
Small Non-Coding RNAs and Their Role in Locoregional Metastasis and Outcomes in Early-Stage Breast Cancer Patients

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Patients
2.2. RNA Sequencing
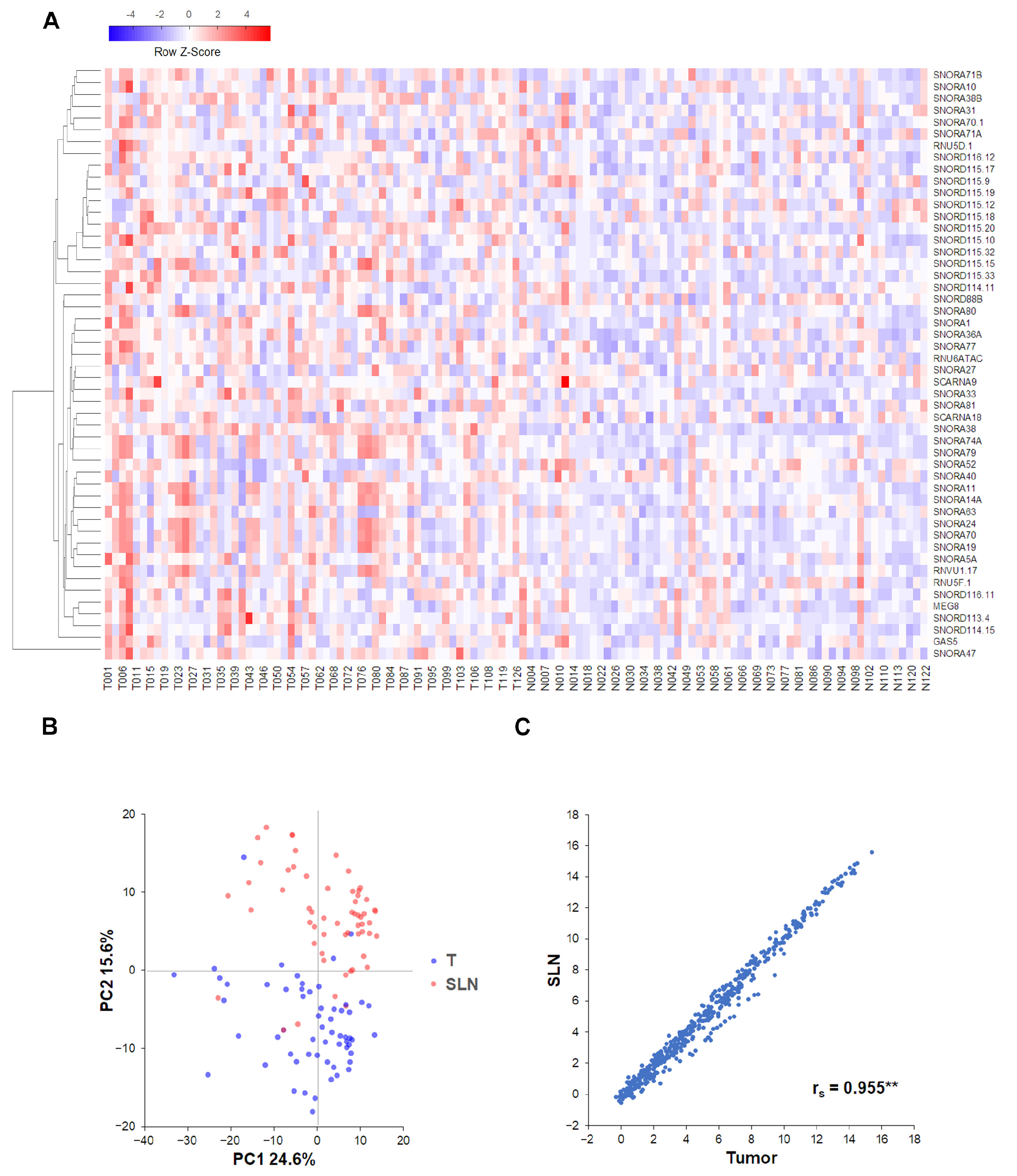
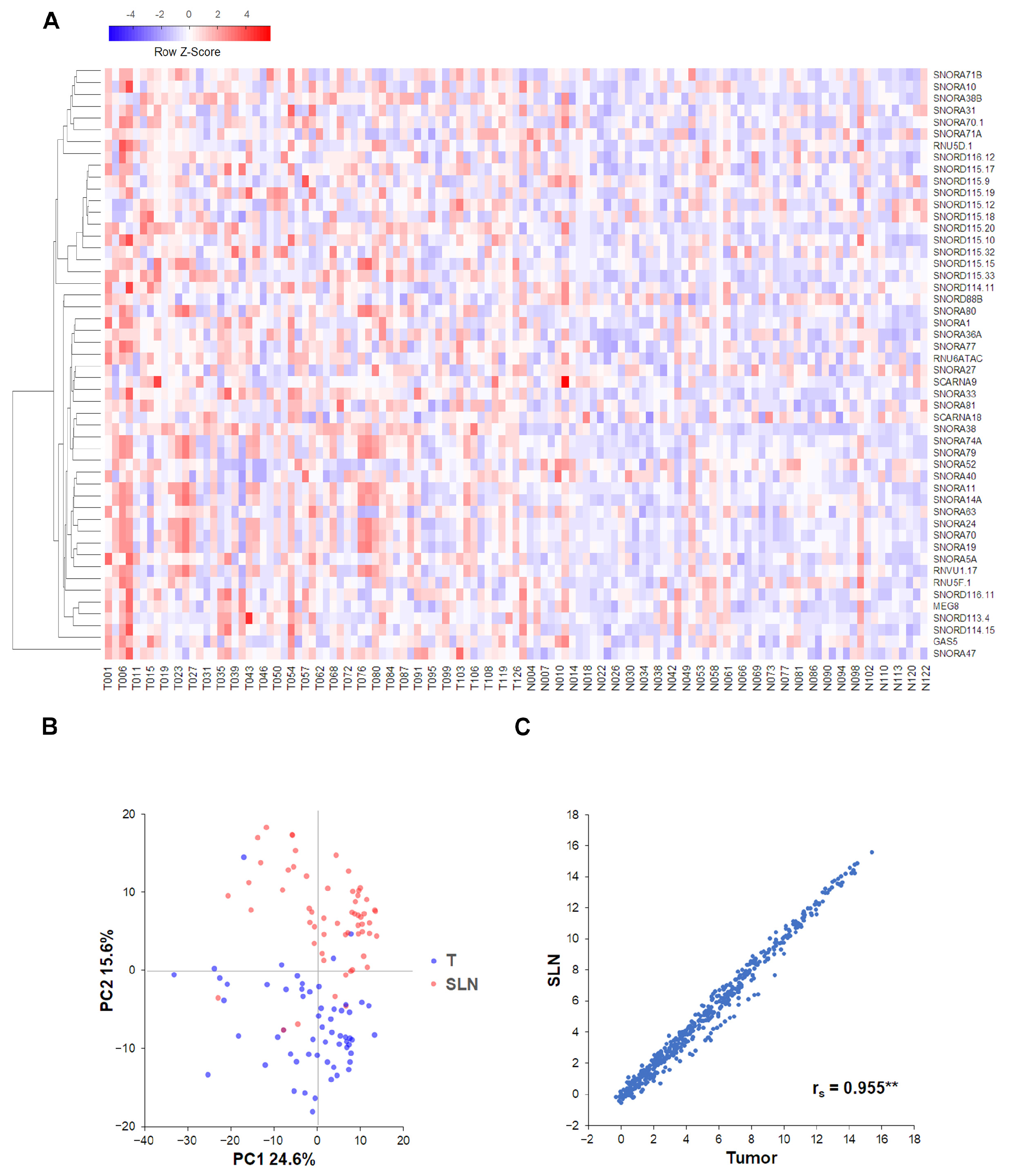
2.3. Correlations and Clustering Analyses
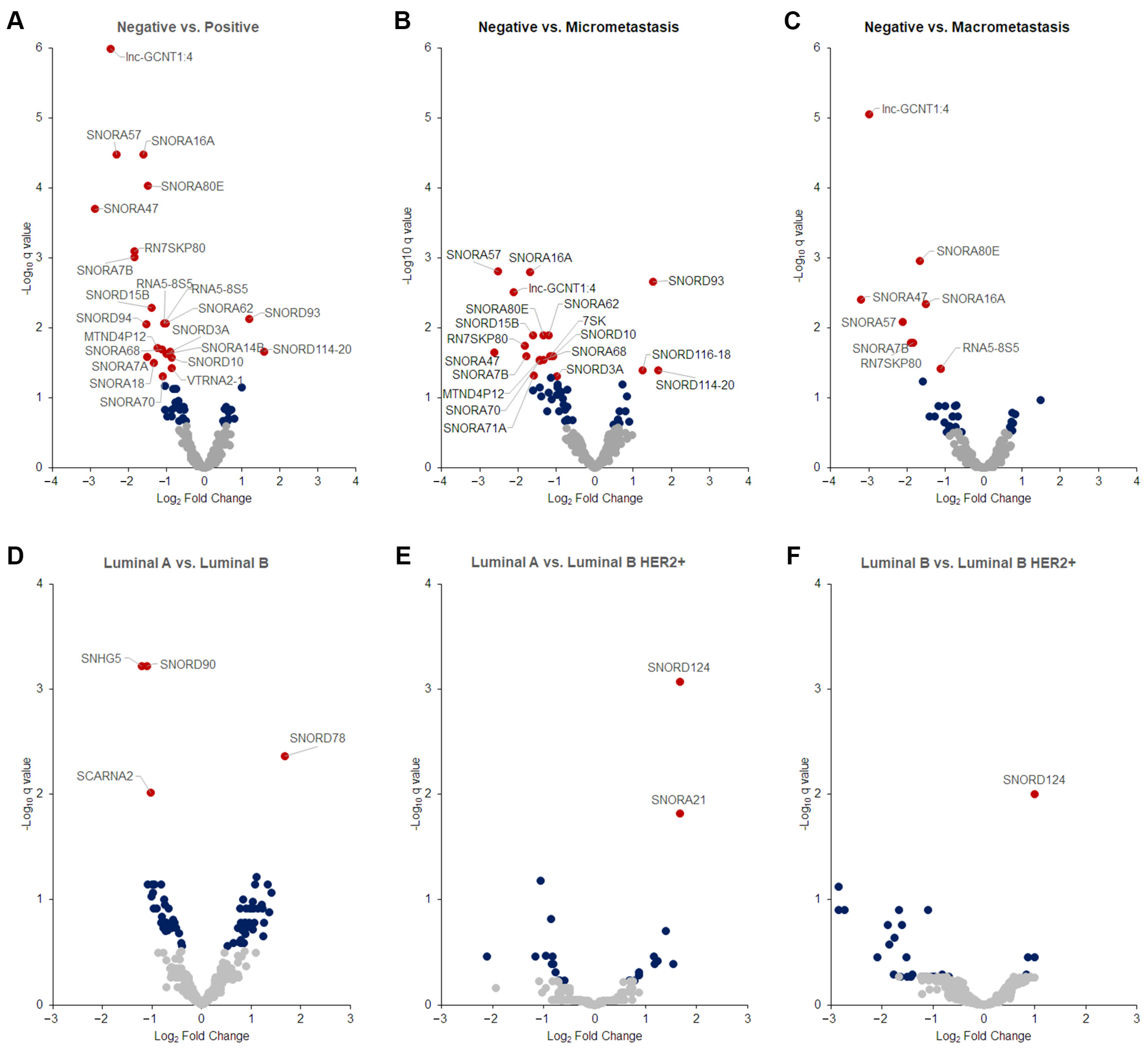
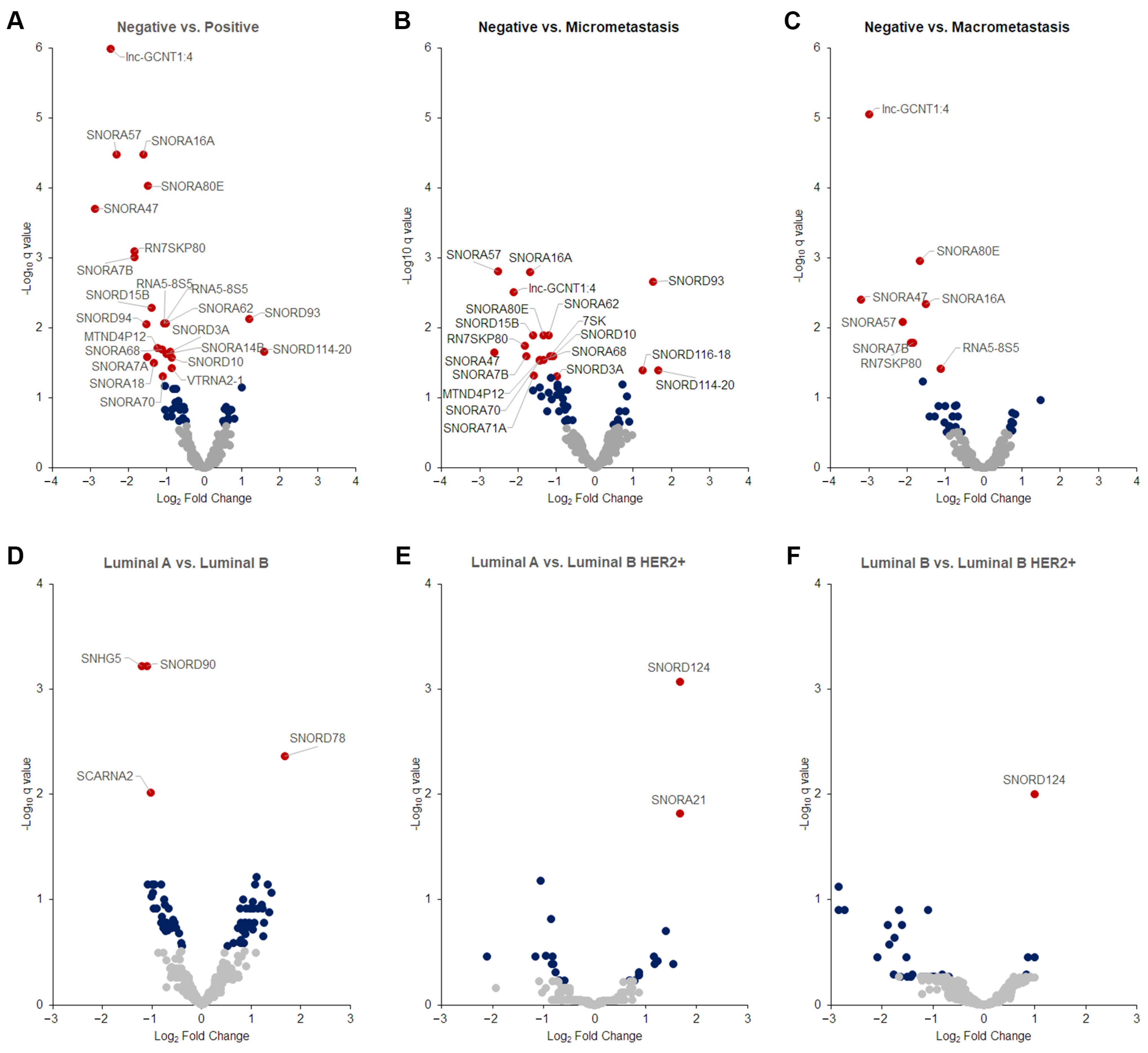
2.4. Differentially Expressed Tumor snoRNAs
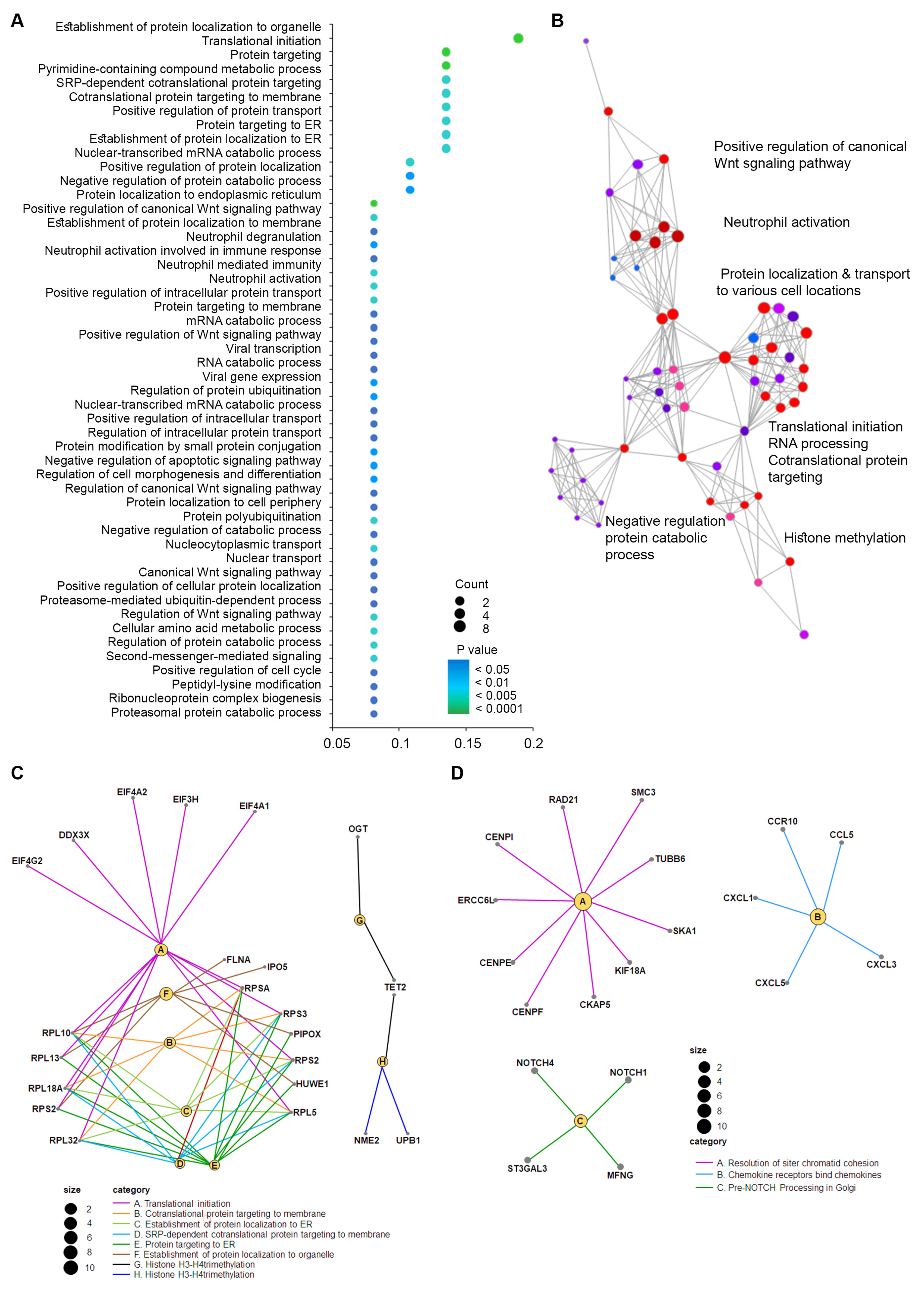
2.5. Biological Significance and Enriched Analysis of sncRNAs
2.6. Clinicopathological Correlation with DE sncRNAs
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huijbers, A.; Velstra, B.; Dekker, T.J.; Mesker, W.E.; van der Burgt, Y.E.; Mertens, B.J.; Deelder, A.M.; Tollenaar, R.A. Proteomic serum biomarkers and their potential application in cancer screening programs. Int. J. Mol. Sci. 2010, 11, 4175–4193. [Google Scholar] [CrossRef] [PubMed]
- Pilvenyte, G.; Ratautaite, V.; Boguzaite, R.; Ramanavicius, A.; Viter, R.; Ramanavicius, S. Molecularly Imprinted Polymers for the Determination of Cancer Biomarkers. Int. J. Mol. Sci. 2023, 24, 4105. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, E.S.; Fournier, M.J. The small nucleolar RNAs. Annu. Rev. Biochem. 1995, 64, 897–934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, C.; Xia, S.; Xiao, F.; Peng, J.; Gao, Y.; Yu, F.; Wang, C.; Chen, X. The emerging role of snoRNAs in human disease. Genes. Dis. 2023, 10, 2064–2081. [Google Scholar] [CrossRef]
