
Protein Disulfide Isomerase 4 Is an Essential Regulator of Endothelial Function and Survival

 ,
,
Abstract
1. Introduction
2. Results
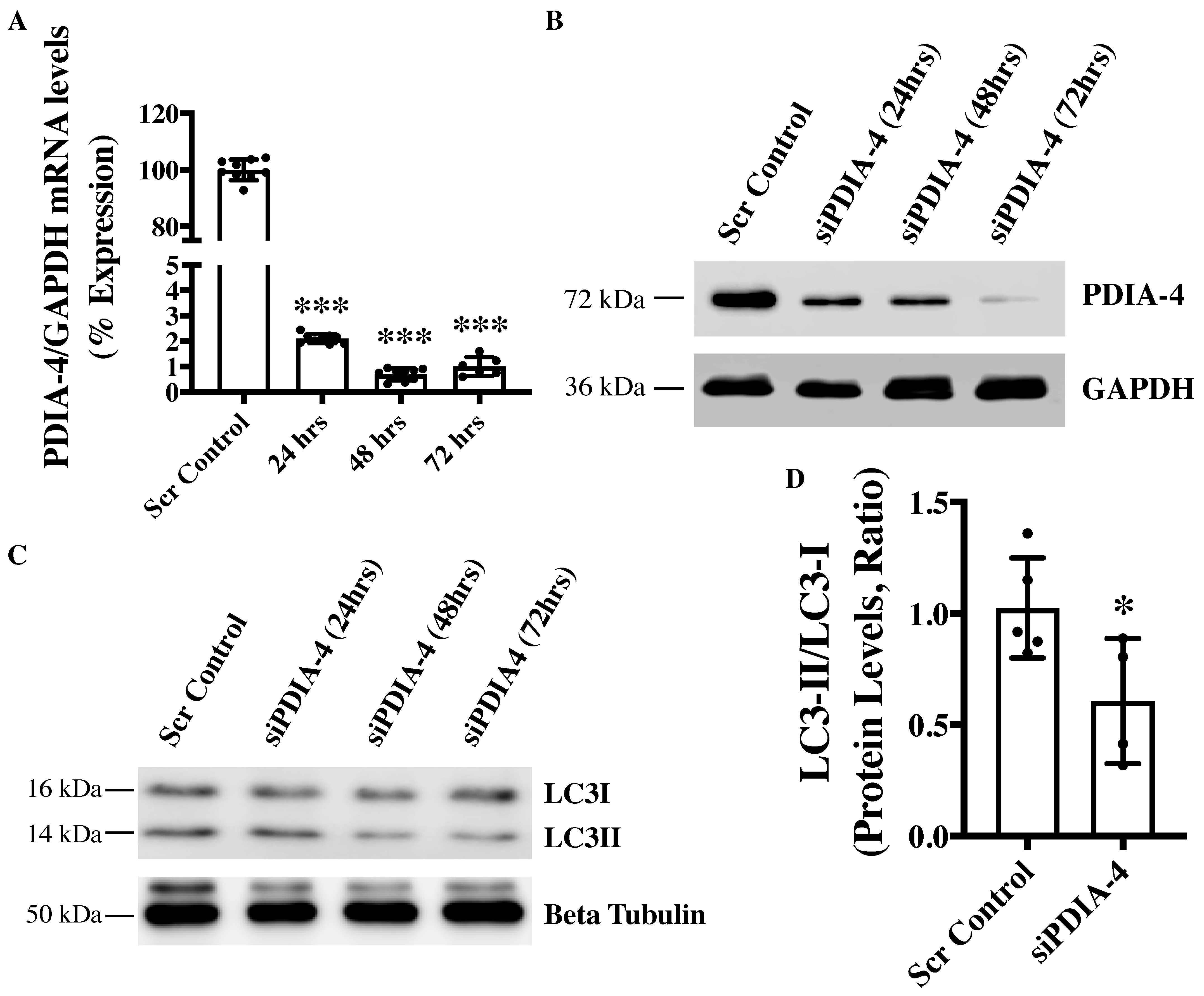
2.1. Loss of PDIA-4 Inhibits Autophagy in Endothelial Cells
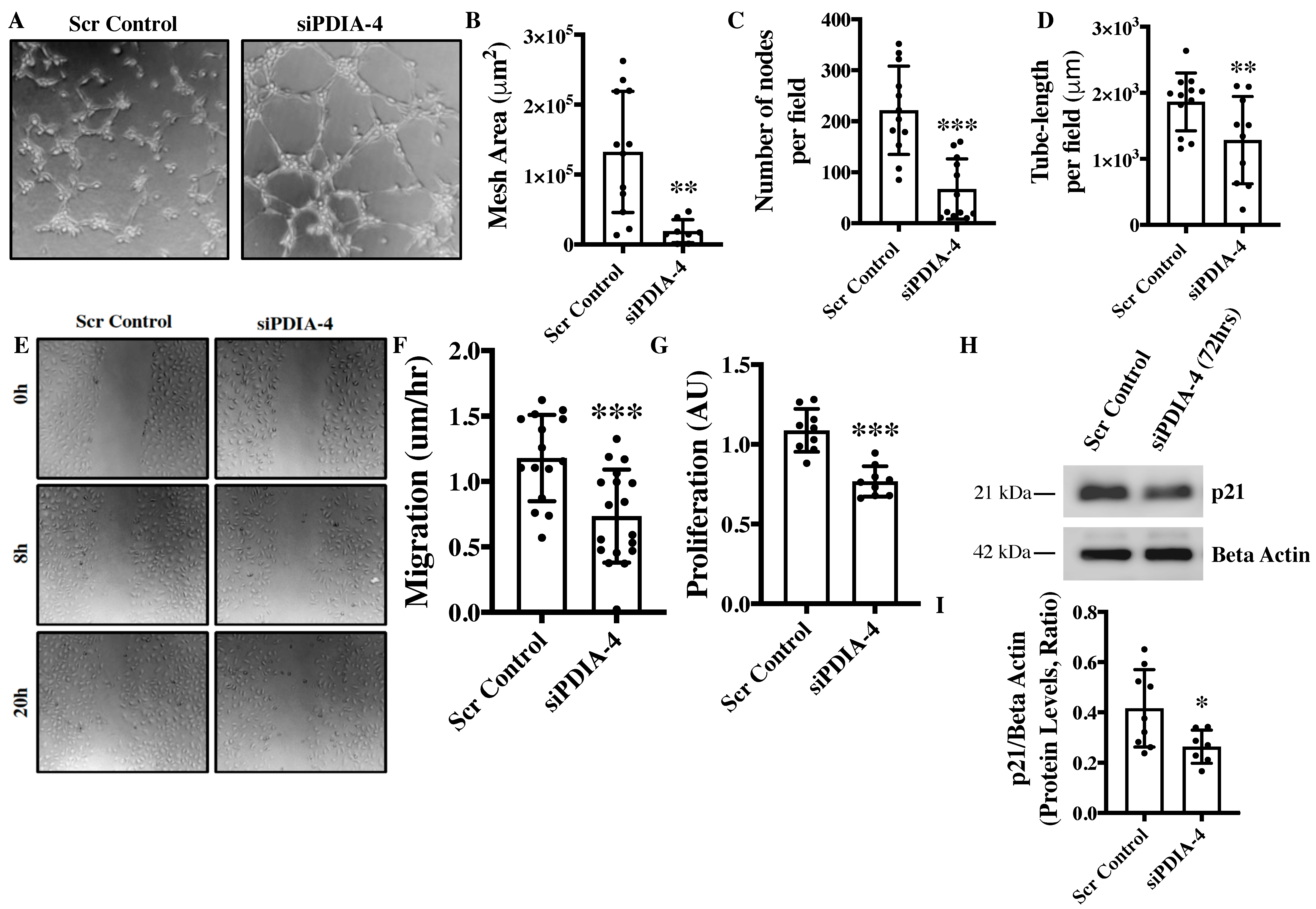
2.2. Loss of PDIA-4 Impairs Endothelial Functions
2.3. Effect of Loss of PDIA-4 on Regulators of Endothelial Functions
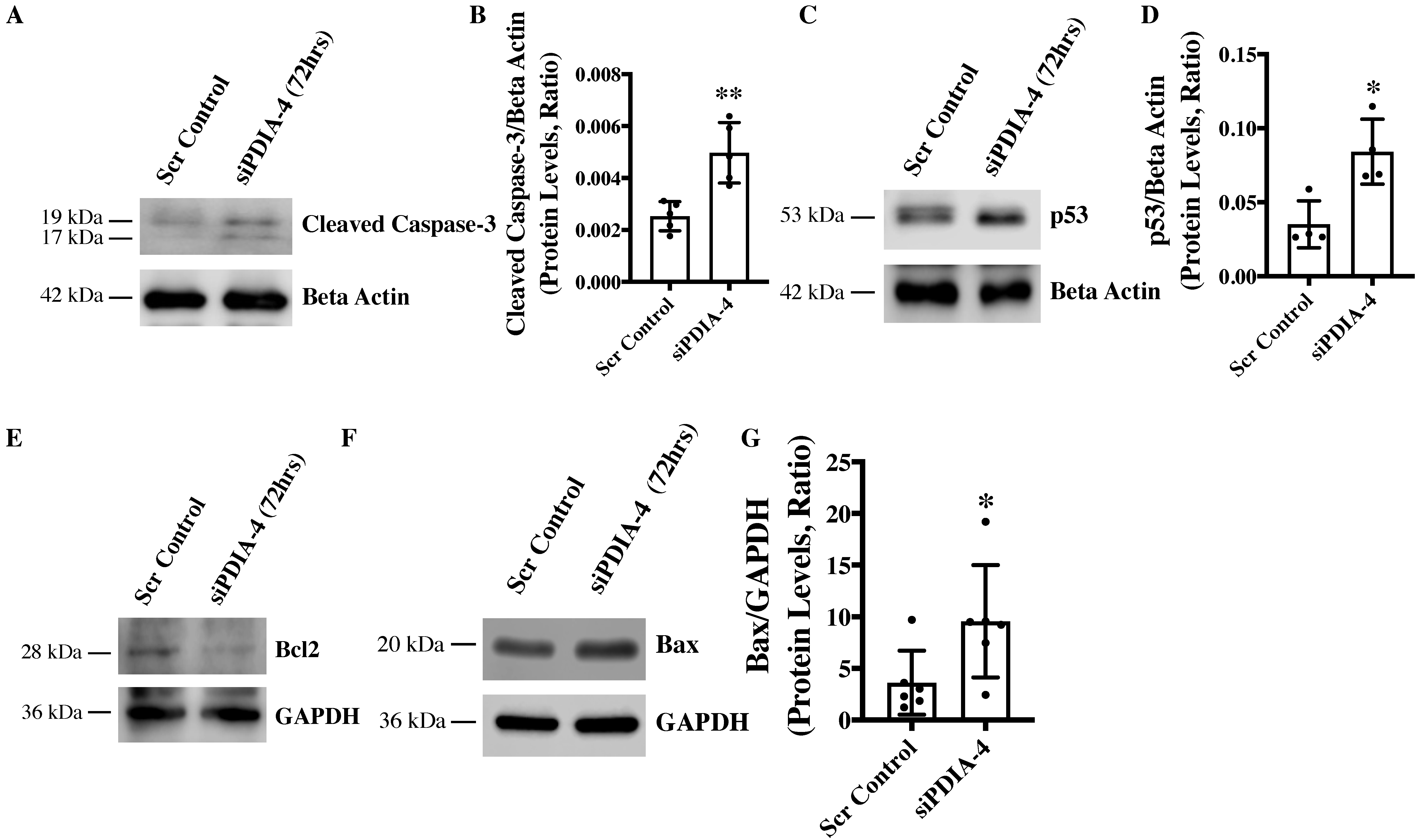
2.4. Loss of PDIA-4 Induced p53-Mediated Apoptosis in Endothelial Cells
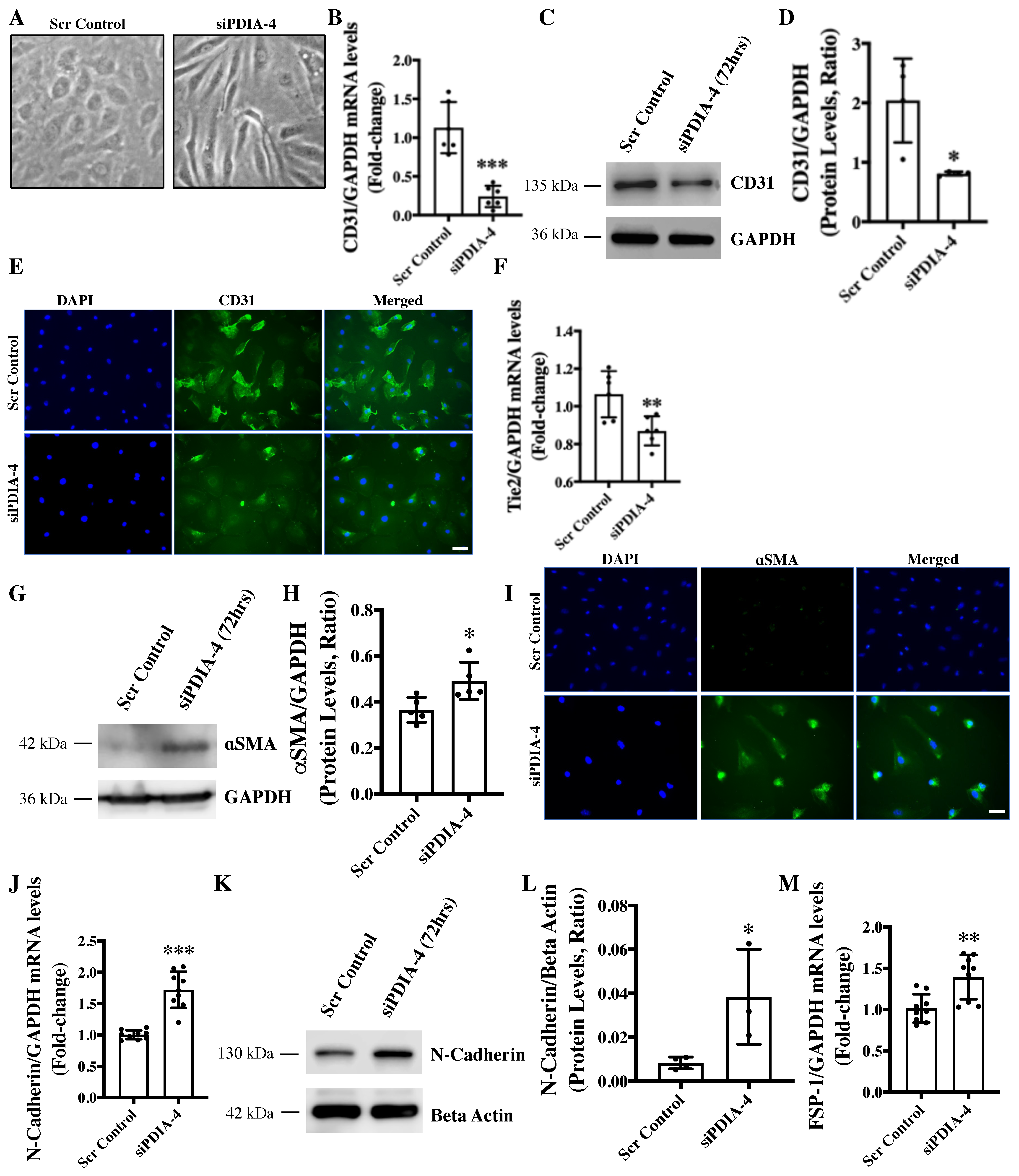
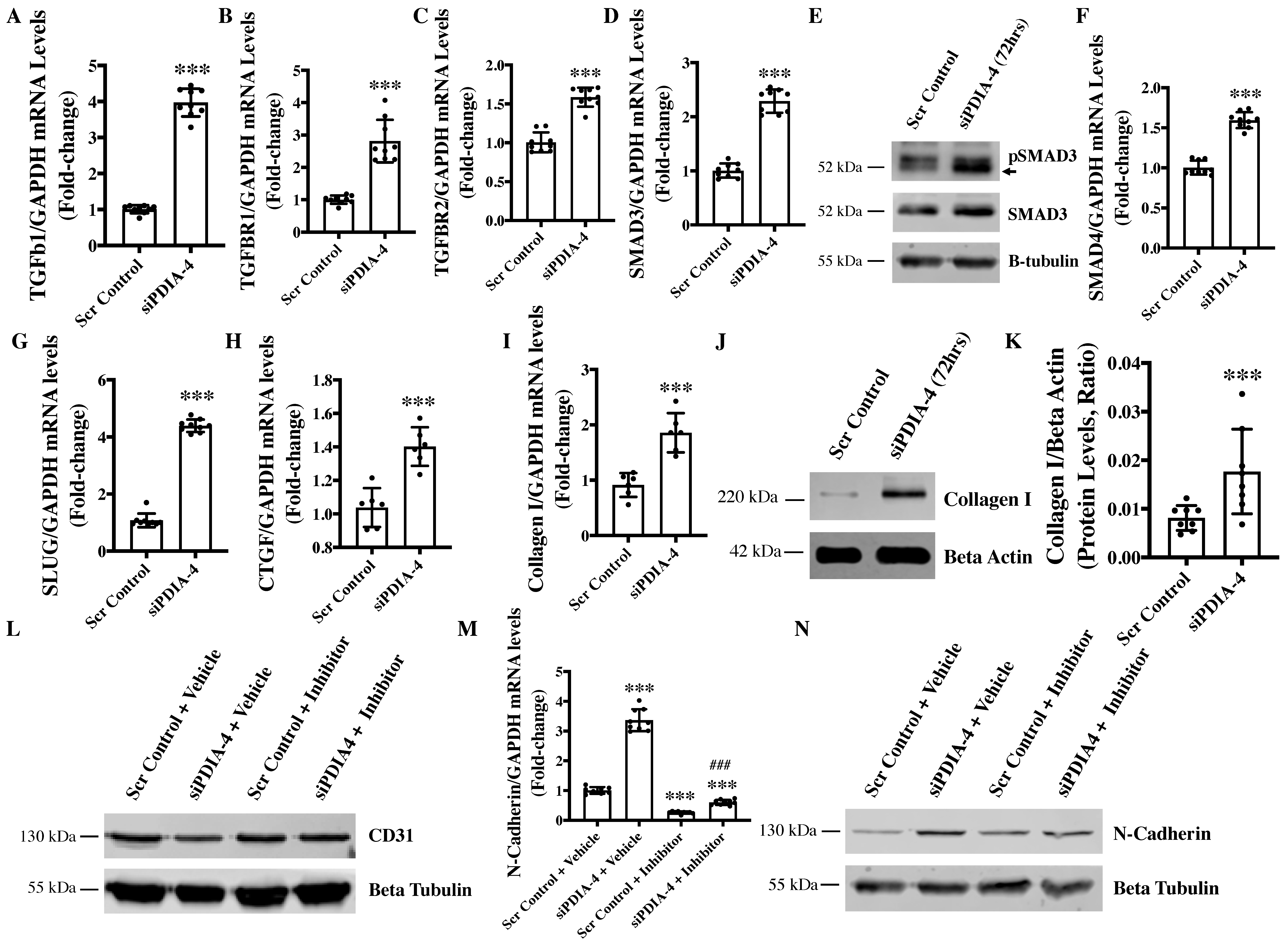
2.5. Loss of PDIA-4 Induced EndMT in Endothelial Cells
2.6. Loss of PDIA-4 Induced EndMT Is Mediated by the TGFβ Pathway
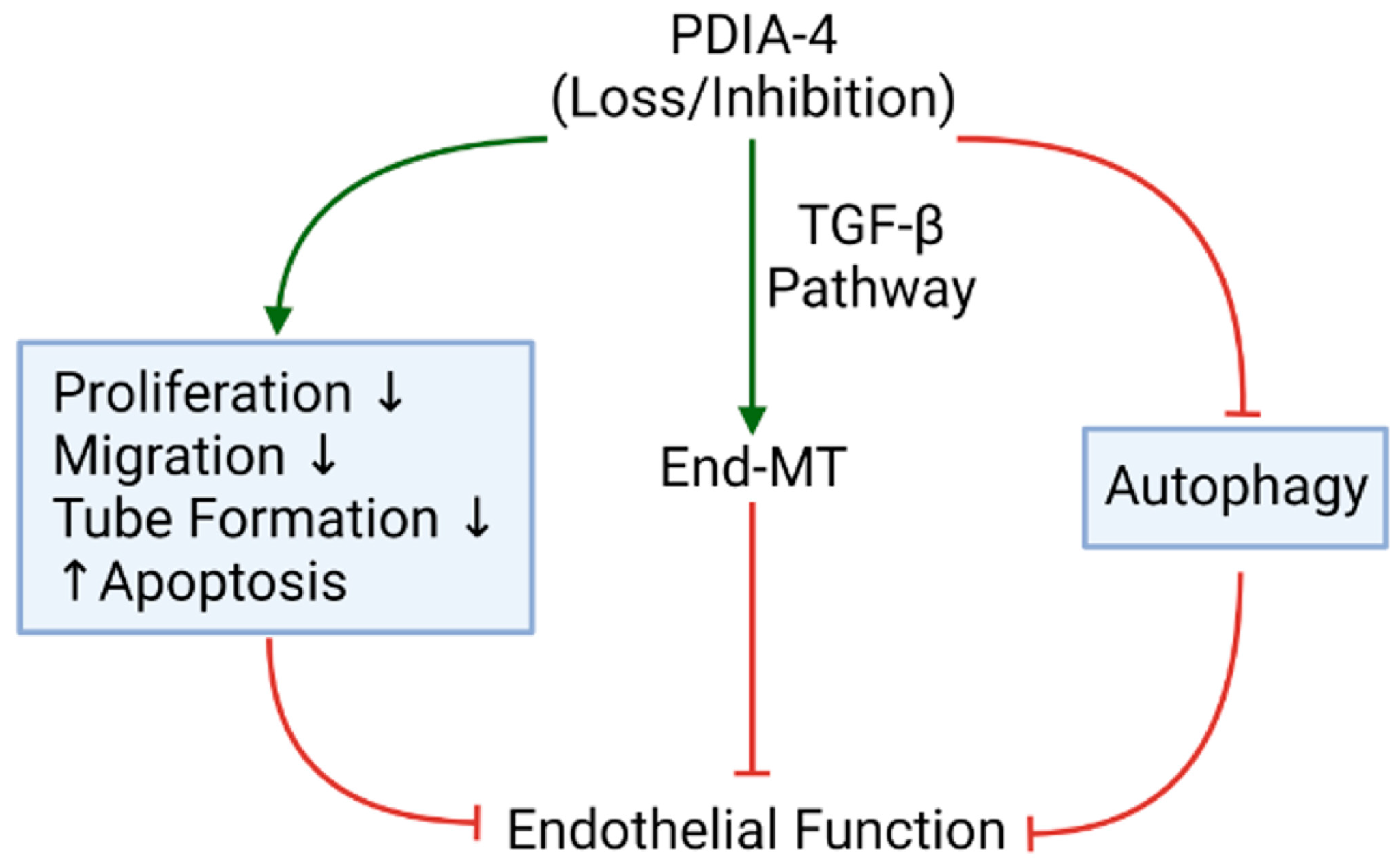
3. Discussion
4. Materials and Methods
4.1. cDNA Synthesis and Real-Time Quantitative PCR
4.2. Western Blotting
4.3. Proliferation Assay
4.4. Migration Assay
4.5. Angiogenesis Assay
4.6. Immunofluorescence
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Blood Vessels and Endothelial Cells. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The Role of Autophagy in Vascular Biology. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef]
- Bu, S.; Singh, K.K. Epigenetic Regulation of Autophagy in Cardiovascular Pathobiology. Int. J. Mol. Sci. 2021, 22, 6544. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Yanagawa, B.; Quan, A.; Wang, R.; Garg, A.; Khan, R.; Pan, Y.; Wheatcroft, M.D.; Lovren, F.; Teoh, H.; et al. Autophagy Gene Fingerprint in Human Ischemia and Reperfusion. J. Thorac. Cardiovasc. Surg. 2014, 147, 1065–1072.e1. [Google Scholar] [CrossRef]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact Endothelial Autophagy Is Required to Maintain Vascular Lipid Homeostasis. Aging Cell 2016, 15, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Bu, S.; Nguyen, H.C.; Michels, D.C.R.; Rasheed, B.; Nikfarjam, S.; Singh, R.; Wang, L.; Patel, D.A.; Singh, S.; Qadura, M.; et al. Transcriptomics of Angiotensin II-Induced Long Noncoding and Coding RNAs in Endothelial Cells. J. Hypertens. 2022, 40, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The Essential Autophagy Gene ATG7 Modulates Organ Fibrosis via Regulation of Endothelial-to-Mesenchymal Transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Adam, M.; Matkar, P.N.; Bugyei-Twum, A.; Desjardins, J.-F.; Chen, H.H.; Nguyen, H.; Bazinet, H.; Michels, D.; Liu, Z.; et al. Endothelial-Specific Loss of IFT88 Promotes Endothelial-to-Mesenchymal Transition and Exacerbates Bleomycin-Induced Pulmonary Fibrosis. Sci. Rep. 2020, 10, 4466. [Google Scholar] [CrossRef] [PubMed]
- Murugavel, S.; Bugyei-Twum, A.; Matkar, P.N.; Al-Mubarak, H.; Chen, H.H.; Adam, M.; Jain, S.; Narang, T.; Abdin, R.M.; Qadura, M.; et al. Valproic Acid Induces Endothelial-to-Mesenchymal Transition-Like Phenotypic Switching. Front. Pharmacol. 2018, 9, 737. [Google Scholar] [CrossRef]
