
Glucagon-like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide, and Glucagon Receptor Agonists in Metabolic Dysfunction-Associated Steatotic Liver Disease: Novel Medication in New Liver Disease Nomenclature

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Incretin Hormones
2.1. Glucagon-like Peptide-1 (GLP-1) Agonism
2.2. Glucose-Dependent Insulinotropic Polypeptide (GIP) Agonism
2.3. Glucagon (GCG) Agonism
Literature Search
3. The Beneficial Effect of Incretin Agonists on Aspects of MASLD/MASH; Evidence from Mouse Studies
3.1. GLP-1 Signaling
3.1.1. Contribution of GIP to GLP-1 Signaling
3.1.2. Contribution of GCG to GLP-1 Signaling
4. GLP-1 in MASLD and MASH: Randomized Clinical Trials
4.1. Liraglutide
4.2. Exenatide
4.3. Dulaglutide
4.4. Semaglutide
5. Combined Incretin Receptor Agonism for MASLD and MASLD-Related Complications
6. Closing Remarks and Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Quek, J.; Chan, K.E.; Wong, Z.Y.; Tan, C.; Tan, B.; Lim, W.H.; Tan, D.J.H.; Tang, A.S.P.; Tay, P.; Xiao, J.; et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2023, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Chrysavgis, L.; Vachliotis, I.D.; Chartampilas, E.; Cholongitas, E. Nonalcoholic fatty liver disease and hepatocellular carcinoma:Insights in epidemiology, pathogenesis, imaging, prevention and therapy. Semin. Cancer Biol. 2023, 93, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Chrysavgis, L.; Giannakodimos, I.; Diamantopoulou, P.; Cholongitas, E. Non-alcoholic fatty liver disease and hepatocellular carcinoma: Clinical challenges of an intriguing link. World J. Gastroenterol. 2022, 28, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.-U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Rinella, M.E.; Neuschwander-Tetri, B.A.; Siddiqui, M.S.; Abdelmalek, M.F.; Caldwell, S.; Barb, D.; Kleiner, D.E.; Loomba, R. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology 2023, 77, 1797–1835. [Google Scholar] [CrossRef]
- Eslam, M.; Sarin, S.K.; Wong, V.W.-S.; Fan, J.-G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.-H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 889–919. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.-F.; Anstee, Q.M.; Bugianesi, E.; Harrison, S.; Loomba, R.; Paradis, V.; Tilg, H.; Wong, V.W.-S.; Zelber-Sagi, S. Current therapies and new developments in NASH. Gut 2022, 71, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Osna, N.A.; Kharbanda, K.K. Treatment options for alcoholic and non-alcoholic fatty liver disease: A review. World J. Gastroenterol. 2017, 23, 6549–6570. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. S1), 5–21. [Google Scholar] [CrossRef]
- Campbell, J.E.; Müller, T.D.; Finan, B.; DiMarchi, R.D.; Tschöp, M.H.; D’Alessio, D.A. GIPR/GLP-1R dual agonist therapies for diabetes and weight loss-chemistry, physiology, and clinical applications. Cell Metab. 2023, 35, 1519–1529. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016, 4, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Hinnen, D. Glucagon-Like Peptide 1 Receptor Agonists for Type 2 Diabetes. Diabetes Spectr. 2017, 30, 202–210. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- Chow, E.; Chan, J.C.N. The emerging role of incretins and twincretins. Nat. Rev. Endocrinol. 2022, 18, 73–74. [Google Scholar] [CrossRef]
- Frias, J.P. Tirzepatide: A glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes. Expert Rev. Endocrinol. Metab. 2020, 15, 379–394. [Google Scholar] [CrossRef]
- Karagiannis, T.; Avgerinos, I.; Liakos, A.; Del Prato, S.; Matthews, D.R.; Tsapas, A.; Bekiari, E. Management of type 2 diabetes with the dual GIP/GLP-1 receptor agonist tirzepatide: A systematic review and meta-analysis. Diabetologia 2022, 65, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Sofogianni, A.; Filippidis, A.; Chrysavgis, L.; Tziomalos, K.; Cholongitas, E. Glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: An update. World J. Hepatol. 2020, 12, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Targher, G. Tirzepatide adds hepatoprotection to its armoury. Lancet Diabetes Endocrinol. 2022, 10, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.A.; D’Alessio, D.A. Physiology of proglucagon peptides: Role of glucagon and GLP-1 in health and disease. Physiol. Rev. 2015, 95, 513–548. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef] [PubMed]
- Yabut, J.M.; Drucker, D.J. Glucagon-like Peptide-1 Receptor-based Therapeutics for Metabolic Liver Disease. Endocr. Rev. 2023, 44, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 721135. [Google Scholar] [CrossRef] [PubMed]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Ørskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef]
- Laurindo, L.F.; Barbalho, S.M.; Guiguer, E.L.; Souza, M.d.S.S.d.; de Souza, G.A.; Fidalgo, T.M.; Araújo, A.C.; Gonzaga, H.F.d.S.; Teixeira, D.d.B.; Ullmann, T.d.O.S.; et al. GLP-1a: Going beyond Traditional Use. Int. J. Mol. Sci. 2022, 23, 739. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef] [PubMed]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef] [PubMed]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 2020, 5, e133429. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Maeda, M.; Matsumura, M.; Shimizu, R.; Banba, N.; Aso, Y.; Yasu, T.; Harasawa, H. Effect of GLP-1 receptor agonist on gastrointestinal tract motility and residue rates as evaluated by capsule endoscopy. Diabetes Metab. 2017, 43, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Gutniak, M.; Ørkov, C.; Holst, J.J.; Ahrén, B.; Efendić, S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Creutzfeldt, W.O.C.; Kleine, N.; Willms, B.; Ørskov, C.; Holst, J.J.; Nauck, M.A. