
Oxidative Effects in Early Stages of Embryo Development Due to Alcohol Consumption


Abstract
1. Introduction
2. Fetal Alcohol Spectrum Disorder (FASD)
- alcohol-related birth defects;
- Alcohol-Related Neurodevelopmental Disorder;
- FAS;
- partial fetal alcohol syndrome;
2.1. Characteristics of Patients with FASD
- Six or more drinks per week throughout two or more weeks of pregnancy;
- Three or more drinks per occasion on two or more occasions;
- Legal or social problems related to alcohol during pregnancy;
- Alcohol intoxication during pregnancy registered in blood, lung (air), or urine;
- Positive alcohol biomarker test during pregnancy. Such tests are considered positive when fatty acid ethyl esters, ethyl glucuronic acid, or phosphatidyl ethanol is detected in the hair, nails, placenta, meconium, or blood.
- Increased prenatal risk associated with alcohol use during pregnancy.
2.2. FASD Subtypes
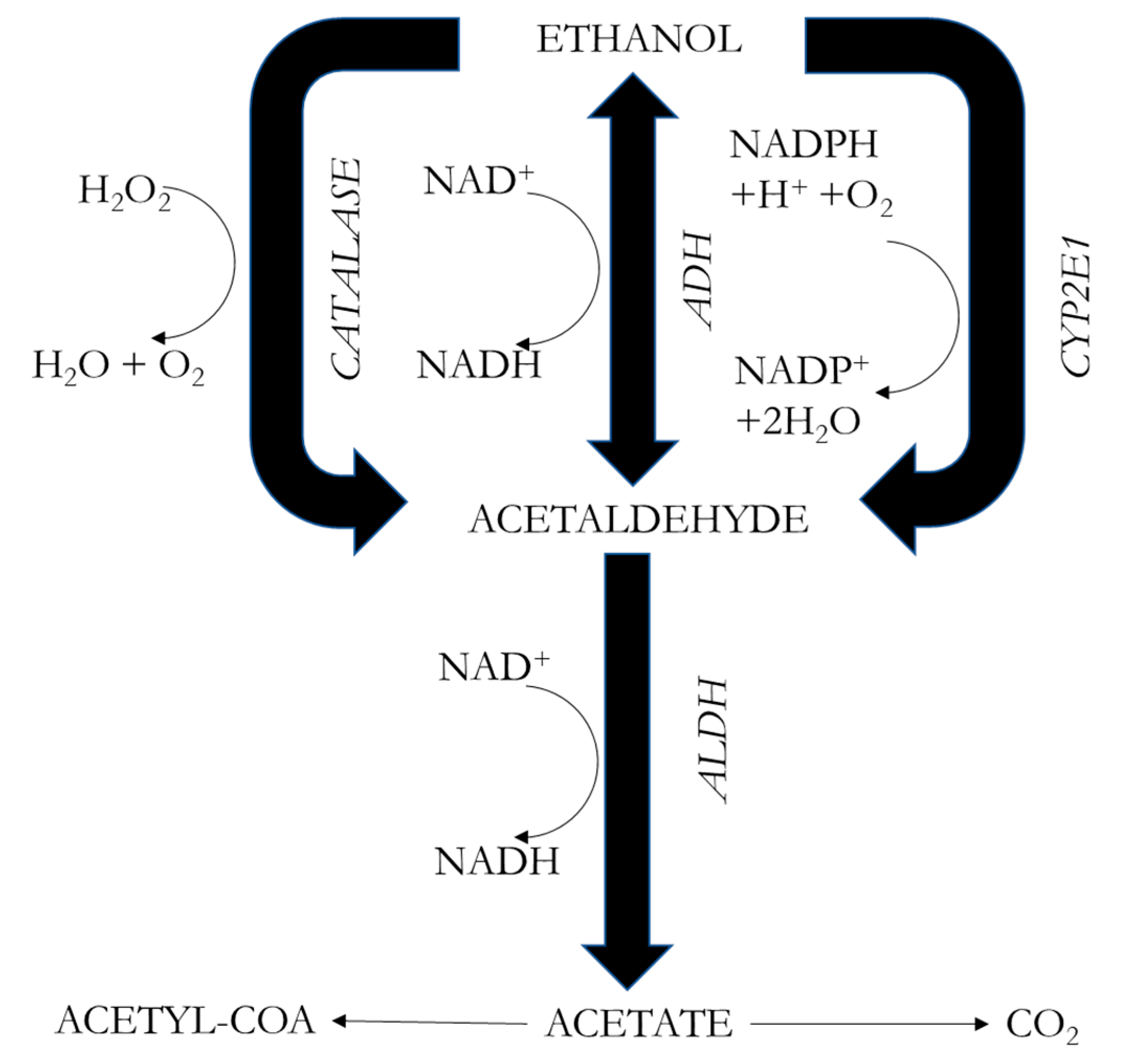
3. Metabolism of Alcohol
3.1. Changes in Metabolism and Blood Alcohol Concentration during Pregnancy
- The absorption of alcohol varies during pregnancy because in this state there is a delay in stomach emptying and a decrease in intestinal motility. Therefore, it can be expected that in pregnant women, the concentrations of alcohol in the blood will be maintained for a longer time, although the serum alcohol peaks will be lower.
- Alcohol, once in the blood, passes to the placenta by simple diffusion. The diffusion balance is based on the water content. During pregnancy, the volume of water increases up to 6 L in all compartments, including the amniotic fluid, the placenta, the uterus, and the fetus. During the different stages of pregnancy, there are some changes in the volumes of water. Therefore, fetal exposure to alcohol varies according to the changes in the amounts of water in the maternal and fetal organisms. During early pregnancy, the fetal water level is very high, and therefore the fetus is highly exposed to alcohol.
- Alcohol is eliminated through oxidation by ADH in the liver. However, the fetus’s liver enzymes do not mature until about the second half of pregnancy, so it cannot metabolize alcohol. The alcohol concentration in the maternal body is more correlated with fetal harm than is the total amount of alcohol administered.
