
Is There a Place for Lewy Bodies before and beyond Alpha-Synuclein Accumulation? Provocative Issues in Need of Solid Explanations

 ,
, 
 , ,
, , 
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Is Alpha-Synuclein the Major Component of LBs?
1.2. Is Alpha-Syn the Natural Seed of LBs?
1.3. Is Alpha-Syn the Unique Guilty Protein in the Onset and Progression of PD?
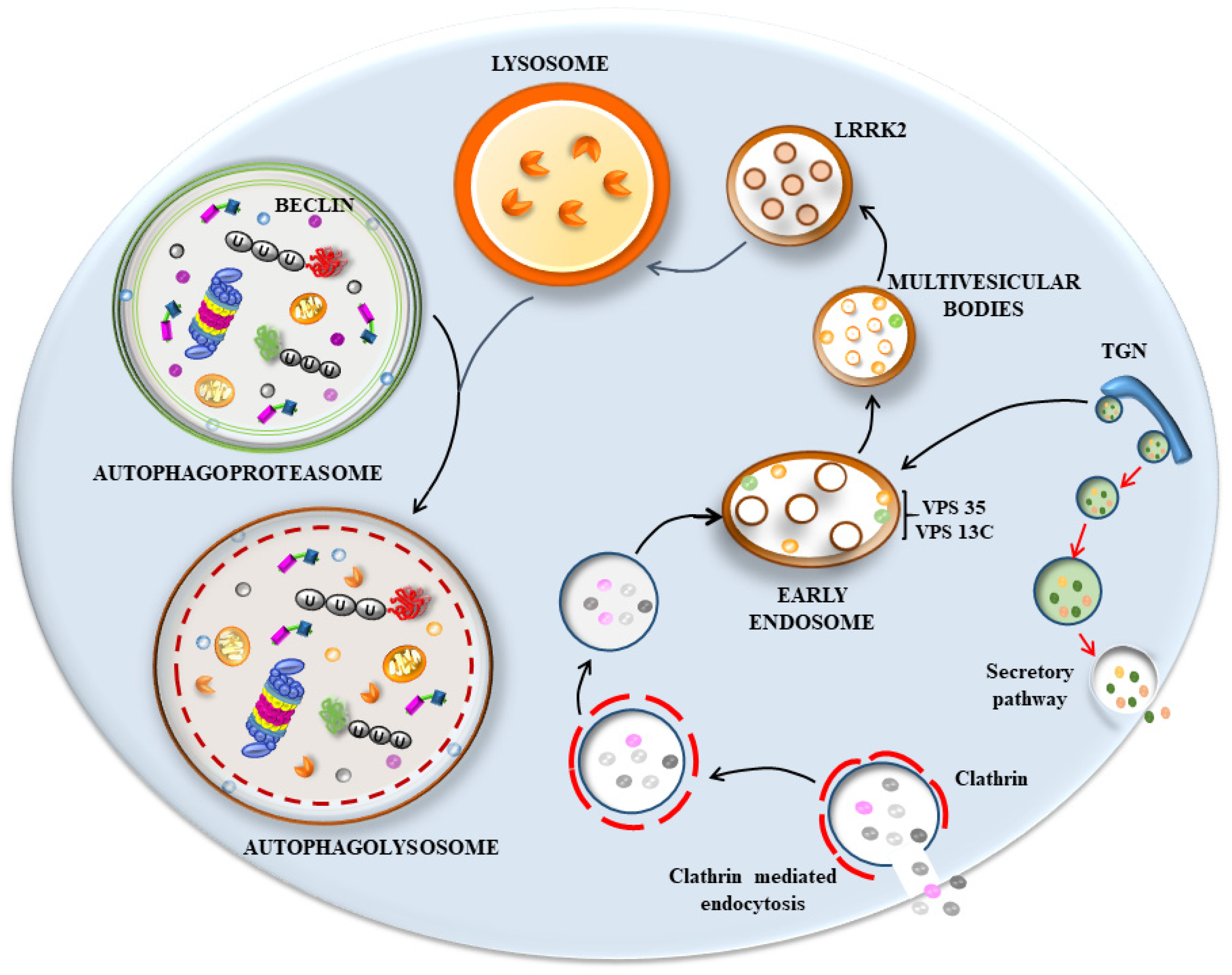
1.4. Is the Endo-Autophago-Lysosomal System Defective in PD?
1.5. How to Reconcile Alpha-Syn with Defective Clearing Systems within LBs and PD Progression?
2. The Starting Evidence Which Led to Define Alpha-Syn as the Major Component of LBs
3. Limitations and Pitfalls about Evidence on the Primary Role of Alpha-Syn in LB
3.1. Is Alpha-Syn the Major Component of LB?
3.2. Is Alpha-Syn the Seed of LBs?
3.3. Is Alpha-Syn the Component of Radiating Filaments within LB?
4. General Doubts
5. Alternative Mechanisms
5.1. The Good and the Bad Copes of Alpha-Syn
5.2. The Good and the Bad Copes of LBs
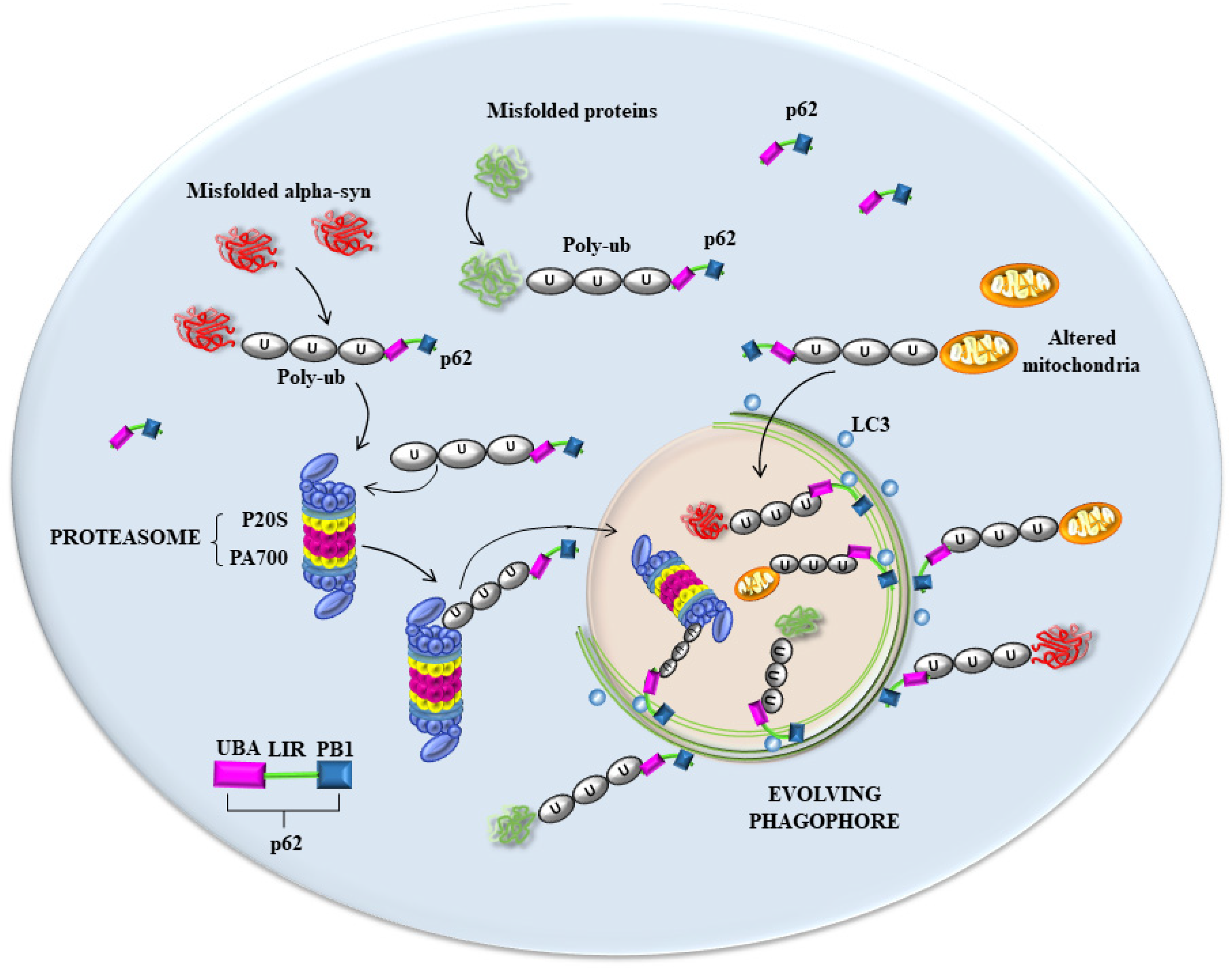
6. The Hypothetical Role of p62 and Poly-Ubiquitin in Seeding Vesicles-Rich Inclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Estaun-Panzano, J.; Arotcarena, M.L.; Bezard, E. Monitoring α-synuclein aggregation. Neurobiol. Dis. 2023, 176, 105966. [Google Scholar] [CrossRef]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef]
- Vidyadhara, D.J.; Lee, J.E.; Chandra, S.S. Role of the endolysosomal system in Parkinson’s disease. J. Neurochem. 2019, 150, 487–506. [Google Scholar] [CrossRef]
- Lashuel, H.A. Do Lewy bodies contain alpha-synuclein fibrils? and Does it matter? A brief history and critical analysis of recent reports. Neurobiol. Dis. 2020, 141, 104876. [Google Scholar] [CrossRef]
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Novello, S. Lewy body-associated proteins: Victims, instigators, or innocent bystanders? The case of AIMP2 and alpha-synuclein. Neurobiol. Dis. 2021, 156, 105417. [Google Scholar] [CrossRef]
- Smith, J.K.; Mellick, G.D.; Sykes, A.M. The role of the endolysosomal pathway in α-synuclein pathogenesis in Parkinson’s disease. Front. Cell. Neurosci. 2023, 16, 1081426. [Google Scholar] [CrossRef]
- Chopra, A.; Outeiro, T.F. Aggregation and beyond: Alpha-synuclein-based biomarkers in synucleinopathies. Brain 2024, 147, 81–90. [Google Scholar] [CrossRef]
- Teixeira, M.; Sheta, R.; Idi, W.; Oueslati, A. Alpha-Synuclein and the Endolysosomal System in Parkinson’s Disease: Guilty by Association. Biomolecules 2021, 11, 1333. [Google Scholar] [CrossRef]
- Kuznetsov, I.A.; Kuznetsov, A.V. Can the lack of fibrillar form of alpha-synuclein in Lewy bodies be explained by its catalytic activity? Math. Biosci. 2022, 344, 108754. [Google Scholar] [CrossRef]
- Kuznetsov, A.V. Lewy body radius growth: The hypothesis of the cube root of time dependency. J. Theor. Biol. 2024, 581, 111734. [Google Scholar] [CrossRef]
- Paine, S.M.; Anderson, G.; Bedford, K.; Lawler, K.; Mayer, R.J.; Lowe, J.; Bedford, L. Pale body-like inclusion formation and neurodegeneration following depletion of 26S proteasomes in mouse brain neurones are independent of α-synuclein. PLoS ONE 2013, 8, e54711. [Google Scholar] [CrossRef]
- Noda, S.; Sato, S.; Fukuda, T.; Ueno, S.; Tada, N.; Hattori, N. Impaired mitochondrial accumulation and Lewy pathology in neuron-specific FBXO7-deficient mice. Mol. Brain 2022, 15, 54. [Google Scholar] [CrossRef]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 18, 2813. [Google Scholar] [CrossRef]
