
Exploring the Gut–Mitochondrial Axis: p66Shc Adapter Protein and Its Implications for Metabolic Disorders


Abstract
1. Introduction
2. Overview of the ShcA Protein Family
3. p66Shc and Oxidative Stress
3.1. Rac1 Activation
3.2. Forkhead-Type Transcription Factors Inactivation
3.3. Mitochondria-Mediated Apoptosis
4. Gut Microbiota and Oxidative Stress
5. Oxidative Stress, Gut Microbiota, and p66Shc
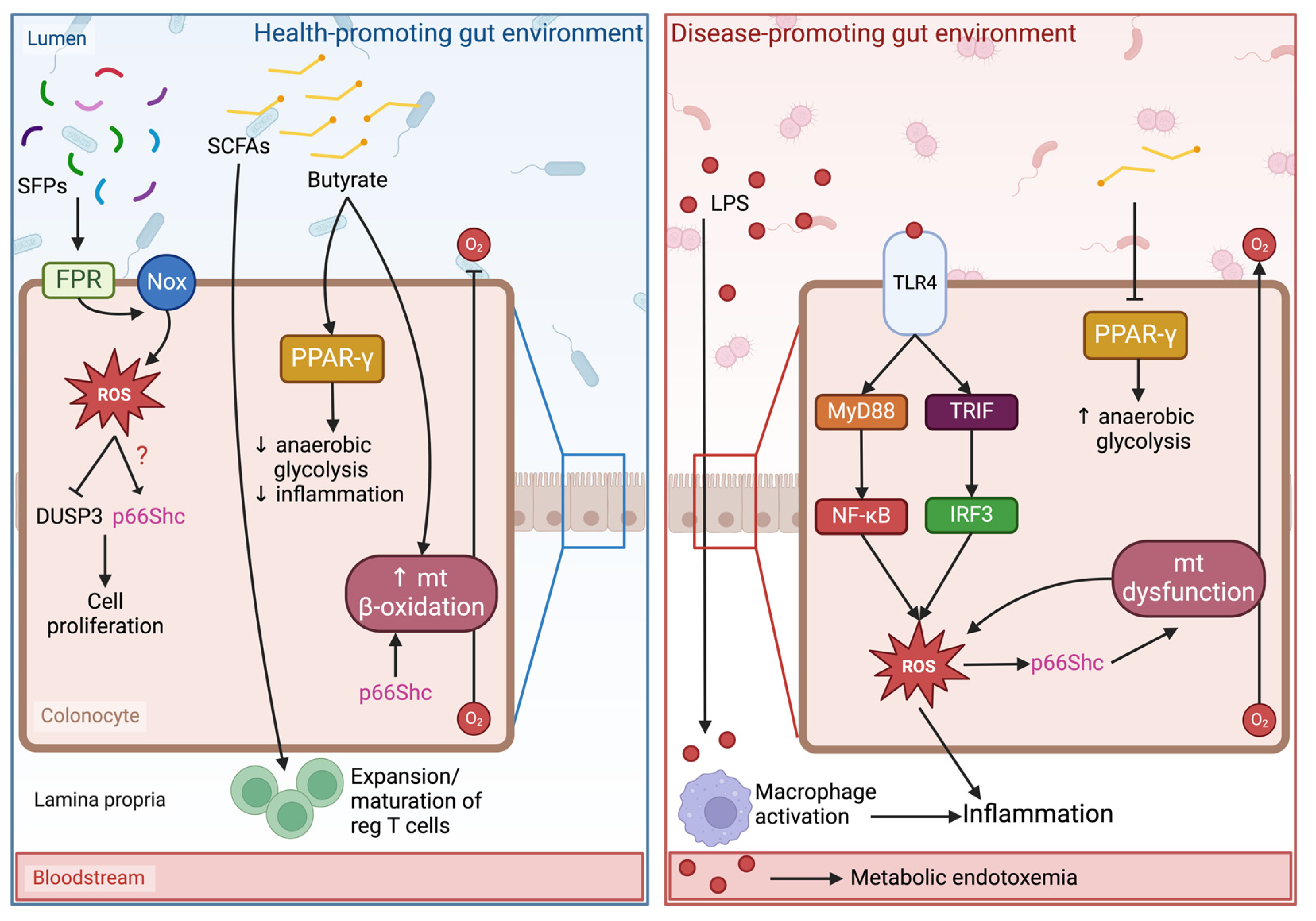
6. Linking Oxidative Stress, Gut Microbiota, and p66Shc to Pathophysiological Outcomes
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Zhunina, O.A.; Yabbarov, N.G.; Grechko, A.V.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Vascular Disease, Tumorigenesis, and Diabetes. Front. Mol. Biosci. 2021, 8, 671908. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41, bnaa005. [Google Scholar] [CrossRef] [PubMed]
- Mir, H.A.; Ali, R.; Mushtaq, U.; Khanday, F.A. Structure-functional implications of longevity protein p66Shc in health and disease. Ageing Res. Rev. 2020, 63, 101139. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Albiero, M.; Campanaro, S.; Poncina, N.; Tedesco, S.; Scattolini, V.; Dalla Costa, F.; Cignarella, A.; Vettore, M.; Di Gangi, I.M.; et al. Interplay between gut microbiota and p66Shc affects obesity-associated insulin resistance. FASEB J. 2018, 32, 4004–4015. [Google Scholar] [CrossRef] [PubMed]
- Illiano, P.; Brambilla, R.; Parolini, C. The mutual interplay of gut microbiota, diet and human disease. FEBS J. 2020, 287, 833–855. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front. Cell Infect. Microbiol. 2021, 11, 625913. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Backhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Ballard, J.W.O.; Towarnicki, S.G. Mitochondria, the gut microbiome and ROS. Cell. Signal. 2020, 75, 109737. [Google Scholar] [CrossRef]
- Uchiyama, J.; Akiyama, M.; Hase, K.; Kumagai, Y.; Kim, Y.G. Gut microbiota reinforce host antioxidant capacity via the generation of reactive sulfur species. Cell Rep. 2022, 38, 110479. [Google Scholar] [CrossRef]
- Galimov, E.R. The Role of p66shc in Oxidative Stress and Apoptosis. Acta Naturae 2010, 2, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, E.; Mele, S.; Salcini, A.E.; Pelicci, G.; Lai, K.M.; Superti-Furga, G.; Pawson, T.; Di Fiore, P.P.; Lanfrancone, L.; Pelicci, P.G. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 1997, 16, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, Y.R.; Vikram, A.; Naqvi, A.; Li, Q.; Kassan, M.; Kumar, V.; Bachschmid, M.M.; Jacobs, J.S.; Kumar, A.; et al. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2017, 114, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Faisal, A.; el-Shemerly, M.; Hess, D.; Nagamine, Y. Serine/threonine phosphorylation of ShcA. Regulation of protein-tyrosine phosphatase-pest binding and involvement in insulin signaling. J. Biol. Chem. 2002, 277, 30144–30152. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Bornbaum, J.; Schluter, K.D.; Schulz, R. P66shc and its role in ischemic cardiovascular diseases. Basic Res. Cardiol. 2019, 114, 29. [Google Scholar] [CrossRef] [PubMed]
- Haslem, L.; Hays, J.M.; Hays, F.A. p66Shc in Cardiovascular Pathology. Cells 2022, 11, 1855. [Google Scholar] [CrossRef]