3.2. Markers of OS during Pregnancy
4. Toxicity of Alcohol
- In 1981, Henderson et al. [24] began to talk about the mutagenicity of ethanol.
- Ten years later, Michaelis discussed the role of interactions between the hypoxic conditions generated by ethanol in the fetus and the mechanisms activated by neurotransmitters in the production of cell damage in developing neurons, as well as abnormalities in calcium-manipulation mechanisms and their effects on migration and neuronal differentiation [25]. It is important to note that modifications in calcium signaling have been related to cell death and apoptosis [35,64,65].
- The latest hypotheses deal with neural death during forebrain maturation due to the blockade of the NMDA-glutamate receptor and the activation of GABA-A receptors [26]. Excitatory amino acids (glutamate) influence the processes of neuronal differentiation and synaptogenesis because they can modulate the organization of neuronal circuits and can opportunely regulate biochemical events related to the phenomenon of neuronal plasticity. Therefore, it is conceivable that if ethanol exposure reduces glutamatergic transmission at critical stages of development, this may play a key role in determining the neurotoxic effects of alcohol abuse.
4.1. Main Mechanisms of Alcohol Toxicity
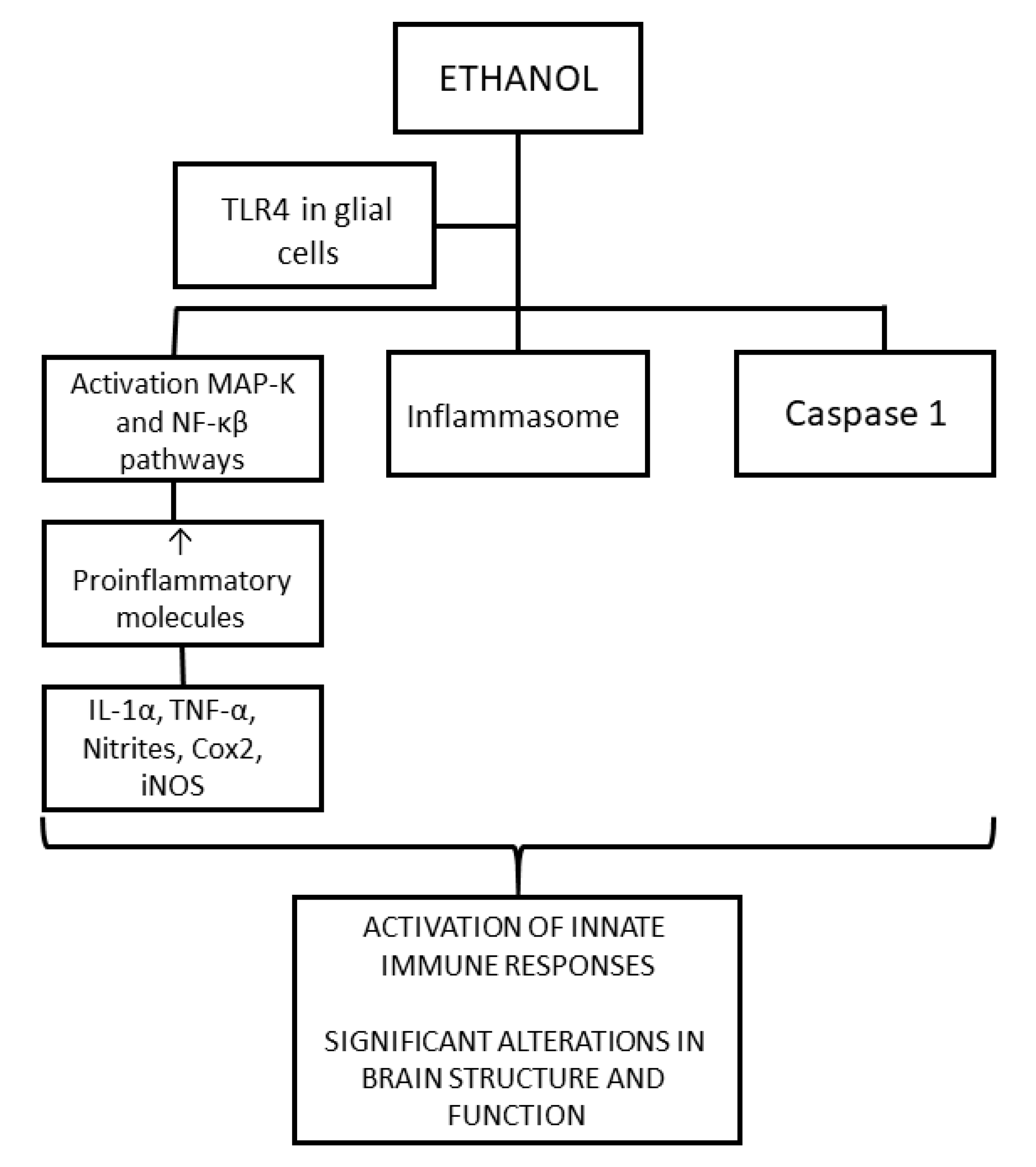
4.1.1. Neuroinflammation
4.1.2. Oxidative Stress
4.1.3. Other Mechanisms of Teratogenesis and Alcohol Toxicity
- Ischemia or hypoxia;
- Dysregulation of mitochondrial bioenergetics [76];
5. Influence of Genetics on the Variability of the Disease
6. Pharmacological Treatments under Study
7. Discussion
- Oxidative stress: Ethanol metabolism produces ROS that cause lipid peroxidation, protein oxidation, and DNA damage. It is well known that these events produce apoptosis and neurodegeneration [35,64]. Within the context of pediatric disorders, such as FASD, OS may have a minor first-order effect [10]. Preclinical research has demonstrated that alcohol consumption during pregnancy impairs the capacity of potassium channels to dilate cerebral arterioles; this disruption appears to be mediated also by elevated levels of OS [139]. Additionally, in a rat model, neonatal ethanol exposure causes deficiencies in context-dependent fear learning and depressive-like behavior, which are linked to higher levels of OS in the hippocampus and prefrontal cortex [140].