- Sato, S.; Noda, S.; Torii, S.; Amo, T.; Ikeda, A.; Funayama, M.; Yamaguchi, J.; Fukuda, T.; Kondo, H.; Tada, N.; et al. Homeostatic p62 levels and inclusion body formation in CHCHD2 knockout mice. Hum. Mol. Genet. 2021, 30, 443–453. [Google Scholar] [CrossRef]
- Sato, S.; Noda, S.; Hattori, N. Pathogenic insights to Parkin-linked model mice. Neurosci. Res. 2020, 159, 47–51. [Google Scholar] [CrossRef]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nature 2013, 498, E4–E6, discussion E6–E7. [Google Scholar] [CrossRef]
- Diao, J.; Burré, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Südhof, T.C.; Brunger, A.T. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2013, 2, e00592. [Google Scholar] [CrossRef]
- Vidyadhara, D.J.; Somayaji, M.; Wade, N.; Yücel, B.; Zhao, H.; Shashaank, N.; Ribaudo, J.; Gupta, J.; Lam, T.T.; Sames, D.; et al. Dopamine transporter and synaptic vesicle sorting defects underlie auxilin-associated Parkinson’s disease. Cell Rep. 2023, 42, 112231. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, H.; Yan, N.; Zhao, P.; Xu, H.; Zheng, W.; Zhang, X.; Wang, T.; Guo, C.; Zhong, M. ATP13A2 Declines Zinc-Induced Accumulation of α-Synuclein in a Parkinson’s Disease Model. Int. J. Mol. Sci. 2022, 23, 8035. [Google Scholar] [CrossRef]
- Yang, B.; Yang, Z.; Liu, H.; Qi, H. Dynamic modelling and tristability analysis of misfolded α-synuclein degraded via autophagy in Parkinson’s disease. Biosystems 2023, 233, 105036. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xu, F.; Lai, H.; Yuan, H.; Li, X.Y.; Hu, J.; Li, W.; Liu, L.; Wang, C. ACO2 deficiency increases vulnerability to Parkinson’s disease via dysregulating mitochondrial function and histone acetylation-mediated transcription of autophagy genes. Commun. Biol. 2023, 6, 1201. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Yan, J.; Huang, P.; Wang, X.; Zhang, R.; Zhang, C.; Wang, W.; Qian, W.; Zhou, J.; Zhao, Y.; et al. miR-214-3p promotes the pathogenesis of Parkinson’s disease by inhibiting autophagy. Biomed. Pharmacother. 2024, 171, 116123. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Bai, L.; Yan, F.; Zhang, S.; Zhang, X.; Deng, R.; Zeng, X.; Sun, B.; Hu, X.; Li, Y.; et al. Thioredoxin-1 decreases alpha-synuclein induced by MPTP through promoting autophagy-lysosome pathway. Cell Death Discov. 2024, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Markham, B.N.; Ramnarine, C.; Kim, S.; Grever, W.E.; Soto-Beasley, A.I.; Heckman, M.; Ren, Y.; Osborne, A.C.; Bhagwate, A.V.; Liu, Y.; et al. miRNA family miR-29 inhibits PINK1-PRKN dependent mitophagy via ATG9A. bioRxiv 2024. [Google Scholar] [CrossRef]
- Matsuki, A.; Watanabe, Y.; Hashimoto, S.; Hoshino, A.; Matoba, S. Cathepsin L prevents the accumulation of alpha-synuclein fibrils in the cell. Genes Cells 2024. [Google Scholar] [CrossRef] [PubMed]
- Skou, L.D.; Johansen, S.K.; Okarmus, J.; Meyer, M. Pathogenesis of DJ-1/PARK7-Mediated Parkinson’s Disease. Cells 2024, 13, 296. [Google Scholar] [CrossRef]
- Volta, M. Roles of neuronal lysosomes in the etiology of Parkinson’s disease. Neural Regen. Res. 2024, 19, 1981–1983. [Google Scholar] [CrossRef]