- Liang, J.; Wen, J.; Huang, Z.; Chen, X.P.; Zhang, B.X.; Chu, L. Small Nucleolar RNAs: Insight into Their Function in Cancer. Front. Oncol. 2019, 9, 587. [Google Scholar] [CrossRef] [PubMed]
- Brameier, M.; Herwig, A.; Reinhardt, R.; Walter, L.; Gruber, J. Human box C/D snoRNAs with miRNA like functions: Expanding the range of regulatory RNAs. Nucleic Acids Res. 2011, 39, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Falaleeva, M.; Welden, J.R.; Duncan, M.J.; Stamm, S. C/D-box snoRNAs form methylating and non-methylating ribonucleoprotein complexes: Old dogs show new tricks. Bioessays 2017, 39, 1600264. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.S.; Ono, M. From snoRNA to miRNA: Dual function regulatory non-coding RNAs. Biochimie 2011, 93, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, T.; Chen, Q. Exploring the expanding universe of small RNAs. Nat. Cell Biol. 2022, 24, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, N. Small nuclear RNA genes: A model system to study fundamental mechanisms of transcription. J. Biol. Chem. 2001, 276, 26733–26736. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Wang, J.; Ju, S.; Cui, M.; Jing, R. Disorders and roles of tsRNA, snoRNA, snRNA and piRNA in cancer. J. Med. Genet. 2022, 59, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, K.L.; Saksena, M.A.; Freer, P.E.; Smith, B.L.; Rafferty, E.A. To do or not to do: Axillary nodal evaluation after ACOSOG Z0011 Trial. Radiographics 2014, 34, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Zurrida, S.; Veronesi, U. Milestones