- Ciciliot, S.; Fadini, G.P. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66(Shc). Int. J. Mol. Sci. 2019, 20, 985. [Google Scholar] [CrossRef]
- Mousavi, S.; Khazeei Tabari, M.A.; Bagheri, A.; Samieefar, N.; Shaterian, N.; Kelishadi, R. The Role of p66Shc in Diabetes: A Comprehensive Review from Bench to Bedside. J. Diabetes Res. 2022, 2022, 7703520. [Google Scholar] [CrossRef]
- Hughes, W.E.; Hockenberry, J.; Miller, B.; Sorokin, A.; Beyer, A.M. Modulation of p66Shc impairs cerebrovascular myogenic tone in low renin but not low nitric oxide models of systemic hypertension. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H1096–H1102. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Gadi, I.; Nazir, S.; Al-Dabet, M.M.; Kohli, S.; Bock, F.; Breitenstein, L.; Ranjan, S.; Fuchs, T.; Halloul, Z.; et al. Activated protein C reverses epigenetically sustained p66(Shc) expression in plaque-associated macrophages in diabetes. Commun. Biol. 2018, 1, 104. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Mitchell, K.; Mohammed, S.A.; Hussain, S.; Gkolfos, C.; Berrino, L.; Volpe, M.; Schwarzwald, C.; Luscher, T.F.; et al. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66(Shc). Int. J. Cardiol. 2018, 268, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Vashistha, H.; Marrero, L.; Reiss, K.; Cohen, A.J.; Malhotra, A.; Javed, T.; Bradley, A.; Abbruscato, F.; Giusti, S.; Jimenez, A.; et al. Aging phenotype(s) in kidneys of diabetic mice are p66ShcA dependent. Am. J. Physiol. Renal Physiol. 2018, 315, F1833–F1842. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Krankel, N.; Cosentino, F.; Luscher, T.F. Reprogramming ageing and longevity genes restores paracrine angiogenic properties of early outgrowth cells. Eur. Heart J. 2016, 37, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Vono, R.; Fuoco, C.; Testa, S.; Pirro, S.; Maselli, D.; Ferland McCollough, D.; Sangalli, E.; Pintus, G.; Giordo, R.; Finzi, G.; et al. Activation of the Pro-Oxidant PKCbetaII-p66Shc Signaling Pathway Contributes to Pericyte Dysfunction in Skeletal Muscles of Patients With Diabetes With Critical Limb Ischemia. Diabetes 2016, 65, 3691–3704. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.; Montecucco, F.; Braunersreuther, V.; Camici, G.G.; Jakob, P.; Reiner, M.F.; Glanzmann, M.; Burger, F.; Paneni, F.; Galan, K.; et al. Genetic deletion of the adaptor protein p66Shc increases susceptibility to short-term ischaemic myocardial injury via intracellular salvage pathways. Eur. Heart J. 2015, 36, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Spescha, R.D.; Klohs, J.; Semerano, A.; Giacalone, G.; Derungs, R.S.; Reiner, M.F.; Rodriguez Gutierrez, D.; Mendez-Carmona, N.; Glanzmann, M.; Savarese, G.; et al. Post-ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur. Heart J. 2015, 36, 1590–1600. [Google Scholar] [CrossRef]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef]