- Ferrucci, M.; Lenzi, P.; Lazzeri, G.; Busceti, C.L.; Frati, A.; Puglisi-Allegra, S.; Fornai, F. Combined light and electron microscopy (CLEM) to quantify methamphetamine-induced alpha-synuclein-related pathology. J. Neural. Transm. 2024. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Gesi, M.; Soldani, P.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; Battaglia, G.; De Blasi, A.; Nicoletti, F.; et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J. Neurochem. 2004, 88, 114–123. [Google Scholar] [CrossRef]
- Quan, L.; Ishikawa, T.; Michiue, T.; Li, D.R.; Zhao, D.; Oritani, S.; Zhu, B.L.; Maeda, H. Ubiquitin-immunoreactive structures in the midbrain of methamphetamine abusers. Leg. Med. 2005, 7, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Gesi, M.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Ruffoli, R.; Soldani, P.; Ruggieri, S.; Alessandri, M.G.; et al. Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J. Neurosci. 2003, 23, 8955–8966. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Schlüter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha- synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Pasquali, L.; Ruggieri, S.; Paparelli, A.; Fornai, F. Alpha-synuclein and autophagy as common steps in neurodegeneration. Park. Relat. Disord. 2008, 14 (Suppl. 2), S180–S184. [Google Scholar] [CrossRef]
- Isidoro, C.; Biagioni, F.; Giorgi, F.S.; Fulceri, F.; Paparelli, A.; Fornai, F. The role of autophagy on the survival of dopamine neurons. Curr. Top. Med. Chem. 2009, 9, 869–879. [Google Scholar] [PubMed]
- Pasquali, L.; Ruggieri, S.; Murri, L.; Paparelli, A.; Fornai, F. Does autophagy worsen or improve the survival of dopaminergic neurons? Park. Relat. Disord. 2009, 15 (Suppl. 4), S24–S27. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [CrossRef]
- Gambardella, S.; Biagioni, F.; Ferese, R.; Busceti, C.L.; Frati, A.; Novelli, G.; Ruggieri, S.; Fornai, F. Vacuolar Protein Sorting Genes in Parkinson’s Disease: A Re- appraisal of Mutations Detection Rate and Neurobiology of Disease. Front. Neurosci. 2016, 10, 532. [Google Scholar] [CrossRef]
- Hattori, N.; Mizuno, Y. Twenty years since the discovery of the parkin gene. J. Neural. Transm. 2017, 124, 1037–1054. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, S.; Ferese, R.; Biagioni, F.; Busceti, C.L.; Campopiano, R.; Griguoli, A.M.P.; Limanaqi, F.; Novelli, G.; Storto, M.; Fornai, F. The Monoamine Brainstem Reticular Formation as a Paradigm for Re-Defining Various Phenotypes of Parkinson’s Disease Owing Genetic and Anatomical Specificity. Front. Cell. Neurosci. 2017, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; López-Otín, C. Autophagy: Molecular mechanisms, physiological functions and relevance in human pathology. Cell. Mol. Life Sci. 2004, 61, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Bursch, W.; Ellinger, A. Autophagy--a basic mechanism and a potential role for neurodegeneration. Folia Neuropathol. 2005, 43, 297–310. [Google Scholar] [PubMed]
- Winslow, A.R.; Rubinsztein, D.C. The Parkinson disease protein α-synuclein inhibits autophagy. Autophagy 2011, 7, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.B.; Jeon, B.S. Protective role of heat shock and heat shock protein 70 in lactacystin-induced cell death both in the rat substantia nigra and PC12 cells. Brain Res. 2006, 1087, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Rahman, T.; Ghosh, S.; Kalmanovich, J.B.; Awuah, A.W.; Zivcevska, M.; Khalifa, S.; Bassey, E.E.; Ali, N.A.; Ferreira, M.M.D.S.; Umar, T.P.; et al. The role of membrane trafficking and retromer complex in Parkinson’s and Alzheimer’s disease. J. Neurosci. Res. 2024, 102, e25261. [Google Scholar] [CrossRef]