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon-like peptide I(7-36) amide in type I diabetic patients. Diabetes Care 1996, 19, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Bifari, F.; Manfrini, R.; Cas, M.D.; Berra, C.; Siano, M.; Zuin, M.; Paroni, R.; Folli, F. Multiple target tissue effects of GLP-1 analogues on non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Pharmacol. Res. 2018, 137, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Mantovani, A.; Byrne, C.D. Mechanisms and possible hepatoprotective effects of glucagon-like peptide-1 receptor agonists and other incretin receptor agonists in non-alcoholic fatty liver disease. Lancet Gastroenterol. Hepatol. 2023, 8, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Muzurović, E.M.; Volčanšek, Š.; Tomšić, K.Z.; Janež, A.; Mikhailidis, D.P.; Rizzo, M.; Mantzoros, C.S. Glucagon-Like Peptide-1 Receptor Agonists and Dual Glucose-Dependent Insulinotropic Polypeptide/Glucagon-Like Peptide-1 Receptor Agonists in the Treatment of Obesity/Metabolic Syndrome, Prediabetes/Diabetes and Non-Alcoholic Fatty Liver Disease-Current Evidence. J. Cardiovasc. Pharmacol. Ther. 2022, 27, 10742484221146371. [Google Scholar]
- Lindquist, P.; Gasbjerg, L.S.; Mokrosinski, J.; Holst, J.J.; Hauser, A.S.; Rosenkilde, M.M. The Location of Missense Variants in the Human GIP Gene Is Indicative for Natural Selection. Front. Endocrinol. 2022, 13, 891586. [Google Scholar] [CrossRef]
- Yip, R.G.C.; Boylan, M.O.; Kieffer, T.J.; Wolfe, M.M. Functional GIP receptors are present on adipocytes. Endocrinology 1998, 139, 4004–4007. [Google Scholar] [CrossRef] [PubMed]
- Bollag, R.J.; Zhong, Q.; Phillips, P.; Min, L.; Zhong, L.; Cameron, R.; Mulloy, A.L.; Rasmussen, H.; Qin, F.; Ding, K.H.; et al. Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology 2000, 141, 1228–1235. [Google Scholar] [CrossRef]
- Harada, N.; Inagaki, N. Role of GIP receptor signaling in beta-cell survival. Diabetol. Int. 2017, 8, 137–138. [Google Scholar] [CrossRef]
- Christensen, M.; Calanna, S.; Sparre-Ulrich, A.H.; Kristensen, P.L.; Rosenkilde, M.M.; Faber, J.; Purrello, F.; van Hall, G.; Holst, J.J.; Vilsbøll, T.; et al. Glucose-dependent insulinotropic polypeptide augments glucagon responses to hypoglycemia in type 1 diabetes. Diabetes 2015, 64, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Gallwitz, B.; Siepmann, N.; Holst, J.J.; Deacon, C.F.; Schmidt, W.E.; Nauck, M.A. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003, 46, 798–801. [Google Scholar] [CrossRef]
- Michałowska, J.; Miller-Kasprzak, E.; Bogdański, P. Incretin Hormones in Obesity and Related Cardiometabolic Disorders: The Clinical Perspective. Nutrients 2021, 13, 351. [Google Scholar] [CrossRef]
- Samms, R.J.; Coghlan, M.P.; Sloop, K.W. How May GIP Enhance the Therapeutic Efficacy of GLP-1? Trends Endocrinol. Metab. 2020, 31, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Pacini, G.; De Michieli, F.; Cassader, M. Prolonged saturated fat-induced, glucose-dependent insulinotropic polypeptide elevation is associated with adipokine imbalance and liver injury in nonalcoholic steatohepatitis: Dysregulated enteroadipocyte axis as a novel feature of fatty liver. Am. J. Clin. Nutr. 2009, 89, 558–567. [Google Scholar] [CrossRef]
- Staub, A.; Sinn, L.; Behrens, O.K. Purification and crystallization of glucagon. J. Biol. Chem. 1955, 214, 619–632. [Google Scholar] [CrossRef]
- Bromer, W.W.; Sinn, L.G.; Staub, A.; Behrens, O.K. The amino acid sequence of glucagon. Diabetes 1957, 6, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [PubMed]
- Müller, T.D.; Finan, B.; Clemmensen, C.; DiMarchi, R.D.; Tschöp, M.H. The New Biology and Pharmacology of Glucagon. Physiol. Rev. 2017, 97, 721–766. [Google Scholar] [CrossRef]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, M.; Sachs, S.; Habegger, K.M.; Hofmann, S.M.; Müller, T.D. Glucagon Regulation of Energy Expenditure. Int. J. Mol. Sci. 2019, 20, 5407. [Google Scholar] [CrossRef]
- Authier, F.; Desbuquois, B. Glucagon receptors. Cell. Mol. Life Sci. 2008, 65, 1880–1899. [Google Scholar] [CrossRef] [PubMed]
- Al-Massadi, O.; Fernø, J.; Diéguez, C.; Nogueiras, R.; Quiñones, M. Glucagon Control on Food Intake and Energy Balance. Int. J. Mol. Sci. 2019, 20, 3905. [Google Scholar] [CrossRef] [PubMed]
- Geary, N.; Kissileff, H.R.; Pi-Sunyer, F.X.; Hinton, V. Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men. Am. J. Physiol. 1992, 262 Pt 2, R975–R980. [Google Scholar] [CrossRef] [PubMed]
- Geary, N. Pancreatic glucagon signals postprandial satiety. Neurosci. Biobehav. Rev. 1990, 14, 323–338. [Google Scholar] [CrossRef]
- Svendsen, B.; Larsen, O.; Gabe, M.B.N.; Christiansen, C.B.; Rosenkilde, M.M.; Drucker, D.J.; Holst, J.J. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep. 2018, 25, 1127–1134.e2. [Google Scholar] [CrossRef]
- Capozzi, M.E.; Wait, J.B.; Koech, J.; Gordon, A.N.; Coch, R.W.; Svendsen, B.; Finan, B.; D’Alessio, D.A.; Campbell, J.E. Glucagon lowers glycemia when beta-cells are active. JCI Insight 2019, 4, e129954. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Albrechtsen, N.J.W. Glucagon Receptor Signaling and Lipid Metabolism. Front. Physiol. 2019, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D.; Pedersen, J.; Kjeldsen, S.A.; Winther-Sørensen, M.; Stojanovska, E.; Vilstrup, H.; Ørskov, C.; Albrechtsen, N.J.W.; Holst, J.J. Glucagon receptor signaling is not required for N-carbamoyl glutamate- and l-citrulline-induced ureagenesis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G912–G927. [Google Scholar] [CrossRef] [PubMed]