in breast cancer treatment. Breast J. 2015, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, U.; Paganelli, G.; Galimberti, V.; Viale, G.; Zurrida, S.; Bedoni, M.; Costa, A.; de Cicco, C.; Geraghty, J.G.; Luini, A.; et al. Sentinel-node biopsy to avoid axillary dissection in breast cancer with clinically negative lymph-nodes. Lancet 1997, 349, 1864–1867. [Google Scholar] [CrossRef] [PubMed]
- Escuin, D.; Lopez-Vilaro, L.; Bell, O.; Mora, J.; Moral, A.; Perez, J.I.; Arqueros, C.; Ramon, Y.C.T.; Lerma, E.; Barnadas, A. MicroRNA-1291 Is Associated with Locoregional Metastases in Patients with Early-Stage Breast Cancer. Front. Genet. 2020, 11, 562114. [Google Scholar] [CrossRef] [PubMed]
- Escuin, D.; Lopez-Vilaro, L.; Mora, J.; Bell, O.; Moral, A.; Perez, I.; Arqueros, C.; Garcia-Valdecasas, B.; Ramon, Y.C.T.; Lerma, E.; et al. Circulating microRNAs in Early Breast Cancer Patients and Its Association with Lymph Node Metastases. Front. Oncol. 2021, 11, 627811. [Google Scholar] [CrossRef] [PubMed]
- Escuin, D.; Lopez-Vilaro, L.; Bell, O.; Mora, J.; Garcia-Valdecasas, B.; Moral, A.; Clos, M.; Boronat, L.; Arqueros, C.; Barnadas, A. Circulating miRNA Expression Is Inversely Correlated with Tumor Tissue or Sentinel Lymph Nodes in Estrogen Receptor-Positive Early Breast Cancer Patients. Int. J. Mol. Sci. 2023, 24, 13293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, J.; Diao, L.; Han, L. Small non-coding RNAs in human cancer: Function, clinical utility, and characterization. Oncogene 