- Herman, M.; Randall, G.W.; Spiegel, J.L.; Maldonado, D.J.; Simoes, S. Endo-lysosomal dysfunction in neurodegenerative diseases: Opinion on current progress and future direction in the use of exosomes as biomarkers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20220387. [Google Scholar] [CrossRef]
- Morris, H.R.; Spillantini, M.G.; Sue, C.M.; Williams-Gray, C.H. The pathogenesis of Parkinson’s disease. Lancet. 2024, 403, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, J.; Moore, D.J. VPS35 and retromer dysfunction in Parkinson’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20220384. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Cullen, P.J.; Cookson, M.; Merchant, K.M.; Small, S.A. Retromer-dependent lysosomal stress in Parkinson’s disease. Philos. Trans. R. Soc. Lond. B Biol Sci. 2024, 379, 20220376. [Google Scholar] [CrossRef] [PubMed]
- Bhore, N.; Bogacki, E.C.; O’Callaghan, B.; Plun-Favreau, H.; Lewis, P.A.; Herbst, S. Common genetic risk for Parkinson’s disease and dysfunction of the endo-lysosomal system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20220517. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, I.; Russo, L.; Torretta, E.; Barbacini, P.; Contarini, G.; Pacinelli, G.; Bizzotto, D.; Moriggi, M.; Braghetta, P.; Papaleo, F.; et al. GBA1 inactivation in oligodendrocytes affects myelination and induces neurodegenerative hallmarks and lipid dyshomeostasis in mice. Mol. Neurodegener. 2024, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Hull, A.; Atilano, M.L.; Gergi, L.; Kinghorn, K.J. Lysosomal storage, impaired autophagy and innate immunity in Gaucher and Parkinson’s diseases: Insights for drug discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20220381. [Google Scholar] [CrossRef] [PubMed]
- Rubilar, J.C.; Outeiro, T.F.; Klein, A.D. The lysosomal β-glucocerebrosidase strikes mitochondria: Implications for Parkinson’s therapeutics. Brain 2024, 4, awae070. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; McDonald, D.; Blain, A.; Mossman, E.; Atkin, K.; Marusich, M.F.; Capaldi, R.; Bone, L.; Smith, A.; Filby, A.; et al. Parkinson’s disease neurons exhibit alterations in mitochondrial quality control proteins. NPJ Park. Dis. 2023, 9, 120. [Google Scholar] [CrossRef]
- Choong, C.J.; Mochizuki, H. Involvement of Mitochondria in Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 17027. [Google Scholar] [CrossRef]
- Liang, Y.; Zhong, G.; Li, Y.; Ren, M.; Wang, A.; Ying, M.; Liu, C.; Guo, Y.; Zhang, D. Comprehensive Analysis and Experimental Validation of the Parkinson’s Disease Lysosomal Gene ACP2 and Pan-cancer. Biochem. Genet. 2024. [Google Scholar] [CrossRef]
- Xu, J.; Deng, Z.; Shang, S.; Wang, C.; Han, H. FUNDC1 collaborates with PINK1 in regulating mitochondrial Fission and compensating for PINK1 deficiency. Biochem. Biophys. Res. Commun. 2023, 687, 149210. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.R.; Nagy, J.K. Misfolding of membrane proteins in health and disease: The lady or the tiger? Curr. Opin. Struct. Biol. 2000, 10, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Heid, H.; Schnoelzer, M.; Kenner, L.; Kleinert, R.; Prinz, M.; Aguzzi, A.; Denk, H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 2002, 160, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Soldani, P.; Lazzeri, G.; di Poggio, A.B.; Biagioni, F.; Fulceri, F.; Batini, S.; Ruggieri, S.; Paparelli, A. Neuronal