- Bischoff, J. Endothelial to Mesenchymal Transition—Purposeful versus Maladaptive Differentiation. Circ. Res. 2019, 124, 1163–1165. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Bu, S.; Joseph, J.J.; Nguyen, H.C.; Ehsan, M.; Rasheed, B.; Singh, A.; Qadura, M.; Frisbee, J.C.; Singh, K.K. MicroRNA miR-378-3p Is a Novel Regulator of Endothelial Autophagy and Function. J. Mol. Cell. Cardiol. Plus 2023, 3, 100027. [Google Scholar] [CrossRef]
- Benham, A.M. The Protein Disulfide Isomerase Family: Key Players in Health and Disease. Antioxid. Redox Signal 2012, 16, 781–789. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Cheng, Q. PDIA4: The Basic Characteristics, Functions and Its Potential Connection with Cancer. Biomed. Pharmacother. 2020, 122, 109688. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic Reticulum Stress Triggers Autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [PubMed]
- Kyani, A.; Tamura, S.; Yang, S.; Shergalis, A.; Samanta, S.; Kuang, Y.; Ljungman, M.; Neamati, N. Discovery and Mechanistic Elucidation of a Class of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma. ChemMedChem 2018, 13, 164–177. [Google Scholar] [CrossRef]
- Logsdon, E.A.; Finley, S.D.; Popel, A.S.; Gabhann, F.M. A Systems Biology View of Blood Vessel Growth and Remodelling. J. Cell. Mol. Med. 2014, 18, 1491–1508. [Google Scholar] [CrossRef]
- Nguyen, H.C.; Bu, S.; Nikfarjam, S.; Rasheed, B.; Michels, D.C.R.; Singh, A.; Singh, S.; Marszal, C.; McGuire, J.J.; Feng, Q.; et al. Loss of Fatty Acid Binding Protein 3 Ameliorates Lipopolysaccharide-Induced Inflammation and Endothelial Dysfunction. J. Biol. Chem. 2023, 299, 102921. [Google Scholar] [CrossRef]