- Neuroinflammation: Ethanol activates the innate immune system in the fetal brain by activating TLR4, producing cytokines and proinflammatory molecules. This activation produces gliosis, neuronal inflammation, alteration of myelin, and neuronal damage.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aros, A.S. Exposición fetal a alcohol. Rev. Chil. Ped. 2008, 79 (Suppl. S1), 46–50. [Google Scholar] [CrossRef][Green Version]
- Esteban, Y.A.; Marín, F.A.; Pina, E.J. Trastornos del Desarrollo Asociados Con la Exposición al Alcohol Durante el Embarazo y la Lactancia; Nau Llibres: Valencia, Spain, 2012; p. 7. [Google Scholar]
- Galicia-Moreno, M.; Gutiérrez-Reyes, G. The role of oxidative stress in the development of alcoholic liver disease. Rev. Gastroenterol. México 2014, 79, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed]
- Souli, A.; Sebai, H.; Chehimi, L.; Rtibi, K.; Tounsi, H.; Boubaker, S.; Sakly, M.; El-Benna, J.; Amri, M. Hepatoprotective effect of carob against acute ethanol-induced oxidative stress in rat. Toxicol. Ind. Health 2015, 31, 802–810. [Google Scholar] [CrossRef]
- Lin, H.Z.; Yang, S.Q.; Zeldin, G.; Diehl, A.M. Chronic ethanol consumption induces the production of tumor necrosis factor-alpha and related cytokines in liver and adipose tissue. Alcohol. Clin. Exp. Res. 1998, 22, 231S–237S. [Google Scholar] [CrossRef] [PubMed]
- Brocardo, P.S.; Gil-Mohapel, J.; Christie, B.R. The Role of Oxidative Stress in Fetal Alcohol Spectrum Disorders. Brain Res. Rev. 2011, 67, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Rashba-Step, J.; Cederbaum, A.I. Stable Expression of Human CYP 2E1 in HepG2 Cells: Characterization of Catalytic Activities and Production of Reactive Oxygen Intermediates. Biochemistry 1993, 32, 6928–6937. [Google Scholar] [CrossRef]
- Rashba-Step, J.; Turro, N.J.; Cederbaum, A.I. Increased NADPH- and NADH-Dependent Production of Superoxide and Hydroxyl Radical after Chronic Ethanol Treatment. Arch. Biochem. Biophys. 1993, 300, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Micangeli, G.; Menghi, M.; Profeta, G.; Tarani, F.; Mariani, A.; Petrella, C.; Barbato, C.; Ferraguti, G.; Ceccanti, M.; Tarani, L.; et al. The Impact of Oxidative Stress on Pediatrics Syndromes. Antioxidants 2022, 11, 1983. [Google Scholar] [CrossRef]
- Lemoine, P.; Haroosseau, H.; Borteryu, J.P.; Menuet, J.C. Les Enfants de Parents Alcooliques. Anomalies Observee a Propos de 127 cas [The Children of Alcoholic Parents: Anomalies Observed in 127 Cases]. Quest Medicale 1968, 21, 476–482. (In French) [Google Scholar]
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 302, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Pérez López, J.A. Tabaco, alcohol y embarazo en Atención Primaria. Med. Integr. 2000, 36, 343–354. [Google Scholar]
- Popova, S.; Lange, S.; Probst, C.; Gmel, G.; Rhem, J. Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Gosdin, L.K.; Deputy, N.P.; Kim, S.Y.; Dang, E.P.; Denny, C.H. Alcohol consumption and binge drinking during pregnancy among adults aged 18–49 years—United States, 2018–2020. Morb. Mortal. Wkly Rep. 2022, 71, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Cebral, E. Implicación del consumo materno de alcohol en el desarrollo embriofetal. Rev. Soc. Arg. Endocrinol. Ginecol. Reprod. 2006, 13, 21–33. [Google Scholar]
- Fuentes Soliz, J.A.; Vidal Lia, O.V.; Fuentes Moya, J.M.; López, E. Consumo de Alcohol durante el Embarazo: Múltiples Efectos Negativos en el Feto. Rev. Cient. Cienc. Méd. 2009, 12, 26–31. [Google Scholar]
- Joya, X.; Garcia-Algar, O.; Salat-Batlle, J.; Pujades, C.; Vall, O. Advances in the development of novel antioxidant therapies as an approach for fetal alcohol syndrome prevention: Antioxidant Therapies for FAS Prevention. Birth Defects Res. A Clin. Mol. Teratol. 2015, 103, 163–177. [Google Scholar] [CrossRef]
- Carito, V.; Ceccanti, M.; Ferraguti, G.; Coccurello, R.; Ciafrè, S.; Tirassa, P.; Fiore, M. NGF and BDNF Alterations by Prenatal Alcohol Exposure. Curr. Neuropharmacol. 2019, 17, 308–317. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Baete, A.; Russo, J.; Elliott, A.J.; Blankenship, J.; Kalberg, W.O.; Buckley, D.; Brooks, M.; Hasken, J.; Abdul-Rahman, O.; et al. Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics 2014, 134, 855–866. [Google Scholar] [CrossRef]
- Chudley, A.E.; Conry, J.; Cook, J.L.; Loock, C.; Rosales, T.; LeBlanc, N. Fetal alcohol spectrum disorder: Canadian guidelines for diagnosis. CMAJ 2005, 172, S1–S21. [Google Scholar] [CrossRef]
- Wozniak, J.R.; Riley, E.P.; Charness, M.E. Clinical presentation, diagnosis, and management of fetal alcohol spectrum disorder. Lancet Neurol. 2019, 18, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Denny, L.; Coles, S.; Blitz, R. Fetal Alcohol Syndrome and Fetal Alcohol Spectrum Disorders. Am. Fam. Physician 2017, 96, 515–522. [Google Scholar] [PubMed]
- Henderson, G.I.; Patwardhan, R.V.; Hoyumpa, A.M.; Schenker, S., Jr. Fetal alcohol syndrome: Overview of pathogenesis. Neurobehav. Toxicol. Teratol. 1981, 3, 73–80. [Google Scholar] [PubMed]
- Anderson, P. The impact of alcoholic beverages on human health. Nutrients 2021, 13, 4417. [Google Scholar] [CrossRef] [PubMed]
- Gaviria, M.M.; Correa Arango, G.; Navas, M.C. Alcohol, cirrosis y predisposición genética. Rev. Col. Gastroenterol. 2016, 31, 27. [Google Scholar] [CrossRef]