- Suppli, M.P.; Bagger, J.I.; Lund, A.; Demant, M.; van Hall, G.; Strandberg, C.; Kønig, M.J.; Rigbolt, K.; Langhoff, J.L.; Albrechtsen, N.J.W.; et al. Glucagon Resistance at the Level of Amino Acid Turnover in Obese Subjects With Hepatic Steatosis. Diabetes 2020, 69, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Albrechtsen, N.J.W.; Pedersen, J.; Knop, F.K. Glucagon and Amino Acids Are Linked in a Mutual Feedback Cycle: The Liver-alpha-Cell Axis. Diabetes 2017, 66, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Nevola, R.; Epifani, R.; Imbriani, S.; Tortorella, G.; Aprea, C.; Galiero, R.; Rinaldi, L.; Marfella, R.; Sasso, F.C. GLP-1 Receptor Agonists in Non-Alcoholic Fatty Liver Disease: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 1703. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Chen, S.; Chen, X.; Ren, Q.; Yue, L.; Pan, X.; Zhao, H.; Li, Z.; Chen, X. Semaglutide ameliorates metabolism and hepatic outcomes in an NAFLD mouse model. Front. Endocrinol. 2022, 13, 1046130. [Google Scholar] [CrossRef]
- Møllerhøj, M.B.; Veidal, S.S.; Thrane, K.T.; Oró, D.; Overgaard, A.; Salinas, C.G.; Madsen, M.R.; Pfisterer, L.; Vyberg, M.; Simon, E.; et al. Hepatoprotective effects of semaglutide, lanifibranor and dietary intervention in the GAN diet-induced obese and biopsy-confirmed mouse model of NASH. Clin. Transl. Sci. 2022, 15, 1167–1186. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, M.; Ren, H.; Hu, H.; Boden, G.; Li, L.; Yang, G. GLP-1 analogue prevents NAFLD in ApoE KO mice with diet and Acrp30 knockdown by inhibiting c-JNK. Liver Int. 2013, 33, 794–804. [Google Scholar] [CrossRef]
- Chiu, H.-Y.; Tsai, S.-C.; Tsai, F.-J.; Lo, Y.-H.; Cheng, C.-C.; Liu, T.-Y.; Jhan, S.-R.; Yang, J.-S.; Chiu, Y.-J. Liraglutide With Metformin Therapy Ameliorates Hepatic Steatosis and Liver Injury in a Mouse Model of Non-alcoholic Steatohepatitis. In Vivo 2023, 37, 1037–1046. [Google Scholar] [CrossRef]
- Shiraishi, D.; Fujiwara, Y.; Komohara, Y.; Mizuta, H.; Takeya, M. Glucagon-like peptide-1 (GLP-1) induces M2 polarization of human macrophages via STAT3 activation. Biochem. Biophys. Res. Commun. 2012, 425, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Heimbürger, S.M.N.; Hoe, B.; Nielsen, C.N.; Bergman, N.C.; Skov-Jeppesen, K.; Hartmann, B.; Holst, J.J.; Dela, F.; Overgaard, J.; Størling, J.; et al. GIP affects hepatic fat and brown adipose tissue thermogenesis but not white adipose tissue transcriptome in type 1 diabetes. J. Clin. Endocrinol. Metab. 2022, 107, 3261–3274. [Google Scholar] [CrossRef]
- Thondam, S.K.; Cuthbertson, D.J.; Wilding, J.P. The influence of Glucose-dependent Insulinotropic Polypeptide (GIP) on human adipose tissue and fat metabolism: Implications for obesity, type 2 diabetes and Non-Alcoholic Fatty Liver Disease (NAFLD). Peptides 2020, 125, 170208. [Google Scholar] [CrossRef] [PubMed]
- Sachs, S.; Niu, L.; Geyer, P.; Jall, S.; Kleinert, M.; Feuchtinger, A.; Stemmer, K.; Brielmeier, M.; Finan, B.; DiMarchi, R.D.; et al. Plasma proteome profiles treatment efficacy of incretin dual agonism in diet-induced obese female and male mice. Diabetes Obes. Metab. 2021, 23, 195–207. [Google Scholar] [CrossRef]
- Kayed, A.; Melander, S.A.; Khan, S.; Andreassen, K.V.; Karsdal, M.A.; Henriksen, K. The Effects of Dual GLP-1/Glucagon Receptor Agonists with Different Receptor Selectivity in Mouse Models of Obesity and Nonalcoholic Steatohepatitis. J. Pharmacol. Exp. Ther. 2023, 384, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; van Eenige, R.; Ge, X.; van Marwijk, C.; Lambooij, J.M.; Guigas, B.; Giera, M.; de Boer, J.F.; Coskun, T.; Qu, H.; et al. Combined GIP receptor and GLP1 receptor agonism attenuates NAFLD in male APOE∗ 3-Leiden. CETP mice. eBioMedicine 2023, 93, 104684. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Lu, W.; Wang, Y.; Qian, P.; Tian, H.; Gao, X.; Yao, W. An oral GLP-1 and GIP dual receptor agonist improves metabolic disorders in high fat-fed mice. Eur. J. Pharmacol. 2022, 914, 174635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Delessa, C.T.; Wolfrum, C. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 2021, 33, 833–844.e5. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, A.E.; Biggs, E.K.; Darwish, T.; Tadross, J.; Sukthankar, T.; Girish, M.; Polex-Wolf, J.; Lam, B.Y.; Zvetkova, I.; Pan, W.; et al. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell Metab. 2019, 30, 987–996.e6. [Google Scholar] [CrossRef]
- Borner, T.; Geisler, C.E.; Fortin, S.M.; Cosgrove, R.; Alsina-Fernandez, J.; Dogra, M.; Doebley, S.; Sanchez-Navarro, M.J.; Leon, R.M.; Gaisinsky, J.; et al. GIP Receptor Agonism Attenuates GLP-1 Receptor Agonist-Induced Nausea and Emesis in Preclinical Models. Diabetes 2021, 70, 2545–2553. [Google Scholar] [CrossRef]
- Marcondes-De-Castro, I.A.; Oliveira, T.F.; Spezani, R.; Marinho, T.S.; Cardoso, L.E.M.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Cotadutide effect in liver and adipose tissue in obese mice. J. Mol. Endocrinol. 2023, 70, e220168. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465. [Google Scholar] [CrossRef]
- Boland, M.L.; Laker, R.C.; Mather, K.; Nawrocki, A.; Oldham, S.; Boland, B.B.; Lewis, H.; Conway, J.; Naylor, J.; Guionaud, S.; et al. Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist cotadutide via modulating mitochondrial function and lipogenesis. Nat. Metab. 2020, 2, 413–431. [Google Scholar] [CrossRef]
- Valdecantos, M.P.; Pardo, V.; Ruiz, L.; Castro-Sánchez, L.; Lanzón, B.; Fernández-Millán, E.; García-Monzón, C.; Arroba, A.I.; González-Rodríguez, Á.; Escrivá, F.; et al. A novel glucagon-like peptide 1/glucagon receptor dual agonist improves steatohepatitis and liver regeneration in mice. Hepatology 2017, 65, 950–968. [Google Scholar] [CrossRef] [PubMed]
- Kannt, A.; Madsen, A.N.; Kammermeier, C.; Elvert, R.; Klöckener, T.; Bossart, M.; Haack, T.; Evers, A.; Lorenz, K.; Hennerici, W.; et al. Incretin combination therapy for the treatment of non-alcoholic steatohepatitis. Diabetes Obes. Metab. 2020, 22, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Nestor, J.J.; Parkes, D.; Feigh, M.; Suschak, J.J.; Harris, M.S. Effects of ALT-801, a GLP-1 and glucagon receptor dual agonist, in a translational mouse model of non-alcoholic steatohepatitis. Sci. Rep. 2022, 12, 6666. [Google Scholar] [CrossRef]