- Bu, S.; Nguyen, H.C.; Nikfarjam, S.; Michels, D.C.R.; Rasheed, B.; Maheshkumar, S.; Singh, S.; Singh, K.K. Endothelial Cell-Specific Loss of eNOS Differentially Affects Endothelial Function. PLoS ONE 2022, 17, e0274487. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Daido, S.; Yamamoto, A.; Kobayashi, R.; Yokoyama, T.; Aoki, H.; Iwado, E.; Shinojima, N.; Kondo, Y.; Kondo, S. Pivotal Role of the Cyclin-Dependent Kinase Inhibitor p21WAF1/CIP1 in Apoptosis and Autophagy. J. Biol. Chem. 2008, 283, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Fan, Y.-J.; Zong, W.-X. The Cellular Decision between Apoptosis and Autophagy. Chin. J. Cancer 2013, 32, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. P53-Dependent Apoptosis Modulates the Cytotoxicity of Anticancer Agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.; Kraft, A.S. Bax-Induced Apoptotic Cell Death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor Suppressor P53 Is a Regulator of Bcl-2 and Bax Gene Expression In Vitro and In Vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar] [PubMed]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial Functions of PECAM-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Wong, A.L.; Haroon, Z.A.; Werner, S.; Dewhirst, M.W.; Greenberg, C.S.; Peters, K.G. Tie2 Expression and Phosphorylation in Angiogenic and Quiescent Adult Tissues. Circ. Res. 1997, 81, 567–574. [Google Scholar] [CrossRef]