- Dorta, D.J. Toxicologia Forense; Edgard Blücher Ltd.: Sao Paulo, Brazil, 2018; p. 751. [Google Scholar]
- Bhatia, S.; Drake, D.M.; Miller, L.; Wells, P.G. Oxidative stress and DNA damage in the mechanism of fetal alcohol spectrum disorders. Birth Defects Res. 2019, 111, 714–748. [Google Scholar] [CrossRef] [PubMed]
- Chater-Diehl, E.J.; Laufer, B.I.; Castellani, C.A.; Alberry, B.L.; Singh, S.M. Alteration of gene expression, DNA methylation, and histone methylation in free radical scavenging networks in adult mouse hippocampus following fetal alcohol exposure. PLoS ONE 2016, 11, e0154836. [Google Scholar] [CrossRef]
- Khalid, O.; Kim, J.J.; Kim, H.S.; Hoang, M.; Tu, T.G.; Elie, O.; Lee, C.; Vu, C.; Horvath, S.; Spigelman, I.; et al. Gene expression signatures affected by alcohol-induced DNA methylomic deregulation in human embryonic stem cells. Stem Cell Res. 2014, 12, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.L.; Cox, M.M. Princípios de bioquímica de Lehninger, 7th ed.; Artmed: Porto Alegre, Brazil, 2017; p. 1227. [Google Scholar]
- Sid, B.; Verrax, J.; Calderon, P.B. Role of oxidative stress in the pathogenesis of alcohol-induced liver disease. Free Radic. Res. 2013, 47, 894–904. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Goldberg, E.M.; Aliani, M. Metabolomics and fetal alcohol spectrum disorder. Biochem. Cell Biol. 2017, 96, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, I.; Espino, J.; González-Flores, D.; Casado, J.G.; Redondo, P.C.; Rosado, J.A.; Barriga, C.; Pariente, J.A.; Rodríguez, A.B. Role of Calcium Signals on Hydrogen Peroxide-Induced Apoptosis in Human Myeloid HL-60 Cells. Int. J. Biomed. Sci. 2009, 5, 246–256. [Google Scholar] [PubMed]
- Gupta, K.K.; Gupta, V.K.; Shirasaka, T. An Update on Fetal Alcohol Syndrome-Pathogenesis, Risks, and Treatment. Alcohol. Clin. Exp. Res. 2016, 40, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Szwajgier, D.; Baranowska-Wójcik, E.; Grzelczyk, J.; Zukiewicz-Sobczak, W. Peripheral Oxidation Markers in Down Syndrome Patients: The Better and the Worse. Dis. Markers 2021, 2021, 5581139. [Google Scholar] [CrossRef] [PubMed]
- Davis-Anderson, K.L.; Wesseling, H.; Siebert, L.M.; Lunde-Young, E.R.; Naik, V.D.; Steen, H.; Ramadoss, J. Fetal regional brain protein signature in FASD rat model. Reprod. Toxicol. 2018, 76, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Memo, L.; Gnoato, E.; Caminiti, S.; Pichini, S.; Tarani, L. Fetal alcohol spectrum disorders and fetal alcohol syndrome: The state of the art and new diagnostic tools. Early Hum. Dev. 2013, 89, S40–S43. [Google Scholar] [CrossRef] [PubMed]
- Brzezinski, M.R.; Boutelet-Bochan, H.; Person, R.E.; Fantel, A.G.; Juchau, M.R. Catalytic activity and quantitation of cytochrome P-450 2E1 in prenatal human brain. J. Pharmacol. Exp. Ther. 1999, 289, 1648–1653. [Google Scholar] [PubMed]
- Mattson, S.N.; Crocker, N.; Nguyen, T.T. Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychol. Rev. 2011, 21, 81–101. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Horn, K.H.; Zhou, F.C. Alcohol teratogenesis: Mechanisms of damage and strategies for intervention. Exp. Biol. Med. 2005, 230, 394–406. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, S14–S22. [Google Scholar] [CrossRef]
- Pascale, A. Consumo de drogas durante el embarazo. Efectos sobre el binomio materno-fetal, recién nacido y primera infancia. In Modalidades Terapéuticas y Estrategias de Prevención; Mujer y Salud en Uruguay: Salto, Uruguay, 2017. [Google Scholar]
- Puig, M.; Arce, A.; De Juana, R.; Leandro, S.V.; Villa-Elízaga, I. Síndrome de alcohol fetal. Rev. Med. Univ. Navar. 1979, 23, 34–44. [Google Scholar] [CrossRef]
- Moraes, L.; Dries, S.S.; Seibert, B.S.; Linden, R.; Perassolo, M.S. Evaluation of oxidative stress markers in ethanol users. Braz. J. Med. Biol. Res. 2023, 56, e12465. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism and signaling mechanisms of malondialdehyde and 4-hidroxy-2-nonenal. Oxidative Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Kattimani, S.; Sridhar, M.G. Oxidative stress during alcohol withdrawal and its relationship with withdrawal severity. Indian J. Psychol. Med. 2015, 37, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Mutlu-Türkoğlu, Ü.; Doğru-Abbasoğlu, S.; Aykac¸-Toker, G.; Mirsal, H.; Beyazyürek, M.; Uysal, M. Increased lipid and protein oxidation and DNA damage in patients with chronic alcoholism. J. Lab. Clin. Med. 2000, 136, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Maturu, P.; Reddy, V.D.; Padmavathi, P.; Varadacharyulu, N. Ethanol induced adaptive changes in blood for the pathological and toxicological effects of chronic ethanol consumption in humans. Exp. Toxicol. Pathol. 2012, 64, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.C.; Tang, S.H.; Huang, M.C.; Chen, C.C.; Kuo, T.L.; Yin, S.J. Oxidative status in patients with alcohol dependence: A clinical study in Taiwan. J. Toxicol. Environ. Health 2005, 68, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.C.; Chen, C.H.; Peng, F.C.; Tang, S.H.; Chen, C.C. Alterations in oxidative stress status during early alcohol withdrawal in alcoholic patients. J. Formos. Med. Assoc. 2009, 108, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Saribal, D.; Hocaoglu-Emre, F.S.; Karaman, F.; Mırsal, H.; Akyolcu, M.C. Trace element levels and oxidant/antioxidant status in patients with alcohol abuse. Biol. Trace Elem. Res. 2020, 193, 7–13. [Google Scholar] [CrossRef]