2021, 40, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; He, Y.; Liu, X.; Zheng, Z.; Zhang, M.; Qin, F.; Lan, X. Small nucleolar RNA 47 promotes tumorigenesis by regulating EMT markers in hepatocellular carcinoma. Minerva Med. 2017, 108, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Pourebrahim, R.; Zhang, Y.; Liu, B.; Gao, R.; Xiong, S.; Lin, P.P.; McArthur, M.J.; Ostrowski, M.C.; Lozano, G. Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes. Dev. 2017, 31, 1847–1857. [Google Scholar] [CrossRef]
- Li, J.L.; Mazar, J.; Zhong, C.; Faulkner, G.J.; Govindarajan, S.S.; Zhang, Z.; Dinger, M.E.; Meredith, G.; Adams, C.; Zhang, S.; et al. Genome-wide methylated CpG island profiles of melanoma cells reveal a melanoma coregulation network. Sci. Rep. 2013, 3, 2962. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Ramirez-Anguita, J.M.; Sauch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jeong, S. Wnt activated beta-catenin and YAP proteins enhance the expression of non-coding RNA component of RNase MRP in colon cancer cells. Oncotarget 2015, 6, 34658–34668. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, E.; Li, Y.; Ye, D.; Cai, Y.; Wang, Q.; Li, Q.; Zhang, X. H/ACA box small nucleolar RNA 7B acts as an oncogene and a potential prognostic biomarker in breast cancer. Cancer Cell Int. 2019, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Schulten, H.J.; Bangash, M.; Karim, S.; Dallol, A.; Hussein, D.; Merdad, A.; Al-Thoubaity, F.K.; Al-Maghrabi, J.; Jamal, A.; Al-Ghamdi, F.; et al. Comprehensive molecular biomarker identification in breast cancer brain metastases. J. Transl. Med. 2017, 15, 269. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.E.; Dowty, J.G.; Milne, R.L.; Wong, E.M.; Dugue, P.A.; English, D.; Hopper, J.L.; Goldgar, D.E.; Giles, G.G.; Southey, M.C. kConFab, Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat. Commun. 2018, 9, 867. [Google Scholar] [CrossRef]
- Karkkainen, E.; Heikkinen, S.; Tengstrom, M.; Kosma, V.M.; Mannermaa, A.; Hartikainen, J.M. Expression profiles of small non-coding RNAs in breast cancer tumors characterize clinicopathological features and show prognostic and predictive potential. Sci. Rep. 2022, 12, 22614. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, L.; Zhao, Y.; Mo, T.; Wang, B.; Lin, J.; Yang, H. Metabolism-regulating non-coding RNAs in breast cancer: Roles, mechanisms and clinical applications. J. Biomed. Sci. 2024, 31, 25. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [PubMed]
- Good, B.M.; Van Auken, K.; Hill, D.P.; Mi, H.; Carbon, S.; Balhoff, J.P.; Albou, L.P.; Thomas, P.D.; Mungall, C.J.; Blake, J.A.; et al. Reactome and the Gene Ontology: Digital convergence of data resources. Bioinformatics 2021, 37, 3343–3348. [Google Scholar] [CrossRef] [PubMed]
- Schweisguth, F. Regulation of notch signaling activity. Curr. Biol. 2004, 14, R129–R138. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Chemokine-chemokine receptor network in immune cell trafficking. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2004, 4, 343–361. [Google Scholar] [CrossRef]