- Nakajima, Y.; Mironov, V.; Yamagishi, T.; Nakamura, H.; Markwald, R.R. Expression of Smooth Muscle Alpha-Actin in Mesenchymal Cells during Formation of Avian Endocardial Cushion Tissue: A Role for Transforming Growth Factor Beta3. Dev. Dyn. 1997, 209, 296–309. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and Characterization of a Fibroblast Marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-Cadherin in Cancer Metastasis, Its Emerging Role in Haematological Malignancies and Potential as a Therapeutic Target in Cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β Receptor Activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 Is a Potent and Specific Inhibitor of Transforming Growth Factor-β Superfamily Type I Activin Receptor-Like Kinase (ALK) Receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy Requires Endoplasmic Reticulum Targeting of the PI3-Kinase Complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER Stress (PERK/eIF2α Phosphorylation) Mediates the Polyglutamine-Induced LC3 Conversion, an Essential Step for Autophagy Formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.-C.; Kim, K.H.; Kim, G.H.; Kim, S.-W.; Kim, H.L.; Lee, M.-K.; et al. Autophagy Deficiency in Beta Cells Leads to Compromised Unfolded Protein Response and Progression from Obesity to Diabetes in Mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Kardideh, B.; Samimi, Z.; Norooznezhad, F.; Kiani, S.; Mansouri, K. Autophagy, Cancer and Angiogenesis: Where Is the Link? Cell Biosci. 2019, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin Causes Autophagy in Endothelial Cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef] [PubMed]