inclusions in degenerative disorders Do they represent static features or a key to understand the dynamics of the disease? Brain Res. Bull. 2005, 65, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Fulceri, F.; Lazzeri, G.; Casini, A.; Ruggieri, S.; Paparelli, A.; Fornai, F. Analysis of single, purified inclusions as a novel approach to understand methamphetamine neurotoxicity. Ann. N. Y. Acad. Sci. 2008, 1139, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, F.; Ferese, R.; Giorgi, F.S.; Modugno, N.; Olivola, E.; Lenzi, P.; Gambardella, S.; Centonze, D.; Ruggieri, S.; Fornai, F. An attempt to dissect a peripheral marker based on cell pathology in Parkinson’s disease. J. Neural Transm. 2021, 128, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Farrer, M.; Kachergus, J.; Forno, L.; Lincoln, S.; Wang, D.S.; Hulihan, M.; Maraganore, D.; Gwinn-Hardy, K.; Wszolek, Z.; Dickson, D.; et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 2004, 55, 174–179. [Google Scholar] [CrossRef]
- Ferese, R.; Modugno, N.; Campopiano, R.; Santilli, M.; Zampatti, S.; Giardina, E.; Nardone, A.; Postorivo, D.; Fornai, F.; Novelli, G.; et al. Four Copies of SNCA Responsible for Autosomal Dominant Parkinson’s Disease in Two Italian Siblings. Park. Dis. 2015, 2015, 546462. [Google Scholar] [CrossRef]
- Nascimento, A.C.; Erustes, A.G.; Reckziegel, P.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. α-Synuclein Overexpression Induces Lysosomal Dysfunction and Autophagy Impairment in Human Neuroblastoma SH-SY5Y. Neurochem. Res. 2020, 45, 2749–2761. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Porat-Shliom, Y.; Pei, Z.; Cheng, Y.; Xiang, L.; Sommers, K.; Li, Q.; Gillardon, F.; Hengerer, B.; Berlinicke, C.; et al. Baicalein reduces E46K alpha-synuclein aggregation in vitro and protects cells against E46K alpha-synuclein toxicity in cell models of familiar Parkinsonism. J. Neurochem. 2010, 114, 419–429. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant α-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701, Erratum in Mol. Neurobiol. 2014, 50, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Yamaguchi, H.; Fujimuro, M.; Yokosawa, H.; Ihara, Y.; Trojanowski, J.Q.; Lee, V.M. Purification and characterization of Lewy bodies from the brains of patients with diffuse Lewy body disease. Am. J. Pathol. 1996, 148, 1517–1529, Erratum in Am. J. Pathol. 1996, 149, 1770–1771; Erratum in Am. J. Pathol. 1997, 150, 2255. [Google Scholar]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Bergersen, L.H.; Storm-Mathisen, J.; Gundersen, V. Immunogold quantification of amino acids and proteins in complex subcellular compartments. Nat. Protoc. 2008, 3, 144–152. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef]
- Lim, K.L.; Dawson, V.L.; Dawson, T.M. Parkin-mediated lysine 63-linked polyubiquitination: A link to protein inclusions formation in Parkinson’s and other conformational diseases? Neurobiol Aging 2006, 27, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Paine, S.; Bedford, L.; Thorpe, J.R.; Mayer, R.J.; Cavey, J.R.; Bajaj, N.; Sheppard, P.W.; Lowe, J.; Layfield, R. Immunoreactivity to Lys63-linked polyubiquitin is a feature of neurodegeneration. Neurosci. Lett. 2009, 460, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Buneeva, O.; Medvedev, A. Atypical Ubiquitination and Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 3705. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ohama, E.; Suzuki, S.; Horikawa, Y.; Ishikawa, A.; Morita, T.; Tsuji, S.; Ikuta, F. Familial juvenile parkinsonism: Clinical and pathologic study in a family. Neurology 1994, 44, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Kondo, T.; Yokochi, M.; Matsumine, H.; Nakagawa-Hattori, Y.; Miyake, T.; Suda, K.; Mizuno, Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 1998, 51, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Wakabayashi, K.; Ishikawa, A.; Nagai, H.; Saito, M.; Maruyama, M.; Takahashi, T.; Ozawa, T.; Tsuji, S.; Takahashi, H. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov. Disord. 2000, 15, 884–888. [Google Scholar] [CrossRef] [PubMed]
- van de Warrenburg, B.P.; Lammens, M.; Lucking, C.B.; Denefle, P.; Wesseling, P.; Booij, J.; Praamstra, P.; Quinn, N.; Brice, A.; Horstink, M.W. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 2001, 56, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.E. Parkin and PINK1 parkinsonism may represent nigral mitochondrial cytopathies distinct from Lewy body Parkinson’s disease. Park. Relat. Disord. 2009, 15, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N.; et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 2002, 160, 1655–1667. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Radwan, N.; Khan, E.; Kitada, T.; Haque, M.E. Parkin Precipitates on Mitochondria via Aggregation and Autoubiquitination. Int. J. Mol. Sci. 2023, 24, 9027. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.Q.; Zheng, R.; Liu, Y.; Ruan, Y.; Lin, Z.H.; Xue, N.J.; Chen, Y.; Zhang, B.R.; Pu, J.L. Parkin regulates microglial NLRP3 and represses neurodegeneration in Parkinson’s disease. Aging Cell 2023, 22, e13834. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Zuo, Y.; Li, L.; Zhan, L.; Xue, J.; Sun, W.; Xu, E. Hypoxic postconditioning restores mitophagy against transient global cerebral ischemia via Parkin-induced posttranslational modification of TBK1. Neurobiol. Dis. 2023, 179, 106043. [Google Scholar] [CrossRef] [PubMed]
- Nogalska, A.; Terracciano, C.; D’Agostino, C.; King Engel, W.; Askanas, V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009, 118, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Ciechanover, A.; Livneh, I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget 2016, 7, 83833–83834. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, H.; Tsuji, A.; Hashimoto, Y.; Takata, M.; Koga, S.; Nishida, K.; Futamura, N.; Kawamoto, M.; Kohara, N.; Dickson, D.W.; et al. Discrepancy between distribution of alpha-synuclein oligomers and Lewy-related pathology in Parkinson’s disease. Acta Neuropathol. Commun. 2022, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.A.; Lavenir, I.; Fan, J.; Masuda-Suzukake, M.; Passarella, D.; DeTure, M.A.; Dickson, D.W.; Ghetti, B.; Goedert, M. α-Synuclein filaments from transgenic mouse and human synucleinopathy-containing brains are major seed-competent species. J. Biol. Chem. 2020, 295, 6652–6664. [Google Scholar] [CrossRef]
- Lohmann, S.; Bernis, M.E.; Tachu, B.J.; Ziemski, A.; Grigoletto, J.; Tamgüney, G. Oral and intravenous transmission of α-synuclein fibrils to mice. Acta Neuropathol. 2019, 138, 515–533. [Google Scholar] [CrossRef]
- Duffy, P.E.; Tennyson, V.M. Phase and electron microscopic observations of lewy bodies and melanin granules in the substantia nigra and locus caeruleus in parkinson’s disease. J. Neuropathol. Exp. Neurol. 1965, 24, 398–414. [Google Scholar] [CrossRef]