- Song, N.; Xu, H.; Liu, J.; Zhao, Q.; Chen, H.; Yan, Z.; Yang, R.; Luo, Z.; Liu, Q.; Ouyang, J.; et al. Design of a highly potent GLP-1R and GCGR dual-agonist for recovering hepatic fibrosis. Acta Pharm. Sin. B 2022, 12, 2443–2461. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Bizino, M.B.; Jazet, I.M.; de Heer, P.; van Eyk, H.J.; Dekkers, I.A.; Rensen, P.C.N.; Paiman, E.H.M.; Lamb, H.J.; Smit, J.W. Placebo-controlled randomised trial with liraglutide on magnetic resonance endpoints in individuals with type 2 diabetes: A pre-specified secondary study on ectopic fat accumulation. Diabetologia 2020, 63, 65–74. [Google Scholar] [CrossRef]
- Yan, J.; Yao, B.; Kuang, H.; Yang, X.; Huang, Q.; Hong, T.; Li, Y.; Dou, J.; Yang, W.; Qin, G.; et al. Liraglutide, Sitagliptin, and Insulin Glargine Added to Metformin: The Effect on Body Weight and Intrahepatic Lipid in Patients With Type 2 Diabetes Mellitus and Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 2414–2426. [Google Scholar] [CrossRef]
- Guo, W.; Tian, W.; Lin, L.; Xu, X. Liraglutide or insulin glargine treatments improves hepatic fat in obese patients with type 2 diabetes and nonalcoholic fatty liver disease in twenty-six weeks: A randomized placebo-controlled trial. Diabetes Res. Clin. Pract. 2020, 170, 108487. [Google Scholar] [CrossRef] [PubMed]
- Khoo, J.; Hsiang, J.C.; Taneja, R.; Koo, S.; Soon, G.; Kam, C.J.; Law, N.; Ang, T. Randomized trial comparing effects of weight loss by liraglutide with lifestyle modification in non-alcoholic fatty liver disease. Liver Int. 2019, 39, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-Y.; Qu, X.-N.; Sun, Z.-Y.; Zhang, Y. Effect of liraglutide therapy on serum fetuin A in patients with type 2 diabetes and non-alcoholic fatty liver disease. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Dutour, A.; Abdesselam, I.; Ancel, P.; Kober, F.; Mrad, G.; Darmon, P.; Ronsin, O.; Pradel, V.; Lesavre, N.; Martin, J.C.; et al. Exenatide decreases liver fat content and epicardial adipose tissue in patients with obesity and type 2 diabetes: A prospective randomized clinical trial using magnetic resonance imaging and spectroscopy. Diabetes Obes. Metab. 2016, 18, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yan, H.; Xia, M.; Zhao, L.; Lv, M.; Zhao, N.; Rao, S.; Yao, X.; Wu, W.; Pan, B.; et al. Efficacy of exenatide and insulin glargine on nonalcoholic fatty liver disease in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 2020, 36, e3292. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Choudhary, N.S.; Singh, M.K.; Wasir, J.S.; Kaur, P.; Gill, H.K.; Bano, T.; Farooqui, K.J.; et al. Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: Randomised controlled trial (D-LIFT trial). Diabetologia 2020, 63, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.; Andersen, G.; Hockings, P.; Johansson, L.; Morsing, A.; Palle, M.S.; Vogl, T.; Loomba, R.; Plum-Mörschel, L. Randomised clinical trial: Semaglutide versus placebo reduced liver steatosis but not liver stiffness in subjects with non-alcoholic fatty liver disease assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2021, 54, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Abdelmalek, M.F.; Armstrong, M.J.; Jara, M.; Kjær, M.S.; Krarup, N.; Lawitz, E.; Ratziu, V.; Sanyal, A.J.; Schattenberg, J.M.; et al. Semaglutide 2.4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: A randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2023, 8, 511–522. [Google Scholar] [CrossRef]
- Alkhouri, N.; Herring, R.; Kabler, H.; Kayali, Z.; Hassanein, T.; Kohli, A.; Huss, R.S.; Zhu, Y.; Billin, A.N.; Damgaard, L.H.; et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J. Hepatol. 2022, 77, 607–618. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Ludvik, B.; Giorgino, F.; Jódar, E.; Frias, J.P.; Landó, L.F.; Brown, K.; Bray, R.; Rodríguez, Á. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): A randomised, open-label, parallel-group, phase 3 trial. Lancet 2021, 398, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Jastreboff, A.M.; Jastreboff, A.M.; Jastreboff, A.M.; Aronne, L.J.; Aronne, L.J.; Aronne, L.J.; Ahmad, N.N.; Ahmad, N.N.; Ahmad, N.N.; Wharton, S.; et al. Tirzepatide Once Weekly for the Treatment of Obesity. N. Engl. J. Med. 2022, 387, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Nahra, R.; Wang, T.; Gadde, K.M.; Oscarsson, J.; Stumvoll, M.; Jermutus, L.; Hirshberg, B.; Ambery, P. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults With Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care 2021, 44, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Tomah, S.; Suschak, J.; Roberts, S.; Yang, J.; He, L.; Georges, B.; Rodwell-Green, L.; Brown, R.; Harris, M.S.; et al. Pemvidutide, a GLP-1/glucagon dual receptor agonist, significantly reduces liver fat, fibro-inflammation, and body weight in patients with non-alcoholic fatty liver disease: A 24-week multicenter, randomized, double-blind, placebo-controlled trial. J. Hepatol. 2023, 78, S54–S55. [Google Scholar] [CrossRef]
- To, D.; Shin, J.; Karanth, S.; Lin, Y.-H.; Sosnovtseva, S.; Bell, A.C. 72-LB: DD01, a Novel Once-Weekly Dual GLP-1/Glucagon Receptor Agonist, Improves Metabolic Health and Achieves Rapid, Clinically Significant Reductions in Hepatic Steatosis following Only Four Weeks of Treatment and without the Need for Significant Weight Loss in Overweight/Diabetic Subjects with NAFLD. Diabetes 2023, 72 (Suppl. S1), 72-LB. [Google Scholar]