- Shi, Y.; Savarese, G.; Perrone-Filardi, P.; Luscher, T.F.; Camici, G.G. Enhanced age-dependent cerebrovascular dysfunction is mediated by adaptor protein p66Shc. Int. J. Cardiol. 2014, 175, 446–450. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Cosentino, F. p66(Shc)-induced redox changes drive endothelial insulin resistance. Atherosclerosis 2014, 236, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Spescha, R.D.; Glanzmann, M.; Simic, B.; Witassek, F.; Keller, S.; Akhmedov, A.; Tanner, F.C.; Luscher, T.F.; Camici, G.G. Adaptor protein p66(Shc) mediates hypertension-associated, cyclic stretch-dependent, endothelial damage. Hypertension 2014, 64, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Naqvi, A.; Hoffman, T.A.; Kumar, A.; Miller, F.J., Jr.; Kim, C.S.; Irani, K. Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc-regulated reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, G.; Zhai, X.; Hu, Y.; Gao, D.; Ma, L.; Yao, J.; Tian, X. Selective inhibition of protein kinase C beta2 attenuates the adaptor P66 Shc-mediated intestinal ischemia-reperfusion injury. Cell Death Dis. 2014, 5, e1164. [Google Scholar] [CrossRef] [PubMed]
- Bellisario, V.; Berry, A.; Capoccia, S.; Raggi, C.; Panetta, P.; Branchi, I.; Piccaro, G.; Giorgio, M.; Pelicci, P.G.; Cirulli, F. Gender-dependent resiliency to stressful and metabolic challenges following prenatal exposure to high-fat diet in the p66(Shc−/−) mouse. Front. Behav. Neurosci. 2014, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Spescha, R.D.; Shi, Y.; Wegener, S.; Keller, S.; Weber, B.; Wyss, M.M.; Lauinger, N.; Tabatabai, G.; Paneni, F.; Cosentino, F.; et al. Deletion of the ageing gene p66(Shc) reduces early stroke size following ischaemia/reperfusion brain injury. Eur. Heart J. 2013, 34, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Laviola, L.; Orlando, M.R.; Incalza, M.A.; Caccioppoli, C.; Melchiorre, M.; Leonardini, A.; Cignarelli, A.; Tortosa, F.; Labarbuta, R.; Martemucci, S.; et al. TNFalpha signals via p66(Shc) to induce E-Selectin, promote leukocyte transmigration and enhance permeability in human endothelial cells. PLoS ONE 2013, 8, e81930. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.; Shahzad, K.; Wang, H.; Stoyanov, S.; Wolter, J.; Dong, W.; Pelicci, P.G.; Kashif, M.; Ranjan, S.; Schmidt, S.; et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc. Natl. Acad. Sci. USA 2013, 110, 648–653. [Google Scholar] [CrossRef]
- Haslem, L.; Hays, J.M.; Schmitz, H.; Matsuzaki, S.; Sjoelund, V.; Byrum, S.D.; Humphries, K.M.; Frazer, J.K.; Demeler, B.; Benbrook, D.M.; et al. p66Shc is an apoptotic rheostat whose targeted ROS inhibition improves MI outcomes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bhat, S.S.; Anand, D.; Khanday, F.A. p66Shc as a switch in bringing about contrasting responses in cell growth: Implications on cell proliferation and apoptosis. Mol. Cancer 2015, 14, 76. [Google Scholar] [CrossRef]
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Luscher, T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Khanday, F.A.; Yamamori, T.; Mattagajasingh, I.; Zhang, Z.; Bugayenko, A.; Naqvi, A.; Santhanam, L.; Nabi, N.; Kasuno, K.; Day, B.W.; et al. Rac1 leads to phosphorylation-dependent increase in stability of the p66shc adaptor protein: Role in Rac1-induced oxidative stress. Mol. Biol. Cell 2006, 17, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH oxidases: The role of Rac proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.E.; Brunet, A. FOXO transcription factors. Curr. Biol. 2007, 17, R113–R114. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Izumiya, Y.; Walsh, K. Forkhead transcription factors and cardiovascular biology. Circ. Res. 2008, 102, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Gertsberg, Z.; Ozgen, N.; Steinberg, S.F. p66Shc links alpha1-adrenergic receptors to a reactive oxygen species-dependent AKT-FOXO3A phosphorylation pathway in cardiomyocytes. Circ. Res. 2009, 104, 660–669. [Google Scholar] [CrossRef]