- Dobbin, K.K.; Zhao, Y.; Simon, R.M. How large a training set is needed to develop a classifier for microarray data? Clin. Cancer Res. 2008, 14, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, M.; Nakabayashi, K.; Yoshidome, K.; Kaneko, T.; Iwase, T.; Akiyama, F.; Kato, Y.; Tsuda, H.; Ueda, S.; Sato, K.; et al. One-step nucleic acid amplification for intraoperative detection of lymph node metastasis in breast cancer patients. Clin. Cancer Res. 2007, 13, 4807–4816. [Google Scholar] [CrossRef] [PubMed]
- Webber, C.; Gospodarowicz, M.; Sobin, L.H.; Wittekind, C.; Greene, F.L.; Mason, M.D.; Compton, C.; Brierley, J.; Groome, P.A. Improving the TNM classification: Findings from a 10-year continuous literature review. Int. J. Cancer 2014, 135, 371–378. [Google Scholar] [CrossRef]
- Elston, C.W.; Ellis, I.O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 2011, 17, 3. [Google Scholar] [CrossRef]
- Bouchard-Bourelle, P.; Desjardins-Henri, C.; Mathurin-St-Pierre, D.; Deschamps-Francoeur, G.; Fafard-Couture, E.; Garant, J.M.; Elela, S.A.; Scott, M.S. snoDB: An interactive database of human snoRNA sequences, abundance and interactions. Nucleic Acids Res. 2020, 48, D220–D225. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- R_Development_Core_Team. R: A Language and Environment for Statistical Computing; The R Foundation for Statistical Computing: Vienna, Austria, 2011; ISBN 3-900051-07-0. Available online: http://www.R-project.org/ (accessed on 26 March 2024).