- Abdelmalek, M.; Choi, J.; Kim, Y.; Seo, K.; Hompesch, M.; Baek, S. HM15211, a novel GLP-1/GIP/Glucagon triple-receptor co-agonist significantly reduces liver fat and body weight in obese subjects with non-alcoholic fatty liver disease: A Phase 1b/2a, multi-center, randomized, placebo-controlled trial. J. Hepatol. 2020, 73, S124. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Sanchez, W.; Lawitz, E.; Filozof, C.; Cho, H.; Baek, E.; Choi, J.; Baek, S. A phase 2, adaptive randomized, double-blind, placebo-controlled, multicenter, 52-week study of HM15211 in patients with biopsy-confirmed non-alcoholic steatohepatitis—Study design and rationale of HM-TRIA-201 study. Contemp. Clin. Trials 2023, 130, 107176. [Google Scholar] [CrossRef]
- Sanyal, A.; Frias, J.P.; Thomas, M.K.; Mather, K.J.; Wu, Q.; Du, Y.; Brouwers, B.; Haupt, A.; Hartman, M.L. Triple hormone receptor agonist retatrutide resolves steatosis in >85% of subjects with MASLD and obesity in association with improved metabolic health. Hepatology 2023, 78, S154–S155. [Google Scholar]
- Blüher, M.; Rosenstock, J.; Hoefler, J.; Manuel, R.; Hennige, A.M. Dose–response effects on HbA1c and bodyweight reduction of survodutide, a dual glucagon/GLP-1 receptor agonist, compared with placebo and open-label semaglutide in people with type 2 diabetes: A randomised clinical trial. Diabetologia 2023, 67, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Bali, T.; Chrysavgis, L.; Cholongitas, E. Metabolic-Associated Fatty Liver Disease and Sarcopenia. Endocrinol. Metab. Clin. N. Am. 2023, 52, 497–508. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Vachliotis, I.D.; Mantzoros, C.S. Sarcopenia, sarcopenic obesity and nonalcoholic fatty liver disease. Metabolism 2023, 147, 155676. [Google Scholar] [CrossRef] [PubMed]
- Volpe, S.; Lisco, G.; Racaniello, D.; Fanelli, M.; Colaianni, V.; Vozza, A.; Triggiani, V.; Sabbà, C.; Tortorella, C.; De Pergola, G.; et al. Once-Weekly Semaglutide Induces an Early Improvement in Body Composition in Patients with Type 2 Diabetes: A 26-Week Prospective Real-Life Study. Nutrients 2022, 14, 2414. [Google Scholar] [CrossRef] [PubMed]
- Karakousis, N.D.; Chrysavgis, L.; Chatzigeorgiou, A.; Papatheodoridis, G.; Cholongitas, E. Frailty in metabolic syndrome, focusing on nonalcoholic fatty liver disease. Ann. Gastroenterol. 2022, 35, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Skladany, L.; Molcan, P.; Vnencakova, J.; Vrbova, P.; Kukla, M.; Laffers, L.; Koller, T. Frailty in Nonalcoholic Fatty Liver Cirrhosis: A Comparison with Alcoholic Cirrhosis, Risk Patterns, and Impact on Prognosis. Can. J. Gastroenterol. Hepatol. 2021, 2021, 5576531. [Google Scholar] [CrossRef]
- Christoffersen, B.; Sanchez-Delgado, G.; John, L.M.; Ryan, D.H.; Raun, K.; Ravussin, E. Beyond appetite regulation: Targeting energy expenditure, fat oxidation, and lean mass preservation for sustainable weight loss. Obesity 2022, 30, 841–857. [Google Scholar] [CrossRef]
- Chrysavgis, L.; Adamantou, M.; Angelousi, A.; Cholongitas, E. The association of testosterone with sarcopenia and frailty in chronic liver disease. Eur. J. Clin. Investig. 2024, 54, e14108. [Google Scholar] [CrossRef]
- Henson, J.B.; Simon, T.G.; Kaplan, A.; Osganian, S.; Masia, R.; Corey, K.E. Advanced fibrosis is associated with incident cardiovascular disease in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2020, 51, 728–736. [Google Scholar] [CrossRef]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of Hyperglycemia in Type 2 Diabetes, 2022. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2022, 45, 2753–2786. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [PubMed]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Care in Diabetes-2023. Diabetes Care 2023, 46 (Suppl. S1), S140–S157. [Google Scholar] [CrossRef] [PubMed]
- Jujić, A.; Nilsson, P.M.; Atabaki-Pasdar, N.; Dieden, A.; Tuomi, T.; Franks, P.W.; Holst, J.J.; Torekov, S.S.; Ravassa, S.; Díez, J.; et al. Glucose-Dependent Insulinotropic Peptide in the High-Normal Range Is Associated with Increased Carotid Intima-Media Thickness. Diabetes Care 2021, 44, 224–230. [Google Scholar] [CrossRef]
- Sattar, N.; McGuire, D.K.; Pavo, I.; Weerakkody, G.J.; Nishiyama, H.; Wiese, R.J.; Zoungas, S. Tirzepatide cardiovascular event risk assessment: A pre-specified meta-analysis. Nat. Med. 2022, 28, 591–598. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Bhatt, D.L.; Buse, J.B.; Del Prato, S.; Kahn, S.E.; Lincoff, A.M.; McGuire, D.K.; Nauck, M.A.; Nissen, S.E.; Sattar, N.; et al. Comparison of tirzepatide and dulaglutide on major adverse cardiovascular events in participants with type 2 diabetes and atherosclerotic cardiovascular disease: SURPASS-CVOT design and baseline characteristics. Am. Heart J. 2024, 267, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Ambery, P. Incretins (GLP-1 receptor agonists and dual/triple agonists) and the liver. J. Hepatol. 2023, 79, 1557–1565. [Google Scholar] [CrossRef]
- Sodhi, M.; Rezaeianzadeh, R.; Kezouh, A.; Etminan, M. Risk of Gastrointestinal Adverse Events Associated With Glucagon-Like Peptide-1 Receptor Agonists for Weight Loss. JAMA 2023, 330, 1795–1797. [Google Scholar] [CrossRef]
- Wishart, J.M.; Horowitz, M.; Morris, H.A.; Jones, K.L.; Nauck, M.A. Relation between gastric emptying of glucose and plasma concentrations of glucagon-like peptide-1. Peptides 1998, 19, 1049–1053. [Google Scholar] [CrossRef]
- Petrovic, A.; Igrec, D.; Rozac, K.; Bojanic, K.; Kuna, L.; Kolaric, T.O.; Mihaljevic, V.; Sikora, R.; Smolic, R.; Glasnovic, M.; et al. The Role of GLP1-RAs in Direct Modulation of Lipid Metabolism in Hepatic Tissue as Determined Using In Vitro Models of NAFLD. Curr. Issues Mol. Biol. 2023, 45, 4544–4556. [Google Scholar] [CrossRef]