- Woźniak, B.; Musiałkiewicz, D.; Woźniak, A.; Drewa, G.; Drewa, T.; Drewa, S.; Mila-Kierzenkowska, C.; Porzych, M.; Musiałkiewicz, M. Lack of changes in the concentration of thiobarbituric acid-reactive substances (TBARS) and in the activities of erythrocyte antioxidant enzymes in alcoholdependent patients after detoxification. Med. Sci. Monit. 2008, 14, CR32–CR36. [Google Scholar]
- Rua, R.M.; Ojeda, M.L.; Nogales, F.; Rubio, J.M.; Romero-Gómez, M.; Funuyet, J.; Murillo, M.L.; Carreras, O. Serum selenium levels and oxidative balance as differential markers in hepatic damage caused by alcohol. Life Sci. 2014, 94, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Akkus, I.; Gültekin, F.; Aköz, M.; Cağlayan, O.; Bahcaci, S.; Can, U.G.; Ay, M.; Gürel, A. Effect of moderate alcohol intake on lipid peroxidation in plasma, erythrocyte and leukocyte and on some antioxidant enzymes. Clin. Chim. Acta 1997, 266, 141–147. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, L.J.; Bair, M.J.; Yao, M.L.; Peng, H.C.; Yang, S.S.; Yang, S.-C. Antioxidative status of patients with alcoholic liver disease in southeastern Taiwan. World J. Gastroenterol. 2011, 17, 1063–1070. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chen, C.Y.; Huang, T.L.; Tsai, M.C. Brain-derived neurotrophic factor and glutathione peroxidase as state biomarkers in alcohol use disorder patients undergoing detoxification. Medicine 2020, 99, e19938. [Google Scholar] [CrossRef]
- Dries, S.S.; Seibert, B.S.; Bastiani, M.F.; Linden, R.; Perassolo, M.S. Evaluation of oxidative stress biomarkers and liver and renal functional parameters in patients during treatment a mental health unit to treat alcohol dependence. Drug Chem. Toxicol. 2022, 45, 861–867. [Google Scholar] [CrossRef]
- Barbosa, K.B.F.; Costa, N.M.B.; Alfenas, R.D.C.G.; De Paula, S.O.; Minim, V.P.R.; Bressan, J. Oxidative stress: Concept, implications and modulating factors. Rev. Nutr. 2010, 23, 629–643. [Google Scholar] [CrossRef]
- Guemouri, L.; Lecomte, E.; Herbeth, B.; Pirollet, P.; Paille, F.; Siest, G.; Artur, Y. Blood activities of antioxidant enzymes in alcoholics before and after withdrawal. J. Stud. Alcohol. 1993, 54, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Bogdanska, J.; Todorova, B.; Labudovic, D.; Korneti, P.G. Eritrocyte antioxidante enzymes in patients with alcohol dependence syndrome. Bratisl. Lek. Listy 2005, 106, 107–113. [Google Scholar]
- Bjørneboe, G.A.; Johnsen, J.; Bjørneboe, A.; Marklund, S.L.; Skylv, N.; Høiseth, A.; Bache-Wiig, J.; Mørland, J.; Drevon, C.A. Some aspects in blood of antioxidant status from alcoholics. Alcohol. Clin. Exp. Res. 1988, 12, 806–810. [Google Scholar] [CrossRef]
- González, D.; Espino, J.; Bejarano, I.; López, J.J.; Rodríguez, A.B.; Pariente, J.A. Caspase-3 and -9 are activated in human myeloid HL-60 cells by calcium signal. Mol. Cell Biochem. 2010, 333, 151–157. [Google Scholar] [CrossRef]
- González-Flores, D.; De Nicola, M.; Bruni, E.; Caputo, F.; Rodríguez, A.B.; Pariente, J.A.; Ghibelli, L. Nanoceria protects from alterations in oxidative metabolism and calcium overloads induced by TNFα and cycloheximide in U937 cells: Pharmacological potential of nanoparticles. Mol. Cell Biochem. 2014, 397, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Nutt, D.; Hayes, A.; Fonville, L.; Zafar, R.; Palmer, E.O.C.; Paterson, L.; Lingford-Hughes, A. Alcohol and the brain. Nutrients 2021, 13, 3938. [Google Scholar] [CrossRef]
- Lebel, C.; Roussotte, F.; Sowell, E.R. Imaging the impact of prenatal alcohol exposure on the structure of the developing human brain. Neuropsychol. Rev. 2011, 21, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Cigana Schenkel, L.; Huang, L.; Smith, A.; Pare, G.; Ainsworth, P.; Boycott, K.M.; Warman-Chardon, J.; Sadikovic, B. Identification of a methylation profile for DNMT1-associated autosomal dominant cerebellar ataxia, deafness, and narcolepsy. Clin. Epigenetics 2016, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.A.; Bourque, P.; Smith, A.M.; Warman Chardon, J. Autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCA-DN) associated with progressive cognitive and behavioral deterioration. Neuropsychol. 2017, 31, 292–303. [Google Scholar] [CrossRef]
- Selzer, M.; Darbinian, N. Oligodendrocyte pathology in fetal alcohol spectrum disorders. Neural Regen. Res. 2022, 17, 497. [Google Scholar] [CrossRef] [PubMed]
- Abbott, C.W.; Kozanian, O.O.; Kanaan, J.; Wendel, K.M.; Huffman, K.J. The impact of prenatal ethanol exposure on neuroanatomical and behavioural development in mice. Alcohol. Clin. Exp. Res. 2016, 40, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Pascual, M.; Montesinos, J.; Montagud-Romero, S.; Forteza, J.; Rodríguez-Arias, M.; Miñarro, J.; Guerri, C. TLR4 response mediates ethanol-induced neurodevelopment alterations in a model of fetal alcohol spectrum disorders. J. Neuroinflammation 2017, 14, 145. [Google Scholar] [CrossRef]