- Poluzzi, C.; Casulli, J.; Goyal, A.; Mercer, T.J.; Neill, T.; Iozzo, R.V. Endorepellin Evokes Autophagy in Endothelial Cells. J. Biol. Chem. 2014, 289, 16114–16128. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Song, Z.; Cheng, Z. High Expression of PDIA4 Promotes Malignant Cell Behavior and Predicts Reduced Survival in Cervical Cancer. Oncol. Rep. 2022, 48, 184. [Google Scholar] [CrossRef]
- Li, H.; Liu, Q.; Xiao, K.; He, Z.; Wu, C.; Sun, J.; Chen, X.; Chen, S.; Yang, J.; Ma, Q.; et al. PDIA4 Correlates with Poor Prognosis and Is a Potential Biomarker in Glioma. Onco Targets Ther. 2021, 14, 125–138. [Google Scholar] [CrossRef]
- Stojak, M.; Milczarek, M.; Kurpinska, A.; Suraj-Prazmowska, J.; Kaczara, P.; Wojnar-Lason, K.; Banach, J.; Stachowicz-Suhs, M.; Rossowska, J.; Kalviņš, I.; et al. Protein Disulphide Isomerase A1 Is Involved in the Regulation of Breast Cancer Cell Adhesion and Transmigration via Lung Microvascular Endothelial Cells. Cancers 2020, 12, 2850. [Google Scholar] [CrossRef]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J.; Davies, P.F. Chronic Endoplasmic Reticulum Stress Activates Unfolded Protein Response in Arterial Endothelium in Regions of Susceptibility to Atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef]
- Coqueret, O. New Roles for P21 and P27 Cell-Cycle Inhibitors: A Function for Each Cell Compartment? Trends Cell Biol. 2003, 13, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.