- Gong, J.; Li, Y.; Liu, C.J.; Xiang, Y.; Li, C.; Ye, Y.; Zhang, Z.; Hawke, D.H.; Park, P.K.; Diao, L.; et al. A Pan-cancer Analysis of the Expression and Clinical Relevance of Small Nucleolar RNAs in Human Cancer. Cell Rep. 2017, 21, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.S.; Cai, T.; Longton, G. Combining predictors for classification using the area under the receiver operating characteristic curve. Biometrics 2006, 62, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tian, L. Optimal linear combinations of multiple diagnostic biomarkers based on Youden index. Stat. Med. 2014, 33, 1426–1440. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Total (%) | LN Negative (%) | LN Positive (%) | |
|---|---|---|---|---|
| Patients (%) | 60 (100) | 20 (100) | 40 (100) | |
| Age (years) | Mean + SD | 63.6 ± 14.2 | 68.8 ± 11.9 | 60.9 ± 14.7 |
| Median (range) | 64.3 (26–88) | 71 (47–87) | 60 (26–88) | |
| <40 years | 3 (5) | 0 (0) | 3 (8) | |
| 40–50 years | 6 10) | 2 (10) | 4 (10) | |
| >50 years | 51 (85) | 18 (90) | 33 (82) | |
| Tumor status | T1c | 32 (53) | 12 (60) | 20 (50) |
| T2 | 28 (47) | 8 (40) | 19 (50) | |
| Node status | N0 | 20 (33) | 20 (100) | 0 (0) |
| N1 | 40 (67) | 0 (0) | 40 (100) | |
| Tumor stage | IA | 12 (20) | 12 (60) | 0 (0) |
| IIA | 28 (47) | 8 (40) | 20 (50) | |
| IIB | 20 (33) | 0 (0) | 20 (50) | |
| Tumor grade | 1 | 3 (5) | 1 (5) | 2 (5) |
| 2 | 34 (57) | 14 (70) | 20 (50) | |
| 3 | 23 (38) | 5 (25) | 18 (45) | |
| Tumor focality | Unifocal | 38 (63) | 13 (65) | 25 (62) |
| Multifocal | 19 (32) | 7 (35) | 12 (30) | |
| Multicentric | 3 (5) | 0 (0) | 3 (8) | |
| Ki67 status | ≤14% | 29 (48) | 7 (35) | 22 (55) |
| >14% | 31 (52) | 13 (65) | 58 (18) | |
| ER status | Negative | 6 (10) | 2 (10) | 4 (10) |
| Positive | 54 (90) | 18 (90) | 36 (90) | |
| PR status | Negative | 22 (37) | 6 (30) | 16 (40) |
| Positive | 38 (63) | 14 (70) | 24 (60) | |
| Molecular subtype | Luminal A | 25 (42) | 6 (30) | 19 (48) |
| Luminal B | 21 (35) | 10 (50) | 11 (28) | |
| Luminal B HER2+ | 8 (13) | 3 (15) | 6 (15) | |
| HER2+ | 0 (0) | 0 (0) | 0 (0) | |
| TN | 5 (8) | 1 (5) | 4 (9) | |
| LVI | Negative | 52 (87) | 19 (95) | 33 (83) |
| Positive | 8 (13) | 1 (5) | 7 (17) | |
| Menopausal status | Premenopausal | 10 (17) | 2 (10) | 8 (20) |
| Postmenopausal | 50 (83) | 18 (90) | 32 (80) | |
| Breast affected | Left | 28 (47) | 10 (50) | 18 (45) |
| Right | 32 (53) | 10 (50) | 22 (55) | |
| Breast surgery | Mastectomy | 18 (32) | 5 (25) | 13 (33) |
| Lumpectomy | 41 (68) | 15 (75) | 27 (67) |
| ID | Description | Ratio | p | sncRNAs | Target Gene |
|---|---|---|---|---|---|
| GO:0090263 | Positive regulation of canonical Wnt signaling pathway | 3/37 | 0.003 | SNORA80E, SNORD10, SNORD15A | PSMD11, LGR5, DDX3X |
| GO:0030177 | Positive regulation of Wnt signaling pathway | 3/37 | 0.005 | SNORA80E, SNORD10, SNORD15A | PSMD11, LGR5, DDX3X |
| GO:0060070 | Canonical Wnt signaling pathway | 3/37 | 0.026 | SNORA80E, SNORD10, SNORD15A | PSMD11, LGR5, DDX3X |
| GO:0016055 | Wnt signaling pathway | 3/37 | 0.083 | SNORA80E, SNORD10, SNORD15A | PSMD11, LGR5, DDX3X |
| GO:0198738 | Cell–cell signaling by wnt | 3/37 | 0.084 | SNORA80E, SNORD10, SNORD15A | PSMD11, LGR5, DDX3X |
| GO:0007219 | Notch signaling pathway | 12/474 | 0.003 | SNORD15B, SNORA68, SNORD10, SNORD93 | DLL1, EGFL7, MFNG, MIB2, NOTCH4, NOTCH1, DNER, FOXA1, FOXC1, YBX1, TBX2, DLGAP5 |
| R-HSA-8951430 | RUNX3 regulates WNT signaling | 2/305 | 0.021 | SNORD93 | RUNX3, TCF7L1 |