- Nemoto, S.; Finkel, T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 2002, 295, 2450–2452. [Google Scholar] [CrossRef]
- Chahdi, A.; Sorokin, A. Endothelin-1 couples betaPix to p66Shc: Role of betaPix in cell proliferation through FOXO3a phosphorylation and p27kip1 down-regulation independently of Akt. Mol. Biol. Cell 2008, 19, 2609–2619. [Google Scholar] [CrossRef]
- Chahdi, A.; Sorokin, A. Endothelin-1 induces p66Shc activation through EGF receptor transactivation: Role of beta(1)Pix/Galpha(i3) interaction. Cell. Signal. 2010, 22, 325–329. [Google Scholar] [CrossRef]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem. 2004, 279, 25689–25695. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Moroni, M.; Contursi, C.; Yano, M.; Pelicci, P.; Giorgio, M.; Migliaccio, E. Regulatory effects of the mitochondrial energetic status on mitochondrial p66Shc. Biol. Chem. 2006, 387, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Hoyles, L. Human microbiome myths and misconceptions. Nat. Microbiol. 2023, 8, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Larke, J.A.; Bacalzo, N.; Castillo, J.J.; Couture, G.; Chen, Y.; Xue, Z.; Alkan, Z.; Kable, M.E.; Lebrilla, C.B.; Stephensen, C.B.; et al. Dietary Intake of Monosaccharides from Foods is Associated with Characteristics of the Gut Microbiota and Gastrointestinal Inflammation in Healthy US Adults. J. Nutr. 2023, 153, 106–119. [Google Scholar] [CrossRef]
- Ikubo, Y.; Sanada, T.J.; Hosomi, K.; Park, J.; Naito, A.; Shoji, H.; Misawa, T.; Suda, R.; Sekine, A.; Sugiura, T.; et al. Altered gut microbiota and its association with inflammation in patients with chronic thromboembolic pulmonary hypertension: A single-center observational study in Japan. BMC Pulm. Med. 2022, 22, 138. [Google Scholar] [CrossRef]
- Walker, R.L.; Vlamakis, H.; Lee, J.W.J.; Besse, L.A.; Xanthakis, V.; Vasan, R.S.; Shaw, S.Y.; Xavier, R.J. Population study of the gut microbiome: Associations with diet, lifestyle, and cardiometabolic disease. Genome Med. 2021, 13, 188. [Google Scholar] [CrossRef]
- Rohm, T.V.; Fuchs, R.; Muller, R.L.; Keller, L.; Baumann, Z.; Bosch, A.J.T.; Schneider, R.; Labes, D.; Langer, I.; Pilz, J.B.; et al. Obesity in Humans Is Characterized by Gut Inflammation as Shown by Pro-Inflammatory Intestinal Macrophage Accumulation. Front. Immunol. 2021, 12, 668654. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Yuan, J.; Li, J.; Li, H.; Yin, K.; Wang, F.; Li, D. Overweight and underweight status are linked to specific gut microbiota and intestinal tricarboxylic acid cycle intermediates. Clin. Nutr. 2020, 39, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Pinero, F.; Vazquez, M.; Bare, P.; Rohr, C.; Mendizabal, M.; Sciara, M.; Alonso, C.; Fay, F.; Silva, M. A different gut microbiome linked to inflammation found in cirrhotic patients with and without hepatocellular carcinoma. Ann. Hepatol. 