-J.; Liu, Z.; ten Dijke, P. TGF-β Signaling in Vascular Biology and Dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-Mesenchymal Transition Contributes to Cardiac Fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in Kidney Fibrosis Emerge via Endothelial-to-Mesenchymal Transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed]
- Moonen, J.-R.A.J.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; van Kooten, T.G.; van Luyn, M.J.A.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-Mesenchymal Transition Contributes to Fibro-Proliferative Vascular Disease and Is Modulated by Fluid Shear Stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Nguyen, H.; Michels, D.; Bazinet, H.; Matkar, P.N.; Liu, Z.; Esene, L.; Adam, M.; Bugyei-Twum, A.; Mebrahtu, E.; et al. BReast CAncer Susceptibility Gene 2 Deficiency Exacerbates Oxidized LDL-Induced DNA Damage and Endothelial Apoptosis. Physiol. Rep. 2020, 8, e14481. [Google Scholar] [CrossRef]
- Singh, S.; Nguyen, H.C.; Ehsan, M.; Michels, D.C.R.; Singh, P.; Qadura, M.; Singh, K.K. Pravastatin-Induced Changes in Expression of Long Non-Coding and Coding RNAs in Endothelial Cells. Physiol. Rep. 2021, 9, e14661. [Google Scholar] [CrossRef]
- Yuan, J.S.; Reed, A.; Chen, F.; Stewart, C.N. Statistical Analysis of Real-Time PCR Data. BMC Bioinform. 2006, 7, 85. [Google Scholar] [CrossRef]
- Protocol Guide: WST-1 Assay for Cell Proliferation and Viability. Available online: https://www.sigmaaldrich.com/CA/en/technical-documents/protocol/cell-culture-and-cell-culture-analysis/cell-counting-and-health-analysis/cell-proliferation-reagent-wst-1 (accessed on 24 January 2023).