- Arai, T.; Atsukawa, M.; Tsubota, A.; Ono, H.; Kawano, T.; Yoshida, Y.; Okubo, T.; Hayama, K.; Nakagawa-Iwashita, A.; Itokawa, N.; et al. Efficacy and safety of oral semaglutide in patients with non-alcoholic fatty liver disease complicated by type 2 diabetes mellitus: A pilot study. JGH Open 2022, 6, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.Q.; Cheng, W.; Gordian, D.; Lee, J.; Paulsen, S.J.; Hansen, S.N.; Egerod, K.L.; Barkholt, P.; Rhodes, C.J.; Secher, A.; et al. A genetic map of the mouse dorsal vagal complex and its role in obesity. Nat. Metab. 2021, 3, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Gelfanov, V.M.; El, K.; Chen, A.; Rohlfs, R.; DuBois, B.; Hansen, A.M.K.; Perez-Tilve, D.; Knerr, P.J.; D’Alessio, D.; et al. Discovery of a potent GIPR peptide antagonist that is effective in rodent and human systems. Mol. Metab. 2022, 66, 101638. [Google Scholar] [CrossRef] [PubMed]
- Killion, E.A.; Wang, J.; Yie, J.; Shi, S.D.-H.; Bates, D.; Min, X.; Komorowski, R.; Hager, T.; Deng, L.; Atangan, L.; et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci. Transl. Med. 2018, 10, eaat3392. [Google Scholar] [CrossRef] [PubMed]
- Mroz, P.A.; Finan, B.; Gelfanov, V.; Yang, B.; Tschöp, M.H.; DiMarchi, R.D.; Perez-Tilve, D. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol. Metab. 2019, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Nørregaard, P.K.; Deryabina, M.A.; Tofteng Shelton, P.; Fog, J.U.; Daugaard, J.R.; Eriksson, P.O.; Larsen, L.F.; Jessen, L. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes. Metab. 2018, 20, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Killion, E.A.; Chen, M.; Falsey, J.R.; Sivits, G.; Hager, T.; Atangan, L.; Helmering, J.; Lee, J.; Li, H.; Wu, B.; et al. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat. Commun. 2020, 11, 4981. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, K.; Yamada, Y.; Ban, N.; Ihara, Y.; Tsukiyama, K.; Zhou, H.; Fujimoto, S.; Oku, A.; Tsuda, K.; Toyokuni, S.; et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 2002, 8, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Naitoh, R.; Miyawaki, K.; Harada, N.; Mizunoya, W.; Toyoda, K.; Fushiki, T.; Yamada, Y.; Seino, Y.; Inagaki, N. Inhibition of GIP signaling modulates adiponectin levels under high-fat diet in mice. Biochem. Biophys. Res. Commun. 2008, 376, 21–25. [Google Scholar] [CrossRef]
- Althage, M.C.; Ford, E.L.; Wang, S.; Tso, P.; Polonsky, K.S.; Wice, B.M. Targeted ablation of glucose-dependent insulinotropic polypeptide-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet. J. Biol. Chem. 2008, 283, 18365–18376. [Google Scholar] [CrossRef]
- Wang, L. Designing a Dual GLP-1R/GIPR Agonist from Tirzepatide: Comparing Residues Between Tirzepatide, GLP-1, and GIP. Drug Des. Devel. Ther. 2022, 16, 1547–1559. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Klen, J.; Dolžan, V. Glucagon-like Peptide-1 Receptor Agonists in the Management of Type 2 Diabetes Mellitus and Obesity: The Impact of Pharmacological Properties and Genetic Factors. Int. J. Mol. Sci. 2022, 23, 3451. [Google Scholar] [CrossRef] [PubMed]
- Sathananthan, A.; Man, C.D.; Micheletto, F.; Zinsmeister, A.R.; Camilleri, M.; Giesler, P.D.; Laugen, J.M.; Toffolo, G.; Rizza, R.A.; Cobelli, C.; et al. Common genetic variation in GLP1R and insulin secretion in response to exogenous GLP-1 in nondiabetic subjects: A pilot study. Diabetes Care 2010, 33, 2074–2076. [Google Scholar] [CrossRef] [PubMed]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The complex link between NAFLD and type 2 diabetes mellitus—mechanisms and treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD as a driver of chronic kidney disease. J. Hepatol. 2020, 72, 785–801. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Tilg, H.; Byrne, C.D. Non-alcoholic fatty liver disease: A multisystem disease requiring a multidisciplinary and holistic approach. Lancet Gastroenterol. Hepatol. 2021, 6, 578–588. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62 (Suppl. S1), S47–S64. [Google Scholar] [CrossRef]
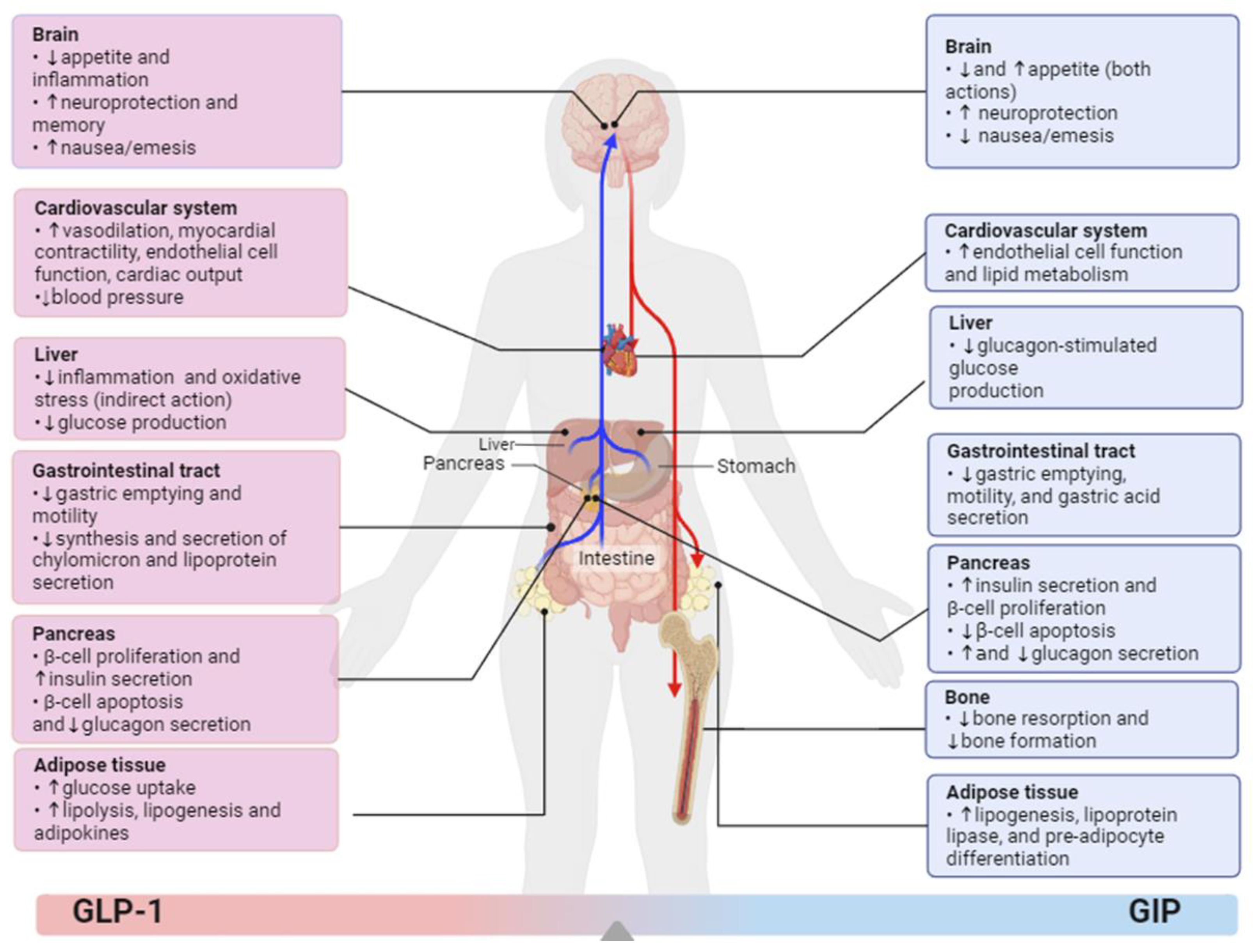
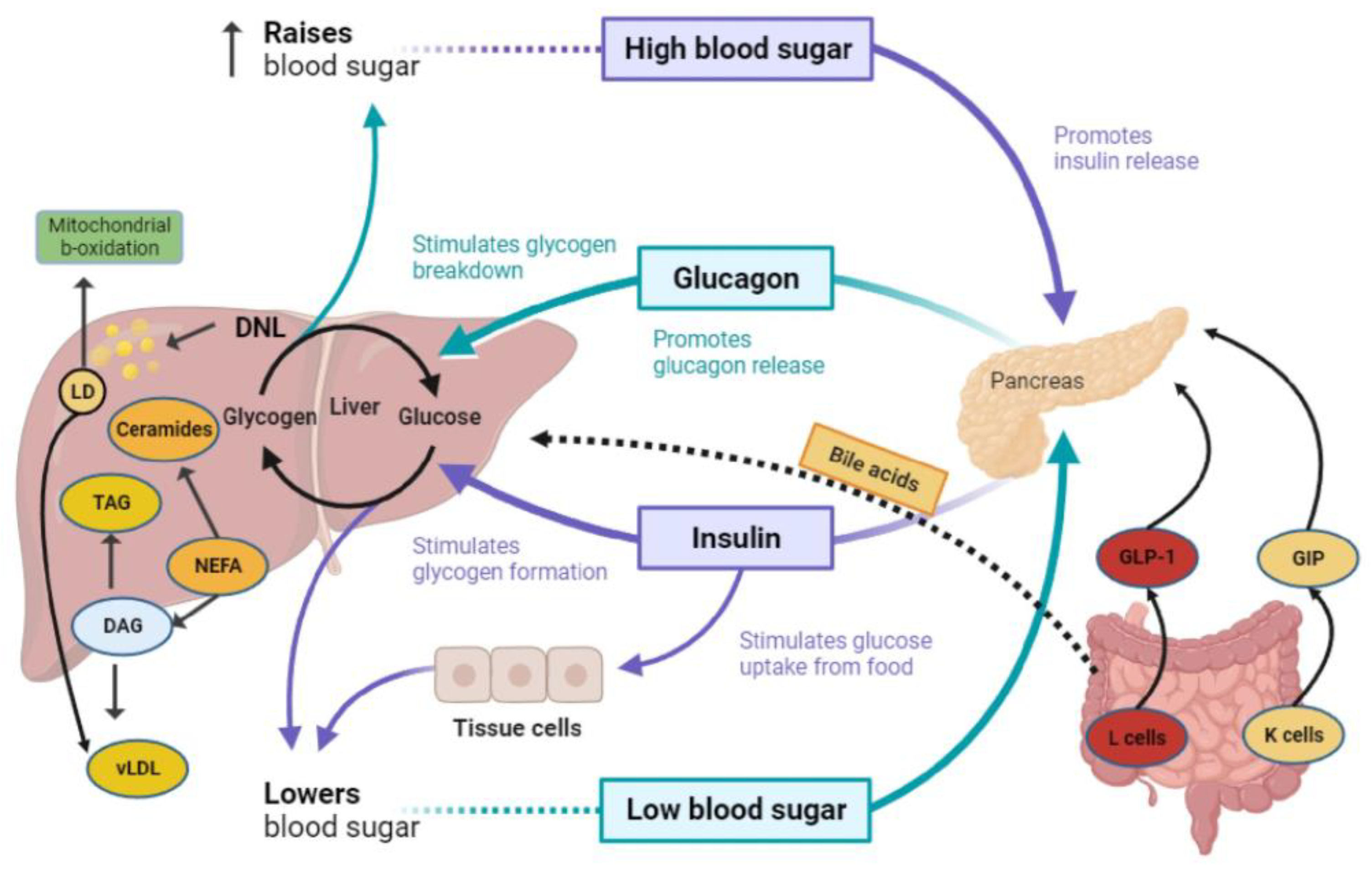

{kind=link}
{kind=link}
| Author/Ref. | Country/Publication Year | Phase and Design of Study | Study Population | Mean Age, Mean BMI, Gender, T2DM (%) | Intervention; Duration; Assessment | Outcomes |
|---|---|---|---|---|---|---|
| Armstrong et al. [88] | United Kingdom, 2016 | Phase 2, double-blind, placebo-controlled | 52 overweight patients with biopsy-confirmed MASH | 51 yo, 36 kg/m2, 60% male, 33% with T2DM | Liraglutide 1.8 mg/day (n = 26) vs. placebo (n = 26); 48 weeks; Liver Biopsy | Greater MASH resolution in the liraglutide group: 39% vs. 9% in the placebo group (p = 0.019). Less liver fibrosis progression with liraglutide: 9% vs. 36% in the placebo group (p = 0.04). |
| Bizino et al. [89] | Netherlands, 2020 | Phase 2, double-blind, placebo-controlled (sub-analysis of the MGNA VICTORIA study) | 49 patients with T2DM | 60 yo, 32 kg/m2, 59% male, 100% with T2DM | Liraglutide 1.8 mg/day (n = 23) vs. placebo (n = 26); 26 weeks; MRI | LFC reduction was not significantly different between groups; liraglutide was associated with significantly greater body weight and subcutaneous fat reduction |
| Yan et al. [90] | China, 2019 | Phase 2, open-label, active-controlled | 75 patients with T2DM and MASLD, with inadequate glycemic control by metformin | 44 yo, 30 kg/m2, 69% male, 100% with T2DM | Liraglutide 1.8 mg/day (n = 24) vs. insulin glargine 0.2 IU/kg/day (n = 24) vs. sitagliptin 100 mg/day (n = 27) (adds-on metformin); 26 weeks; MRI | When combined with metformin, both sitagliptin and liraglutide but not insulin glargine resulted in a significant decrease in LFC; liraglutide group: from 15.4% [SD 5.6] to 12.5% [SD 6.4] (p < 0·001). |