| R-HSA-157118 | Signaling by NOTCH | 14/305 | 0.008 | SNORD15B, SNORA68, SNORD10, SNORD93 | DLL1, HDAC7, MFNG, MIB2, NOTCH4, PSMD12, YWHAZ, DNER, NOTCH1, TLE4, YBX1, FLT4, DLGAP5, ST3GAL3 |
| R-HSA-9012852 | Signaling by NOTCH3 | 5/305 | 0.013 | SNORD15B, SNORA68, SNORD10, SNORD93 | DLL1, NOTCH1, YBX1, DLGAP5, MIB2 |
| R-HSA-2691230 | Signaling by NOTCH1 HD Domain Mutants in Cancer | 3/305 | 0.008 | SNORD15B, SNORA68, SNORD10, SNORD93 | DLL1, NOTCH1, MIB2 |
| R-HSA-2691232 | Constitutive Signaling by NOTCH1 HD Domain Mutants | 3/305 | 0.008 | SNORD15B, SNORA68, SNORD10, SNORD93 | DLL1, NOTCH1, MIB2 |
| R-HSA-9013695 | NOTCH4 Intracellular Domain Regulates Transcription | 3/305 | 0.019 | SNORD15B, SNORA68, SNORD10, SNORD93 | NOTCH4, NOTCH1, FLT4 |
| R-HSA-9013507 | NOTCH3 Activation and Transmission of Signal to the Nucleus | 3/305 | 0.034 | SNORD15B, SNORA68, SNORD10, SNORD93 | DLL1, YBX1, MIB2 |
| R-HSA-350054 | Notch-HLH transcription pathway | 3/305 | 0.045 | SNORD15B, SNORA68, SNORD10, SNORD93 | HDAC7, NOTCH4, NOTCH1 |
| R-HSA-1912399 | Pre-NOTCH Processing in the Endoplasmic Reticulum | 2/305 | 0.011 | SNORD15B, SNORA68, SNORD10, SNORD93 | NOTCH4, NOTCH1 |
| R-HSA-2660825 | Signaling by NOTCH1 t(7;9) (M1580_K2555) Translocation Mutant | 2/305 | 0.016 | SNORD15B, SNORA68, SNORD10/SNORD93 | DLL1, NOTCH1 |
| R-HSA-2660826 | Constitutive Signaling by NOTCH1 t(7;9)(M1580_K2555) Translocation Mutant | 2/305 | 0.016 | SNORD15B, SNORA68, SNORD10/SNORD93 | DLL1, NOTCH1 |
| R-HSA-9013700 | NOTCH4 Activation of Signal to the Nucleus | 2/305 | 0.038 | SNORD15B, SNORA68, SNORD10 | NOTCH4, YWHAZ |
| Variable | N | sncRNA Name |
|---|---|---|
| Tumor grade | 134 | SNORD105B, SNORD19C, SNORD101, SNORD102, SNORD107, SNORD114-3, SNORD126, SNORD18A, SNORD61, SNORD64 |
| Lymphovascular invasion | 20 | RNU5D-1, RNU5E-1, RNU7-1, SNORA15B-1, SNORA2B, SNORA36B, SNORA36C, SNORA38B, SNORA41, SNORA53 |
| Tumor focality | 12 | RNU5D-1, RNU5E-1, RNU7-1, SNORA15B-1, SNORA2B, SNORA36B, SNORA36C, SNORA38B, SNORA41 |
| Menopausal status | 2 | RNU5E-1, RNU5D-1 |
| Tumor stage | 1 | SNORD115-8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escuin, D.; Bell, O.; García-Valdecasas, B.; Clos, M.; Larrañaga, I.; López-Vilaró, L.; Mora, J.; Andrés, M.; Arqueros, C.; Barnadas, A. Small Non-Coding RNAs and Their Role in Locoregional Metastasis and Outcomes in Early-Stage Breast Cancer Patients. Int. J. Mol. Sci. 2024, 25, 3982. https://doi.org/10.3390/ijms25073982
Escuin D, Bell O, García-Valdecasas B, Clos M, Larrañaga I, López-Vilaró L, Mora J, Andrés M, Arqueros C, Barnadas A. Small Non-Coding RNAs and Their Role in Locoregional Metastasis and Outcomes in Early-Stage Breast Cancer Patients. International Journal of Molecular Sciences. 2024; 25(7):3982. https://doi.org/10.3390/ijms25073982
Chicago/Turabian StyleEscuin, Daniel, Olga Bell, Bárbara García-Valdecasas, Montserrat Clos, Itziar Larrañaga, Laura López-Vilaró, Josefina Mora, Marta Andrés, Cristina Arqueros, and Agustí Barnadas. 2024. "Small Non-Coding RNAs and Their Role in Locoregional Metastasis and Outcomes in Early-Stage Breast Cancer Patients" International Journal of Molecular Sciences 25, no. 7: 3982. https://doi.org/10.3390/ijms25073982
APA StyleEscuin, D., Bell, O., García-Valdecasas, B., Clos, M., Larrañaga, I., López-Vilaró, L., Mora, J., Andrés, M., Arqueros, C., & Barnadas, A. (2024). Small Non-Coding RNAs and Their Role in Locoregional Metastasis and Outcomes in Early-Stage Breast Cancer Patients. International Journal of Molecular Sciences, 25(7), 3982. https://doi.org/10.3390/ijms25073982