2019, 18, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.; Cohen, N.A.; Shalev, V.; Uzan, A.; Koren, O.; Maharshak, N. Psoriatic patients have a distinct structural and functional fecal microbiota compared with controls. J. Dermatol. 2019, 46, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Troseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjorndal, B.; Halvorsen, B.; et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015, 277, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3–4 chronic kidney disease. Arch. Med. Res. 2014, 45, 309–317. [Google Scholar] [CrossRef]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive oxygen production induced by the gut microbiota: Pharmacotherapeutic implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef]
- Jones, R.M.; Luo, L.; Ardita, C.S.; Richardson, A.N.; Kwon, Y.M.; Mercante, J.W.; Alam, A.; Gates, C.L.; Wu, H.; Swanson, P.A.; et al. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 2013, 32, 3017–3028. [Google Scholar] [CrossRef]
- Jones, R.M.; Neish, A.S. Redox signaling mediated by the gut microbiota. Free Radic. Biol. Med. 2017, 105, 41–47. [Google Scholar] [CrossRef]
- Jeong, Y.S.; Bae, Y.S. Formyl peptide receptors in the mucosal immune system. Exp. Mol. Med. 2020, 52, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Wu, H.; Collier-Hyams, L.S.; Hansen, J.M.; Li, T.; Yamoah, K.; Pan, Z.Q.; Jones, D.P.; Neish, A.S. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007, 26, 4457–4466. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J. Bacterial-modulated signaling pathways in gut homeostasis. Sci. Signal. 2008, 1, pe24. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Chavez, F.; Zhang, L.F.; Faber, F.; Lopez, C.A.; Byndloss, M.X.; Olsan, E.E.; Xu, G.; Velazquez, E.M.; Lebrilla, C.B.; Winter, S.E.; et al. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe 2016, 19, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Farhana, A.; Khan, Y. Biochemistry, Lipopolysaccharide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- McGuckin, M.A.; Linden, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2020, 11, 594150. [Google Scholar] [CrossRef]
- Anhe, F.F.; Barra, N.G.; Cavallari, J.F.; Henriksbo, B.D.; Schertzer, J.D. Metabolic endotoxemia is dictated by the type of lipopolysaccharide. Cell Rep. 2021, 36, 109691. [Google Scholar] [CrossRef]
- Steimle, A.; Autenrieth, I.B.; Frick, J.S. Structure and function: Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Takeuchi, S.; Kubota, K.; Kobayashi, Y.; Kozakai, S.; Ukai, I.; Shichiku, A.; Okubo, M.; Numasaki, M.; Kanemitsu, Y.; et al. Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1-IKKϵ-IRF3 axis activation. J. Biol. Chem. 2018, 293, 10186–10201. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R., Jr. Metabolic endotoxemia: A molecular link between obesity and cardiovascular risk. J. Mol. Endocrinol. 2013, 51, R51–R64. [Google Scholar] [CrossRef]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013, 23, 129–134. [Google Scholar] [CrossRef]