- Roy, S.; Wolman, L. Ultrastructural observations in Parkinsonism. J. Pathol. 1969, 99, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Zalon, A.J.; Quiriconi, D.J.; Pitcairn, C.; Mazzulli, J.R. α-Synuclein: Multiple pathogenic roles in trafficking and proteostasis pathways in Parkinson’s disease. Neuroscientist 2024, 10738584241232963. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernández-Chacón, R.; Schlüter, O.M.; Südhof, T.C. Alpha- synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, O.M.; Fornai, F.; Alessandrí, M.G.; Takamori, S.; Geppert, M.; Jahn, R.; Südhof, T.C. Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine- induced parkinsonism in mice. Neuroscience 2003, 118, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Machida, Y.; Chiba, T.; Takayanagi, A.; Tanaka, Y.; Asanuma, M.; Ogawa, N.; Koyama, A.; Iwatsubo, T.; Ito, S.; Jansen, P.H.; et al. Common anti-apoptotic roles of parkin and alpha-synuclein in human dopaminergic cells. Biochem. Biophys. Res. Commun. 2005, 332, 233–240, Erratum in Biochem. Biophys. Res. Commun. 2005, 334, 970. [Google Scholar] [CrossRef]
- Wei, J.; Ho, G.; Takamatsu, Y.; Masliah, E.; Hashimoto, M. Therapeutic Potential of α-Synuclein Evolvability for Autosomal Recessive Parkinson’s Disease. Park. Dis. 2021, 2021, 6318067. [Google Scholar] [CrossRef]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef]
- Westphal, C.H.; Chandra, S.S. Monomeric synucleins generate membrane curvature. J. Biol. Chem. 2013, 288, 1829–1840. [Google Scholar] [CrossRef]
- Anwar, S.; Peters, O.; Millership, S.; Ninkina, N.; Doig, N.; Connor-Robson, N.; Threlfell, S.; Kooner, G.; Deacon, R.M.; Bannerman, D.M.; et al. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. 2011, 31, 7264–7274. [Google Scholar] [CrossRef]
- Riederer, P.; Nagatsu, T.; Youdim, M.B.H.; Wulf, M.; Dijkstra, J.M.; Sian-Huelsmann, J. Lewy bodies, iron, inflammation and neuromelanin: Pathological aspects underlying Parkinson’s disease. J. Neural. Transm. 2023, 130, 627–646. [Google Scholar] [CrossRef]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Tokuda, T.; El-Agnaf, O.M.A.; Ohmichi, T.; Mori, Y.; Asano, M.; Kanemoto, M.; Baba, Y.; Tsukie, T.; Ikeuchi, T.; et al. IgG index of cerebrospinal fluid can reflect pathophysiology associated with Lewy bodies in Parkinson’s disease. J. Neurol. Sci. 2023, 452, 120760. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Monoranu, C.; Strobel, S.; Riederer, P. Lewy Bodies: A Spectator or Salient Killer? CNS Neurol. Disord. Drug Targets 2015, 14, 947–955. [Google Scholar] [CrossRef]
- Geibl, F.F.; Henrich, M.; Xie, Z.; Zampese, E.; Tkatch, T.; Wokosin, D.L.; Nasir, E.; Grotmann, C.A.; Dawson, V.L.; Dawson, T.M.; et al. α-Synuclein pathology disrupts mitochondrial function in dopaminergic and cholinergic neurons at-risk in Parkinson’s disease. bioRxiv. 2023. [Google Scholar] [CrossRef]
- Mubariz, F.; Saadin, A.; Lingenfelter, N.; Sarkar, C.; Banerjee, A.; Lipinski, M.M.; Awad, O. Deregulation of mTORC1-TFEB axis in human iPSC model of GBA1-associated Parkinson’s disease. Front. Neurosci. 2023, 17, 1152503. [Google Scholar] [CrossRef]
- Qiao, L.; Hu, J.; Qiu, X.; Wang, C.; Peng, J.; Zhang, C.; Zhang, M.; Lu, H.; Chen, W. LAMP2A, LAMP2B and LAMP2C: Similar structures, divergent roles. Autophagy 2023, 19, 2837–2852. [Google Scholar] [CrossRef] [PubMed]