- González-Flores, D.; Rodríguez, A.B.; Pariente, J.A. TNFα-induced apoptosis in human myeloid cell lines HL-60 and K562 is dependent of intracellular ROS generation. Mol. Cell Biochem. 2014, 390, 281–287. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Hörster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Maya-Enero, S.; Ramis-Fernández, S.M.; Astals-Vizcaino, M.; García-Algar, Ó. Neurocognitive and behavioral profile of fetal alcohol spectrum disorder. An. Pediatr. 2021, 95, 208.e1–208.e9. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, F.; Roozen, S.; Verbeek, J.; Koek, G.; Kok, G.; van Kranen, H.; Evelo, C.T.; Curfs, L.M.G. Review and gap analysis: Molecular pathways leading to fetal alcohol spectrum disorders. Mol. Psychiatry 2019, 24, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Sulik, K.K.; Chen, S.-Y. The role of NOX enzymes in ethanol-induced oxidative stress and apoptosis in mouse embryos. Toxicol. Lett. 2010, 193, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Shukuri, M.; Uchino, M.; Sakamaki, T.; Onoe, S.; Hosoi, R.; Todoroki, K.; Arano, Y.; Sakai, T.; Akizawa, H.; Inoue, O. Ex vivo imaging and analysis of ROS generation correlated with microglial activation in rat model with acute neuroinflammation induced by intrastriatal injection of LPS. Biochem. Biophys, Res. Commun. 2021, 584, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yao, X.; Jiang, W.; Li, W.; Zhu, S.; Liao, C.; Zou, L.; Ding, R.; Chen, J. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J. Neuroinflammation 2020, 17, 90. [Google Scholar] [CrossRef]
- Ramos Quiroga, J.A.; Sánchez Mora, C.; Corominas Roso, M.; Martínez, I.; Barrau Alonso, V.; Prats Torres, L.; Brugué, M.C.; Haro, M.R. Factores neurotróficos y su trascendencia en el trastorno por déficit de atención/hiperactividad. Rev. Neurol. 2014, 58 (Suppl. S1), S19–S24. [Google Scholar] [CrossRef] [PubMed]
- González-Flores, D.; Grilo, A.A.; Rodríguez, A.B.; Franco, L. Consequences of enriched diet of glucose on oncologic patients. Appl. Sci. 2023, 13, 2757. [Google Scholar] [CrossRef]
- Hicks, S.D.; Middleton, F.A.; Miller, M.W. Ethanol-induced methylation of cell cycle genes in neural stem cells: Ethanol affects gene expression in neural stem cells. J. Neurochem. 2010, 114, 1767–1780. [Google Scholar] [CrossRef]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; A Gifford, C.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B.S. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3A, and DNA methylation. J. Neurochem. 2015, 132, 429–442. [Google Scholar] [CrossRef]
- Lussier, A.A.; Weinberg, J.; Kobor, M.S. Epigenetics studies of fetal alcohol spectrum disorder: Where are we now? Epigenomics 2017, 9, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Sundermann, A.C.; Zhao, S.; Young, C.L.; Lam, L.; Jones, S.H.; Velez Edwards, D.R.; Hartmann, K.E. Alcohol use in pregnancy and miscarriage: A systematic review and meta-analysis. Alcohol. Clin. Exp. Res. 2019, 43, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Lovely, C.B. Animal models of gene-alcohol interactions. Birth Defects Res. 2020, 112, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Sambo, D.; Goldman, D. Genetic Influences on Fetal Alcohol Spectrum Disorder. Genes 2023, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Popova, S.; Lange, S.; Shield, K.; Mihic, A.; Chudley, A.E.; Mukherjee, R.A.S.; Bekmuradov, D.; Rehm, J. Comorbidity of fetal alcohol spectrum disorder: A systematic review and meta-analysis. Lancet 2016, 387, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Bingol, N.; Fuchs, M.; Iosub, S.; Kumar, S.; Stone, R.K.; Gromisch, D.S. Fetal alcohol syndrome associated with trisomy 21. Alcohol. Clin. Exp. Res. 1987, 11, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Romke, C.; Heyne, K.; Stewens, J.; Schwinger, E. Erroneous diagnosis of fetal alcohol syndrome in a patient with ring chromosome 6. Eur. J. Pediatr. 1987, 146, 443. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Cobet, G.; Laske, G.; Degen, B.; Grauel, C.; Lehmann, K. Partial monosomy 21 or fetal alcohol embryopathy in a retarded boy? Padiatr. Grenzgeb. 1993, 31, 313–319. [Google Scholar] [PubMed]
- Weyrauch, D.; Schwartz, M.; Hart, B.; Klug, M.G.; Burd, L. Comorbid Mental Disorders in Fetal Alcohol Spectrum Disorders: A Systematic Review. J. Dev. Behav. Pediatr. 2017, 38, 283–291. [Google Scholar] [CrossRef]
- Gavazzi, G.; Faury, G. NOX- and ROS-Driven Hypertension in Elastin Insufficiency. Function 2021, 2, zqab035. [Google Scholar] [CrossRef]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target. 2015, 23, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, W.H.; Nunes, A.K.; Franca, M.E.; Santos, L.A.; Los, D.B.; Rocha, S.W.; Barbosa, K.P.; Rodrigues, G.B.; Peixoto, C.A. Effects of metformin on inflammation and short-term memory in streptozotocin-induced diabetic mice. Brain Res. 2016, 1644, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gallagher, D.; DeVito, L.M.; Cancino, G.I.; Tsui, D.; He, L.; Keller, G.M.; Frankland, P.W.; Kaplan, D.R.; Miller, F.D. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem. Cell 2012, 11, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Ullah, N.; Naseer, M.I.; Lee, H.Y.; Kim, M.O. Neuroprotection with metformin and thymoquinone against ethanol-induced apoptotic neurodegeneration in prenatal rat cortical neurons. BMC Neurosci. 2012, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Roesler, E.; Niehof, M.L.; Roby, D.A.; Mckee, A.; Morley, J.E. Metformin improves learning and memory in the SAMP8 mouse model of Alzheimer’s disease. J Alzheimers Dis. 2019, 68, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Jain, P.; Ghumatkar, P.