- Wentworth, C.C.; Alam, A.; Jones, R.M.; Nusrat, A.; Neish, A.S. Enteric commensal bacteria induce extracellular signal-regulated kinase pathway signaling via formyl peptide receptor-dependent redox modulation of dual specific phosphatase 3. J. Biol. Chem. 2011, 286, 38448–38455. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Sanchez-Alcazar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Trinei, M.; Berniakovich, I.; Pelicci, P.G.; Giorgio, M. Mitochondrial DNA copy number is regulated by cellular proliferation: A role for Ras and p66(Shc). Biochim. Biophys. Acta Bioenerg. 2006, 1757, 624–630. [Google Scholar] [CrossRef]
- Blank, H.M.; Li, C.; Mueller, J.E.; Bogomolnaya, L.M.; Bryk, M.; Polymenis, M. An increase in mitochondrial DNA promotes nuclear DNA replication in yeast. PLoS Genet. 2008, 4, e1000047. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Baumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Luche, E.; Cousin, B.; Garidou, L.; Serino, M.; Waget, A.; Barreau, C.; Andre, M.; Valet, P.; Courtney, M.; Casteilla, L.; et al. Metabolic endotoxemia directly increases the proliferation of adipocyte precursors at the onset of metabolic diseases through a CD14-dependent mechanism. Mol. Metab. 2013, 2, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Postigo, M.; Oliva-Olivera, W.; Coin-Araguez, L.; Ramos-Molina, B.; Giraldez-Perez, R.M.; Lhamyani, S.; Alcaide-Torres, J.; Perez-Martinez, P.; El Bekay, R.; Cardona, F.; et al. Metabolic endotoxemia promotes adipose dysfunction and inflammation in human obesity. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E319–E332. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, S.C.; Fusco, S.; Pani, G. p66(ShcA): Linking mammalian longevity with obesity-induced insulin resistance. Vitam. Horm. 2013, 91, 219–241. [Google Scholar] [CrossRef] [PubMed]
- Berniakovich, I.; Trinei, M.; Stendardo, M.; Migliaccio, E.; Minucci, S.; Bernardi, P.; Pelicci, P.G.; Giorgio, M. p66Shc-generated oxidative signal promotes fat accumulation. J. Biol. Chem. 2008, 283, 34283–34293. [Google Scholar] [CrossRef]
- Ciciliot, S.; Albiero, M.; Menegazzo, L.; Poncina, N.; Scattolini, V.; Danesi, A.; Pagnin, E.; Marabita, M.; Blaauw, B.; Giorgio, M.; et al. p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans. Diabetologia 2015, 58, 2352–2360. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef]
- Park, J.C.; Im, S.H. Of men in mice: The development and application of a humanized gnotobiotic mouse model for microbiome therapeutics. Exp. Mol. Med. 2020, 52, 1383–1396. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Paz, K.; Hemi, R.; LeRoith, D.; Karasik, A.; Elhanany, E.; Kanety, H.; Zick, Y. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 29911–29918. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Ranieri, S.C.; Fusco, S.; Panieri, E.; Labate, V.; Mele, M.; Tesori, V.; Ferrara, A.M.; Maulucci, G.; De Spirito, M.; Martorana, G.E.; et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 13420–13425. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Moreira, A.P.; Texeira, T.F.; Ferreira, A.B.; Peluzio Mdo, C.; Alfenas Rde, C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 2012, 108, 801–809. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef]
- Freeman, A.M.; Acevedo, L.A.; Pennings, A. Insulin Resistance. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507839/ (accessed on 14 February 2024).