| Guo et al. [91] | China, 2020 | Phase 2, open-label, placebo-controlled | 96 patients with T2DM and MASLD with inadequate glycemic control by metformin | 52 yo, 29 kg/m2, 56% male, 100% with T2DM | Liraglutide 1.8 mg/day (n = 32) vs. insulin glargine once daily (n = 32) vs. placebo (n = 32) (adds-on metformin); 26 weeks; MRI | When combined with metformin, liraglutide significantly reduced steatosis from 26.4% [SD 3.2] to 20.6% [SD 3.9] (p < 0·05). |
| Khoo et al. [92] | Singapore, 2019 | Phase 2, open-label, active-controlled | 30 patients with obesity and MASLD | 41 yo, 33 kg/m2, 90% male, 0% with T2DM | Liraglutide 3 mg/day (n = 15) vs. lifestyle modifications: diet and exercise (n = 15); 26 weeks; MRI | Both liraglutide and lifestyle modifications resulted in significant hepatic fat reduction vs. baseline: −7.0% [SD 7.1] and −8.1% [SD 13.2], respectively. These benefits were not sustained in the liraglutide group in a 6-month period. |
| Zhang et al. [93] | China, 2020 | Phase 2, open-label, active-controlled | 60 patients with T2DM and MASLD | 51 yo, 27 kg/m2, 47% male, 100% with T2DM | Liraglutide 1.2 mg/day (n = 30) vs. pioglitazone 30 mg/day (n = 30) (add-on to usual care); 24 weeks; MRI | The addition of liraglutide was associated with a significant reduction in LFC from 24.1% [SD 3.0] to 20.1% [SD 3.8] (p < 0·05). This reduction was significantly greater compared with the addition of pioglitazone. |
| Dutour et al. [94] | France, 2016 | Phase 2, open-label, active-controlled | 44 patients with obesity and T2DM, with inadequate glycemic control by oral antidiabetic therapy (MASLD in 95% of them) | 52 yo, 36 kg/m2, 48% male, 100% with T2DM | Exenatide 5–10 μg twice/day (n = 22) vs. reference treatment according to local guidelines (n = 22); 26 weeks; MRI | Exenatide resulted in a significant decrease in hepatic triglyceride content: −23.8% [SD 9.5] vs. +12.5% [SD 9.6] in the placebo group (p = 0.007). |
| Liu et al. [95] | China, 2020 | Phase 2, open-label, active-controlled | 76 patients with newly diagnosed T2DM and MASLD. Age: 48, BMI: 28, 50% male | 48 yo, 28 kg/m2, 50% male, 100% with T2DM | Exenatide 5–10 μg twice/day (n = 38) vs. insulin glargine 0.2 IU/kg/day (n = 38); 24 weeks; MRI and Fibroscan | Exenatide and insulin glargine both significantly reduced LFC, but exenatide induced a greater reduction in body weight, visceral adiposity, liver enzymes, and Fibrosis-4 (FIB-4) index. |
| Kuchay et al. [96] | India, 2020 | Phase 2, open-label, active-controlled | 64 patients with T2DM and MASLD | 47 yo, 30 kg/m2, 70% male, 100% with T2DM | Dulaglutide 1.5 mg/week adds-on usual care (n = 32) vs. usual care (n = 32); 24 weeks; MRI and Fibroscan | Addition of dulaglutide resulted in a 2.6-fold greater reduction in LFC and a significant improvement in serum GGT level vs. control group. Changes in liver stiffness on Fibroscan, serum AST, and ALT levels were not significant. |
| Newsome et al. [97] | Multicenter, 2020 | Phase 2, double-blind, placebo-controlled | 320 patients with biopsy-confirmed MASH and liver fibrosis of stage F1, F2, or F3 | 55 yo, 36 kg/m2, 41% male, 62% with T2DM | Semaglutide 0.1 mg/day (n = 80) vs. semaglutide 0,2 mg/day (n = 78) vs. semaglutide 0.4 mg/day (n = 82) vs. placebo (n = 80); 72 weeks; liver biopsy | The proportion of patients on semaglutide 0.4 mg/day with resolution of MASH without worsening of fibrosis was significantly higher compared with the placebo group: 59% vs. 17% (p < 0.001). Improvement of liver fibrosis was not significantly different between groups. |
| Flint et al. [98] | Germany, 2021 | Phase 1, double-blind, placebo-controlled | 67 patients with MASLD (assessed by MRI-PDFF and MRE) | 60 yo, >30 kg/m2, 70% male, 73% with T2DM | Semaglutide 0·4 mg/day (n = 34) vs. placebo (n = 33); 72 weeks, MRI | In the semaglutide group, hepatic steatosis presented a significantly greater decrease vs. placebo group at week 24 (−36% vs. −9%, p < 0.001), week 48 (−58% vs. −11%, p < 0.001), and week 72 (−58% vs. −17%, p < 0.001); no significant difference between groups was observed in changes of liver stiffness. |
| Loomba et al. [99] | Multicenter, 2023 | Phase 2, double-blind, placebo-controlled | 71 patients with biopsy-confirmed MASH-related cirrhosis and BMI ≥ 27 kg/m2 | 60 yo, 35 kg/m2, 31% male, 75% with T2DM | Semaglutide 2.4 mg/week (n = 47) vs. placebo (n = 24); 48 weeks, liver biopsy | Neither the proportion of patients with MASH resolution nor the proportion of patients with liver fibrosis improvement without worsening of MASH differed significantly between groups. |
| Alkhouri et al. [100] | USA, 2022 | Phase 2, open-label, active-controlled | 108 patients with MASH (assessed by liver biopsy or by MRI-PDFF ≥10% and Fibroscan measured liver stiffness ≥7 kPa) | 56 yo, 35 kg/m2, 30% male, 55% with T2DM | Semaglutide 2.4 mg/week (n = 21) vs. semaglutide 2.4 mg/week + cilofexor 30 mg/day (n = 22) vs. semaglutide 2.4 mg/week + cilofexor 100 mg/day (n = 22) vs. semaglutide 2.4 mg/week + firsocostat 20 mg/day (n = 22) vs. semaglutide 2.4 mg/week + cilofexor 30 mg/day + firsocostat 20 mg/day (n = 21); 24 weeks; MRI and Fibroscan | Overall, combination therapies resulted in a larger reduction in LFC and greater improvement in liver enzymes and liver fibrosis (as assessed by Fibroscan) than semaglutide alone. When compared to semaglutide monotherapy, the only treatment group with significantly different change in liver steatosis was semaglutide + firsocostat: −11% vs. −8% in the semaglutide monotherapy group (p = 0.035). |
| Author/Ref. | Country/Publication Year | Phase and Design of Study | Study Population | Mean Age, Mean BMI, Gender, T2DM (%) | Intervention; Duration; Assessment | Outcomes |
|---|---|---|---|---|---|---|