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Algire, C.; Moiseeva, O.; Deschênes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.E.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; Van Der Valk, P.; Amor, S. Infammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.-M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-infammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.; Selvakumar, G.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.; Zaheer, A. Neuroinfammation induces neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Sabzali, M.; Eidi, A.; Khaksari, M.; Khastar, H. Anti-inflammatory, Antioxidant, and Antiapoptotic Action of Metformin Attenuates Ethanol Neurotoxicity in the Animal Model of Fetal Alcohol Spectrum Disorders. Neurotox. Res. 2022, 40, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Kadiyala, R.; Peter, R.; Okosieme, O.E. Thyroid dysfunction in patients with diabetes: Clinical implications and screening strategies. Int. J. Clin. Pract. 2010, 64, 1130–1139. [Google Scholar] [CrossRef]
- Harper, K.M.; Tunc-Ozcan, E.; Graf, E.N.; Redei, E.E. Intergenerational effects of prenatal ethanol on glucose tolerance and insulin response. Physiol. Genom. 2014, 46, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, Z.; Gong, J.; Zhang, L.; Wang, L.; Magdalou, J.; Chen, L.; Wang, H. Prenatal ethanol exposure programs an increased susceptibility of non-alcoholic fatty liver disease in female adult offspring rats. Toxicol. Appl. Pharmacol. 2014, 274, 263–273. [Google Scholar] [CrossRef] [PubMed]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradi, A.; Radyushkin, K.; Rossner, M.J. Enhanced memory consolidation in mice lacking the circadian modulators Sharp1 and -2 caused by elevated Igf2 signaling in the cortex. Proc. Natl. Acad. Sci. USA 2015, 112, E3582–E3589. [Google Scholar] [CrossRef]
- Schmeisser, M.J.; Baumann, B.; Johannsen, S.; Vindedal, G.F.; Jensen, V.; Hvalby, O.C.; Sprengel, R.; Seither, J.; Maqbool, A.; Magnutzki, A.; et al. IkappaB kinase/nuclear factor kappaB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J. Neurosci. 2012, 32, 5688–5703. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Role of central nervous system insulin resistance in fetal alcohol spectrum disorders. J. Popul. Ther. Clin. Pharmacol. 2010, 17, e390–e404. [Google Scholar] [PubMed]
- Haycock, P.C.; Ramsay, M. Exposure of mouse embryos to ethanol during preimplantation development: Effect on DNA methylation in the h19 imprinting control region. Biol. Reprod. 2009, 81, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Johnson, T.E.; Larson, C.; Leakey, T.I.; Siegfried, R.N.; Rafferty, T.M.; Cooney, C.A. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: Effects of a methyl-supplemented diet. Alcohol 2011, 45, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Stern, S.A.; Garcia-Osta, A.; Saunier-Rebori, B.; Pollonini, G.; Bambah-Mukku, D.; Blitzer, R.D.; Alberini, C.M. A critical role for IGF-II in memory consolidation and enhancement. Nature 2011, 469, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C.; Borai, A. Insulin-like growth factor-II: Its role in metabolic and endocrine disease. Clin. Endocrinol. 2014, 80, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Tunc-Ozcan, E.; Wert, S.L.; Lim, P.H.; Ferreira, A.; Redei, E.E. Hippocampus-dependent memory and allele-specific gene expression in adult offspring of alcohol-consuming dams after neonatal treatment with thyroxin or metformin. Mol. Psychiatry 2017, 23, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Constancia, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.E.; Lin, S.P.; Ito, M.; Takagi, N.; Takada, S.; Ferguson-Smith, A.C. Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr. Biol. 2002, 12, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Sittig, L.J.; Shukla, P.K.; Herzing, L.B.; Redei, E.E. Strain-specific vulnerability to alcohol exposure in utero via hippocampal parent-of-origin expression of deiodinase-III. FASEB J. 2011, 25, 2313–2324. [Google Scholar] [CrossRef]
- Harper, K.M.; Tunc-Ozcan, E.; Graf, E.N.; Herzing, L.B.; Redei, E.E. Intergenerational and parent of origin effects of maternal calorie restriction on Igf2 expression in the adult rat hippocampus. Psychoneuroendocrinology 2014, 45, 187–191. [Google Scholar] [CrossRef][Green Version]
- Edwards, C.A.; Ferguson-Smith, A.C. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 2007, 19, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Garro, A.J.; McBeth, D.L.; Lima, V.; Lieber, C.S. Ethanol consumption inhibits fetal DNA methylation in mice: Implications for the fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1991, 15, 395–398. [Google Scholar] [CrossRef]
- Haycock, P.C. Fetal alcohol spectrum disorders: The epigenetic perspective. Biol. Reprod. 2009, 81, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 2013, 37, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Tunc-Ozcan, E.; Harper, K.M.; Graf, E.N.; Redei, E.E. Thyroxine administration prevents matrilineal intergenerational consequences of in utero ethanol exposure in rats. Horm. Behav. 2016, 82, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ouko, L.A.; Shantikumar, K.; Knezovich, J.; Haycock, P.; Schnugh, D.J.; Ramsay, M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: Implications for fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2009, 33, 1615–1627. [Google Scholar] [CrossRef]