- Brooker, S.M.; Naylor, G.E.; Krainc, D. Cell biology of Parkinson’s disease: Mechanisms of synaptic, lysosomal, and mitochondrial dysfunction. Curr. Opin. Neurobiol. 2024, 85, 102841. [Google Scholar] [CrossRef]
- Dos Santos, J.C.C.; Mano, G.B.C.; da Cunha Barreto-Vianna, A.R.; Garcia, T.F.M.; de Vasconcelos, A.V.; Sá, C.S.G.; de Souza Santana, S.L.; Farias, A.G.P.; Seimaru, B.; Lima, M.P.P.; et al. The Molecular Impact of Glucosylceramidase Beta 1 (Gba1) in Parkinson’s Disease: A New Genetic State of the Art. Mol. Neurobiol. 2024. [Google Scholar] [CrossRef]
- Furderer, M.L.; Berhe, B.; Chen, T.C.; Wincovitch, S.; Jiang, X.; Tayebi, N.; Sidransky, E.; Han, T.U. A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease. Int. J. Mol. Sci. 2024, 25, 1827. [Google Scholar] [CrossRef] [PubMed]
- Nithianandam, V.; Sarkar, S.; Feany, M.B. Pathways controlling neurotoxicity and proteostasis in mitochondrial complex I deficiency. Hum. Mol. Genet. 2024. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.V.; Campbell, P.; Schapira, A.H.V.; Morris, H.R.; Taanman, J.W. The PINK1-Parkin mitophagy signalling pathway is not functional in peripheral blood mononuclear cells. PLoS ONE 2021, 16, e0259903. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.; Wolozin, B. Pathological proteins in Parkinson’s disease: Focus on the proteasome. J. Mol. Neurosci. 2004, 24, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, K.; Gan-Or, Z. Autophagy lysosomal pathway dysfunction in Parkinson’s disease; evidence from human genetics. Park. Relat. Disord. 2020, 73, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Erb, M.L.; Moore, D.J. LRRK2 and the Endolysosomal System in Parkinson’s Disease. J. Park. Dis. 2020, 10, 1271–1291. [Google Scholar] [CrossRef]
- Gu, X.; Li, C.; Chen, Y.; Ou, R.; Cao, B.; Wei, Q.; Hou, Y.; Zhang, L.; Song, W.; Zhao, B.; et al. Mutation screening and burden analysis of VPS13C in Chinese patients with early-onset Parkinson’s disease. Neurobiol. Aging 2020, 94, 311.e1–311.e4. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenzi, P.; Lazzeri, G.; Ferrucci, M.; Scotto, M.; Frati, A.; Puglisi-Allegra, S.; Busceti, C.L.; Fornai, F. Is There a Place for Lewy Bodies before and beyond Alpha-Synuclein Accumulation? Provocative Issues in Need of Solid Explanations. Int. J. Mol. Sci. 2024, 25, 3929. https://doi.org/10.3390/ijms25073929
Lenzi P, Lazzeri G, Ferrucci M, Scotto M, Frati A, Puglisi-Allegra S, Busceti CL, Fornai F. Is There a Place for Lewy Bodies before and beyond Alpha-Synuclein Accumulation? Provocative Issues in Need of Solid Explanations. International Journal of Molecular Sciences. 2024; 25(7):3929. https://doi.org/10.3390/ijms25073929
Chicago/Turabian StyleLenzi, Paola, Gloria Lazzeri, Michela Ferrucci, Marco Scotto, Alessandro Frati, Stefano Puglisi-Allegra, Carla Letizia Busceti, and Francesco Fornai. 2024. "Is There a Place for Lewy Bodies before and beyond Alpha-Synuclein Accumulation? Provocative Issues in Need of Solid Explanations" International Journal of Molecular Sciences 25, no. 7: 3929. https://doi.org/10.3390/ijms25073929
APA StyleLenzi, P., Lazzeri, G., Ferrucci, M., Scotto, M., Frati, A., Puglisi-Allegra, S., Busceti, C. L., & Fornai, F. (2024). Is There a Place for Lewy Bodies before and beyond Alpha-Synuclein Accumulation? Provocative Issues in Need of Solid Explanations. International Journal of Molecular Sciences, 25(7), 3929. https://doi.org/10.3390/ijms25073929