Visualization of Regulatory Elements and Variations of the EPAS1 Gene in Human. Genes 2021, 12, 1793. [Google Scholar] [CrossRef] [PubMed]
- EPAS1. Endothelial PAS Domain Protein 1. Available online: https://atlasgeneticsoncology.org/gene/44088/epas1-(endothelial-pas-domain-protein-1) (accessed on 20 January 2024).
- Scheuermann, T.H.; Tomchick, D.R.; Machius, M.; Guo, Y.; Bruick, R.K.; Gardner, K.H. Artificial ligand binding within the HIF2α PAS-B domain of the HIF2 transcription factor. Proc. Natl. Acad. Sci. USA 2009, 106, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Alshalalfeh, M.; Sun, N.; Moraes, A.H.; Utani, A.P.A.; Xu, Y. Conformational Distributions of Phenyl β-D-Glucopyranoside and Gastrodin in Solution by Vibrational Optical Activity and Theoretical Calculations. Molecules 2023, 28, 4013. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Li, Y.; de Vries, A.C.; Li, P.; Peppelenbosch, M.P.; Pan, Q. Seasonal coronavirus infections trigger NLRP3 inflammasome activation in macrophages but is therapeutically targetable. Antivir. Res. 2023, 216, 105674. [Google Scholar] [CrossRef]
- Pang, Y.-J.; Feng, H.; Wen, S.-Y.; Qiao, Q.-C.; Zhang, J.; Yang, N. Orexin enhances firing activities in the gigantocellular reticular nucleus through the activation of non-selective cationic conductance. Neurosci. Lett. 2020, 733, 135088. [Google Scholar] [CrossRef]
- Palakurthi, B.; Fross, S.R.; Guldner, I.H.; Aleksandrovic, E.; Liu, X.; Martino, A.K.; Wang, Q.; Neff, R.A.; Golomb, S.M.; Lewis, C.; et al. Targeting CXCL16 and STAT1 augments immune checkpoint blockade therapy in triple-negative breast cancer. Nat. Commun. 2023, 14, 2109. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, Q.; Hong, Y.; Gao, Y.; Hu, J.; Lang, M.; Zhang, H.; Zhou, Y.; Luo, H.; Zhang, X.; et al. β2-Microglobulin coaggregates with Aβ and contributes to amyloid pathology and cognitive deficits in Alzheimer’s disease model mice. Nat. Neurosci. 2023, 26, 1170–1184. [Google Scholar] [CrossRef] [PubMed]
- Ngoc, L.N.T.; Park, S.J.; Huong, T.T.; Lee, K.H.; Kang, H. N4-methylcytidine ribosomal RNA methylation in chloroplasts is crucial for chloroplast function, development, and abscisic acid response in Arabidopsis. J. Integr. Plant Biol. 2020, 63, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-H.; Bao, Y.; Wang, X.; Yan, F.; Guo, S.; Ma, Y.; Xu, D.; Jin, L.; Xu, J.; Wang, J. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 2018, 32, 6694–6705. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Landrum, G.A. RDKit: Open-Source Cheminformatics. 2014. Available online: https://www.bibsonomy.org/bibtex/28d01fceeccd6bf2486e47d7c4207b108/salotz (accessed on 1 March 2014).
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742. [Google Scholar] [CrossRef] [PubMed]
- Ghojogh, B.; Crowley, M.; Karray, F.; Ghodsi, A. Uniform Manifold Approximation and Projection (UMAP). In Elements of Dimensionality Reduction and Manifold Learning; Springer: Cham, Switzerland, 2023. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, A.K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Nir, H.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356, 593007. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Zhen, Q.; Zhang, Y.; Gao, L.; Wang, R.; Chu, W.; Zhao, X.; Li, Z.; Li, H.; Zhang, B.; Lv, B.; et al. EPAS1 promotes peritoneal carcinomatosis of non-small-cell lung cancer by enhancing mesothelial–mesenchymal transition. Strahlenther. Onkol. 2020, 197, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhao, J.; Zhao, L. EPAS1/HIF-2α Acts as an Unanticipated Tumor-Suppressive Role in Papillary Thyroid Carcinoma. Int. J. Gen. Med. 2023, 16, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Recktenwald, M.; Hutt, E.; Fuller, S.; Briggs, M.; Goel, A.; Daringer, N. Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy. Cancers 2022, 14, 1259. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Hua, J.; Fu, Y.; An, J.; Chen, X.; Wang, C.; Zheng, Y.; Wang, F.; Ji, Y.; Li, Q. Updated perspective of EPAS1 and the role in pulmonary hypertension. Front. Cell Dev. Biol. 2023, 11, 1125723. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Maemura, K.; Imai, Y.; Harada, T.; Kawanami, D.; Nojiri, T.; Manabe, I.; Nagai, R. Endothelial PAS domain protein 1 gene promotes angiogenesis through the transactivation of both vascular endothelial growth factor and its receptor, Flt-1. Circ. Res. 2004, 95, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Hoefflin, R.; Harlander, S.; Schäfer, S.; Metzger, P.; Kuo, F.; Schönenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.-Y.; et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Jimenez, C.; Armaiz-Pena, G.; Arenillas, C.; Capdevila, J.; Dahia, P.L.M. Hypoxia-Inducible Factor 2 Alpha (HIF2α) Inhibitors: Targeting Genetically Driven Tumor Hypoxia. Endocr. Rev. 2023, 44, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Simoens, S.; Huys, I. R&D Costs of New Medicines: A Landscape Analysis. Front. Med. 2021, 8, 760762. [Google Scholar] [CrossRef]
- Chong, C.R.; Sullivan, D.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Sun, C.; Tang, M.; Wang, Y.; Ma, J.; Yu, J.; Wei, J.; Ma, J.; Wang, B.; Xie, Q.; et al. Single-cell multiomics analysis reveals regulatory programs in clear cell renal cell carcinoma. Cell Discov. 2022, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, S.; Hu, Y.; Huang, W. Single-cell RNA transcriptome reveals the intra-tumoral heterogeneity and regulators underlying tumor progression in metastatic pancreatic ductal adenocarcinoma. Cell Death Discov. 2021, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Chari, T.; Pachter, L. The specious art of single-cell genomics. PLoS Comput Biol. 2023, 19, e1011288. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, X.; Tang, N.; Guo, M.; Ai, D. Boosting Clear Cell Renal Carcinoma-Specific Drug Discovery Using a Deep Learning Algorithm and Single-Cell Analysis. Int. J. Mol. Sci. 2024, 25, 4134. https://doi.org/10.3390/ijms25074134
Wang Y, Chen X, Tang N, Guo M, Ai D. Boosting Clear Cell Renal Carcinoma-Specific Drug Discovery Using a Deep Learning Algorithm and Single-Cell Analysis. International Journal of Molecular Sciences. 2024; 25(7):4134. https://doi.org/10.3390/ijms25074134
Chicago/Turabian StyleWang, Yishu, Xiaomin Chen, Ningjun Tang, Mengyao Guo, and Dongmei Ai. 2024. "Boosting Clear Cell Renal Carcinoma-Specific Drug Discovery Using a Deep Learning Algorithm and Single-Cell Analysis" International Journal of Molecular Sciences 25, no. 7: 4134. https://doi.org/10.3390/ijms25074134
APA StyleWang, Y., Chen, X., Tang, N., Guo, M., & Ai, D. (2024). Boosting Clear Cell Renal Carcinoma-Specific Drug Discovery Using a Deep Learning Algorithm and Single-Cell Analysis. International Journal of Molecular Sciences, 25(7), 4134. https://doi.org/10.3390/ijms25074134