| Ludvik et al./ Hartman et al. [102,104] | Multinational (13 countries), 2021 | Phase 3, randomized, open-label, parallel-group, multicenter | 1444 overweight patients with T2DM | 57 yo, 33 kg/m2, 56% male, 100% with T2DM | Tirzepatide (5, 10, 15 mg) (n = 358, 360, 359) vs. insulin degludec (n = 360); 52 weeks; HbA1c and bodyweight reduction | Greater reduction in HbA1c vs. baseline [1.93%, 2.2%, 2.37% for 5, 10, and 15 mg, respectively (p = 0.05)] and body weight and lower risk of hypoglycemia; pooled tirzepatide group (10 mg and 15 mg) induced a greater LFC reduction compared with the insulin degludec group (−8.09% vs. −3.38%, p < 0.0001) |
| Hartman et al. [104] | USA/2020 | Phase 2, post-hoc analysis | 316 patients with T2DM | 57 yo, 32.6 kg/m2, 53% male | Tirzepatide (1, 5, 10, 15 mg) (n = 52, n = 55, n = 51, n = 53) vs. dulaglutide (n = 54) or placebo (n = 51); 26 weeks; hepatic dysfunction parameters | Greater decrease in ALT (−6.8 units/L and −6.4 units/L for tirzepatide 10 mg and 15 mg, respectively vs. dulaglutide, p < 0.05), K-18 (−135.2 units/L in the tirzepatide 10 mg vs. placebo group, p < 0.015) pro-C3 (−2.1 ng/mL in the tirzepatide 15 mg vs. placebo group, p = 0.041) |
| Nahra et al. [106] | Multinational (8 countries), 2021 | Phase 2b, double-blind, placebo-controlled | 834 patients with T2DM and BMI ≥ 25 kg/m2 | ~56 yo, ~35 kg/m2, ~45% male, 100% with T2DM | Cotadutide 100 μg (n = 100), 200 μg (n = 256), or 300 μg (n = 256) vs. placebo (n = 110) or liraglutide 1.8 mg (n = 110); 54 weeks; HbA1c, body weight, hepatic parameters for liver fibrosis | Cotadutide (100, 200, or 300 mg) compared with the placebo group achieved greater reductions in AST (−1.77%, −6.22%, −9.14%, and 5.65%, respectively, p < 0.009), ALT (−7.52%, −12.01%, −14.15%, and 0.93%, respectively, p < 0.009), PRO-C3 (−0.38% in 300 mg cotadutide group vs. 13.04 in the placebo group, p = 0.0034), total and LDL cholesterol, triglycerides, and GGT levels, as well as in fatty liver index (−8.08, −6.73, −8.18, and −1.62, respectively, p < 0.001). |
| Haririson et al. [107] | United States, 2023 | Randomized, double-blind, placebo-controlled | 94 patients with MASLD | 36 kg/m2, 29% with T2DM | Pemvidutide 1.2 mg, 1.8 mg, and 2.4 mg vs. placebo; 24 weeks; reduction in LFC, Ct1, ALT, body weight | Pemvidutide reduced LFC at 24 weeks vs. baseline, compared with the placebo group (−56.3%, −75.2%, −76.4%, and −14%, respectively). Dose-dependent reduction in ALT levels (−13.3 IU/L, −13.7 IU/L, −15.2 IU/L, −2.2 IU/L, respectively). |
| To et al. [108] | United States, 2023 | Phase 1 | 18 overweight/obese patients with T2DM and MASLD | 100% with T2DM | DD01 1–80 mg (four once-weekly doses) vs. placebo; 36 days; MRI | Rapid reductions in hepatic steatosis assessed by MRI, HbA1c and greater weight loss. Over the four-week period of the study, patients treated with DD01 had a mean LFC reduction of 52% versus a 2.8% reduction in the placebo group. |
| Abdelmalek et al. [109,110] | United States, 2020 | Phase 1b/2a, multicenter, randomized, placebo-controlled | 66 non-diabetic obese patients with MASLD | 46 yo; 50% men; mean BMI: 36 kg/m2, 0% with T2DM | HM15211 0.01, 0.02, 0.04, 0.06, and 0.08 mg/day vs. placebo; 12 weeks; MRI-PDFF | HM15211 reduced LFC vs. placebo in a dose-dependent manner (mean relative changes from baseline in liver fat at week 12 vs. baseline: −19.6% for 0.01 mg/kg, −36% for 0.02 mg/kg, −38% for 0.04 mg/kg, −59.3% for 0.06 mg/kg, and −5.7% for the placebo group, p < 0.05). HM15211 reduced body weight across all treatment dose groups compared with the placebo arm at week 12 vs. baseline [(placebo-corrected % reduction of body weight was −1.9%, −3.4%, −2.1%, −3.8%, and −5.1%) in 0.01 to 0.08 mg/kg dose cohorts, respectively, p < 0.05)] |
| Sanyal et al. [111] | United States, 2023 | Phase 2 | 338 obese MASLD patients | 46.6 yo, 38.4 kg/m2, 53.1% males, 0% with T2DM | Retatrutide 1, 4, 8, and 12 mg/day vs. placebo; 48 weeks; liver fat change | Retatrutide reduced mean relative LFC in comparison to placebo at 24 and 48 weeks of treatment vs. baseline [change from baseline at 24 weeks was −42.9% (1 mg), −57.0% (4 mg), −81.4% (8 mg), −82.4% (12 mg) and +0.3% (placebo), and at 48 weeks was −51.3% (1 mg), −59.0% (4 mg), −81.7% (8 mg), −86.0% (12 mg) and −4.6% (placebo), all p < 0.001 vs. placebo) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrysavgis, L.G.; Kazanas, S.; Bafa, K.; Rozani, S.; Koloutsou, M.-E.; Cholongitas, E. Glucagon-like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide, and Glucagon Receptor Agonists in Metabolic Dysfunction-Associated Steatotic Liver Disease: Novel Medication in New Liver Disease Nomenclature. Int. J. Mol. Sci. 2024, 25, 3832. https://doi.org/10.3390/ijms25073832
Chrysavgis LG, Kazanas S, Bafa K, Rozani S, Koloutsou M-E, Cholongitas E. Glucagon-like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide, and Glucagon Receptor Agonists in Metabolic Dysfunction-Associated Steatotic Liver Disease: Novel Medication in New Liver Disease Nomenclature. International Journal of Molecular Sciences. 2024; 25(7):3832. https://doi.org/10.3390/ijms25073832
Chicago/Turabian StyleChrysavgis, Lampros G., Spyridon Kazanas, Konstantina Bafa, Sophia Rozani, Maria-Evangelia Koloutsou, and Evangelos Cholongitas. 2024. "Glucagon-like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide, and Glucagon Receptor Agonists in Metabolic Dysfunction-Associated Steatotic Liver Disease: Novel Medication in New Liver Disease Nomenclature" International Journal of Molecular Sciences 25, no. 7: 3832. https://doi.org/10.3390/ijms25073832
APA StyleChrysavgis, L. G., Kazanas, S., Bafa, K., Rozani, S., Koloutsou, M.-E., & Cholongitas, E. (2024). Glucagon-like Peptide 1, Glucose-Dependent Insulinotropic Polypeptide, and Glucagon Receptor Agonists in Metabolic Dysfunction-Associated Steatotic Liver Disease: Novel Medication in New Liver Disease Nomenclature. International Journal of Molecular Sciences, 25(7), 3832. https://doi.org/10.3390/ijms25073832