- Wylie, A.A.; Murphy, S.K.; Orton, T.C.; Jirtle, R.L. Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res. 2000, 10, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Ethen, M.K.; Ramadhani, T.A.; Scheuerle, A.E.; Canfield, M.A.; Wyszynski, D.F.; Druschel, C.M.; Romitti, P.A. Alcohol consumption by women before and during pregnancy. Matern. Child Health J. 2009, 13, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.X.; Jonsson, E.; Salmon, A.; Sebastianski, M. Incidence and prevalence of fetal alcohol spectrum disorder by sex and age group in Alberta, Canada. J. Popul. Ther. Clin. Pharmacol. 2014, 21, e395–e404. [Google Scholar]
- Varlinskaya, E.I.; Mooney, S.M. Acute exposure to ethanol on gestational day 15 affects social motivation of female offspring. Behav. Brain Res. 2014, 261, 106–109. [Google Scholar] [CrossRef][Green Version]
- Hellemans, K.G.; Verma, P.; Yoon, E.; Yu, W.K.; Young, A.H.; Weinberg, J. Prenatal alcohol exposure and chronic mild stress differentially alter depressive- and anxiety-like behaviors in male and female offspring. Alcohol. Clin. Exp. Res. 2010, 34, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Rehman, R.; Abidi, S.H.; Alam, F. Metformin, Oxidative Stress, and Infertility: A Way Forward. Front. Physiol. 2018, 9, 1722. [Google Scholar] [CrossRef]
- Mobasher, M.A.; El-Tantawi, H.G.; El-Said, K.S. Metformin Ameliorates Oxidative Stress Induced by Diabetes Mellitus and Hepatocellular Carcinoma in Rats. Rep. Biochem. Mol. Biol. 2020, 9, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Bestry, M.; Larcombe, A.N.; Kresoje, N.; Chivers, E.K.; Bakker, C.; Fitzpatrick, J.P.; Elliott, E.J.; Craig, J.M.; Muggli, E.; Halliday, J.; et al. The influence of prenatal alcohol exposure and maternal diet on offspring DNA methylation: A cross-species study. bioRxiv 2023, 12, RP92135. [Google Scholar]
- Saha, P.S.; Knecht, T.M.; Arrick, D.M.; Watt, M.J.; Scholl, J.L.; Mayhan, W.G. Prenatal exposure to alcohol impairs responses of cerebral arterioles to activation of potassium channels: Role of oxidative stress. Alcohol. Clin. Exp. Res. 2023, 47, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Lopatynska-mazurek, M.; Komsta, L.; Gibula-tarlowska, E.; Kotlinska, J.H. Aversive learning deficits and depressive-like behaviors are accompanied by an increase in oxidative stress in a rat model of fetal alcohol spectrum disorders: The protective effect of rapamycin. Int. J. Mol. Sci. 2021, 22, 7083. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Guo, L.; Davis, M.A.; Lambert, J.C.; Beier, J.I.; Duveau, I.; Luyendyk, J.P.; Roth, R.A.; Arteel, G.E. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterol. 2006, 130, 2099–2112. [Google Scholar] [CrossRef]
- García-Algar, O.; Astals Vizcaino, M.; González Cochón, P.; Andreu Fernández, V. Informe Sobre Alcohol, Embarazo Y Trastorno del Espectro Alcohólico Fetal (TEAF). 2021. Available online: https://www.sanidad.gob.es/areas/promocionPrevencion/alcohol/embarazo/docs/Informe_AlcoholEmbarazo_TEAF.pdf (accessed on 2 March 2024).
- Adebiyi, B.O.; Mukumbang, F.C.; Beytell, A.-M. A guideline for the prevention and management of Fetal Alcohol Spectrum Disorder in South Africa. BMC Health Serv. Res. 2019, 19, 809. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Enzyme | Control | Alcohol | p |
|---|---|---|---|
| CAT | 1.67 | 0.95 | - |
| FRAP | 1143.14 | 1344 | - |
| GPx | 23.07 | 6.9 | - |
| MDA | 1.25 | 1.47 | * |
| SOD | 93.9 | 433 | * |
| Enzyme | Low | High | p |
|---|---|---|---|
| CAT | 0.56 | 1.12 | * |
| FRAP | 1515 | 1292 | - |
| GPx | 12.34 | 4.07 | - |
| MDA | 1.81 | 1.37 | - |
| SOD | 533 | 417 | - |
| Enzyme | Spearman’s Correlation | p |
|---|---|---|
| CAT | 0.167 | - |
| FRAP | −0.299 | * |
| GPx | −0.188 | - |
| MDA | 0.065 | - |
| SOD | 0.033 | - |
| Treatment | SOD | GPx | MDA | TNFα |
|---|---|---|---|---|
| Milk + Saline | - | - | - | - |
| Ethanol | ↓↓ | ↓↓↓ | ↑↑↑ | ↑↑↑ |
| Ethanol + Met20 | ↓↓ | ↓↓ | ↑↑↑ | ↑↑↑ |
| Ethanol + Met40 | ↓ | ↓ | ↑ | ↑ |
| Treatment | Ctcf | Dio3 | Dnmt1 | Igf2 |
|---|---|---|---|---|
| FAE + metformin | ↓ | ↓ | ↑ | ↑ |
| FAE + T4 | - | ↓ | ↑ | ↑ |
| 5-Aza + metformin | ↓ | ↓ | ↑ | ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Flores, D.; Márquez, A.; Casimiro, I. Oxidative Effects in Early Stages of Embryo Development Due to Alcohol Consumption. Int. J. Mol. Sci. 2024, 25, 4100. https://doi.org/10.3390/ijms25074100
González-Flores D, Márquez A, Casimiro I. Oxidative Effects in Early Stages of Embryo Development Due to Alcohol Consumption. International Journal of Molecular Sciences. 2024; 25(7):4100. https://doi.org/10.3390/ijms25074100
Chicago/Turabian StyleGonzález-Flores, David, Antonia Márquez, and Ilda Casimiro. 2024. "Oxidative Effects in Early Stages of Embryo Development Due to Alcohol Consumption" International Journal of Molecular Sciences 25, no. 7: 4100. https://doi.org/10.3390/ijms25074100
APA StyleGonzález-Flores, D., Márquez, A., & Casimiro, I. (2024). Oxidative Effects in Early Stages of Embryo Development Due to Alcohol Consumption. International Journal of Molecular Sciences, 25(7), 4100. https://doi.org/10.3390/ijms25074100