- Jonkman, J.E.N.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An Introduction to the Wound Healing Assay Using Live-Cell Microscopy. Cell Adh. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Arnedo, A.; Torres Figueroa, F.; Clavijo, C.; Arbeláez, P.; Cruz, J.C.; Muñoz-Camargo, C. An Image J Plugin for the High Throughput Image Analysis of in Vitro Scratch Wound Healing Assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequences (5′-3′) |
|---|---|
| Hsa-PDIA-4-F | 5′-CACGCTTGTGTTGACCAAAGA-3′ |
| Hsa-PDIA-4-R | 5′-AATTGGAGGAGAACGCTTGCT-3′ |
| Hsa-TGFB1-F | 5′-TACAGCACGGTATGCAAGCC-3′ |
| Hsa-TGFB1-R | 5′-GCAACCGATCTAGCTCACAGAG-3r′ |
| Hsa-SMAD3-F | 5′-TGGACGCAGGTTCTCCAAAC-3′ |
| Hsa-SMAD3-R | 5′-CCGGCTC GCAGTAGGAAC-3′ |
| Hsa-SMAD4-F | 5′-CTCATGTGATCTATGCCCGTC-3′ |
| Hsa-SMAD4-R | 5′-AGGTGATACAACTCGTTCGTAGT-3′ |
| Hsa-CD31-F | 5′-TCCTGAGGGTCAAGGTAATAGC-3′ |
| Hsa-CD31-R | 5′-CTCCAGACTGTACATCGTTACCC-3′ |
| Hsa-VE-Cadherin-F | 5′-GAGTCTTCAGCTCACGGACA-3′ |
| Hsa-VE-Cadherin-R | 5′-CAGCTCTGTGAGCCTCTGC-3′ |
| Hsa-SLUG-F | 5′-ATCTGCGCiCAAGGCGTTTTCCA-3′ |
| Hsa-SLUG-R | 5′-GAGCCCTCAGATTTGACCTGTC-3′ |
| Hsa-TGFBR1-F | 5′-TACCTGAACCCGTGTTGCTCTC-3′ |
| Hsa-TGFBR1-R | 5′-GTTGCTGAGGTATCGCCAGGAA-3′ |
| Hsa-TGFBR2-F | 5′-GTCTGTGGATGACCTGGCTAAC-3′ |
| Hsa-TGFBR2-R | 5′-GACATCGGTCTGCTTGAAGGAC-3′ |
| Hsa-FSP1-F | 5′-TCTTGGTCTGGTCTCAACGG-5′ |
| Hsa-FSP1-R | 5′-TGTCACCCTCTTTGCCTGAG-3′ |
| Hsa-N-Cadherin-F | 5′-CCTCCAGAGTTTACTGCCATGAC-3′ |
| Hsa-N-Cadherin-R | 5′-GTAGGATCTCCGCCACTGATTC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bu, S.; Singh, A.; Nguyen, H.C.; Peddi, B.; Bhatt, K.; Ravendranathan, N.; Frisbee, J.C.; Singh, K.K. Protein Disulfide Isomerase 4 Is an Essential Regulator of Endothelial Function and Survival. Int. J. Mol. Sci. 2024, 25, 3913. https://doi.org/10.3390/ijms25073913
Bu S, Singh A, Nguyen HC, Peddi B, Bhatt K, Ravendranathan N, Frisbee JC, Singh KK. Protein Disulfide Isomerase 4 Is an Essential Regulator of Endothelial Function and Survival. International Journal of Molecular Sciences. 2024; 25(7):3913. https://doi.org/10.3390/ijms25073913
Chicago/Turabian StyleBu, Shuhan, Aman Singh, Hien C. Nguyen, Bharatsinai Peddi, Kriti Bhatt, Naresh Ravendranathan, Jefferson C. Frisbee, and Krishna K. Singh. 2024. "Protein Disulfide Isomerase 4 Is an Essential Regulator of Endothelial Function and Survival" International Journal of Molecular Sciences 25, no. 7: 3913. https://doi.org/10.3390/ijms25073913
APA StyleBu, S., Singh, A., Nguyen, H. C., Peddi, B., Bhatt, K., Ravendranathan, N., Frisbee, J. C., & Singh, K. K. (2024). Protein Disulfide Isomerase 4 Is an Essential Regulator of Endothelial Function and Survival. International Journal of Molecular Sciences, 25(7), 3913. https://doi.org/10.3390/ijms25073913