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Van Hul, M.; Cani, P.D. The gut microbiota in obesity and weight management: Microbes as friends or foe? Nat. Rev. Endocrinol. 2023, 19, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 128, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Tomilov, A.A.; Ramsey, J.J.; Hagopian, K.; Giorgio, M.; Kim, K.M.; Lam, A.; Migliaccio, E.; Lloyd, K.C.; Berniakovich, I.; Prolla, T.A.; et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011, 10, 55–65. [Google Scholar] [CrossRef]
- Soliman, M.A.; Abdel Rahman, A.M.; Lamming, D.W.; Birsoy, K.; Pawling, J.; Frigolet, M.E.; Lu, H.; Fantus, I.G.; Pasculescu, A.; Zheng, Y.; et al. The adaptor protein p66Shc inhibits mTOR-dependent anabolic metabolism. Sci. Signal. 2014, 7, ra17. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author | Year | Study Population or Model | Pathophysiological Condition |
| W. E. Hughes [21] | 2021 | Animal (rats) | Hypertension |
| K. Shahzad [22] | 2018 | Animal (mice) | Hyperglycemia-induced atherosclerosis |
| S. Costantino [23] | 2018 | Animal (mice) Cell culture (human cardiomyocytes) | Diabetes-related cardiomyopathy |
| H. Vashistha [24] | 2018 | Animal (mice) Cell culture (Sca-1+ mesenchymal stem cells) | Diabetes-related renal dysfunction |
| F. Paneni [25] | 2016 | Cell culture (early outgrowth cells) | Age-related impaired vascular repair |
| R. Vono [26] | 2016 | Humans (patients with diabetes undergoing major limb amputation) | Diabetes-related critical limb ischemia |
| A. Akhmedov [27] | 2015 | Animal (mice) | Cardiac ischemia and reperfusion |
| R. D. Spescha [28] | 2015 | Animal (mice) Cell culture (primary HBMVECs) Human (acute ischemic stroke patients) | Ischemia/reperfusion brain injury; stroke |
| A. Natalicchio [29] | 2015 | Animal (mice) Cell culture (rat INS-1E cells; murine, human, and mouse islets) | Hyperglycemia |
| Y. Shi [30] | 2014 | Animal (mice) | Age-related cerebrovascular impairment |
| F. Paneni [31] | 2014 | Animal (mice) | Obesity-induced endothelial insulin resistance |
| R. D. Spescha [32] | 2014 | Cell culture (primary human AECs and rat AECs) | Hypertension |
| A. Vikram [33] | 2014 | Animal (mice) Cell culture (various) | Endothelial dysfunction |
| Z. Chen [34] | 2014 | Animal (mice) Cell culture (Caco-2 cells) | Ischemia/reperfusion intestinal injury |
| V. Bellisario [35] | 2014 | Animal (mice) | Detrimental developmental programming |
| R. D. Spescha [36] | 2013 | Animal (mice) | Ischemia/reperfusion brain injury; stroke |
| L. Laviola [37] | 2013 | Cell culture (HUVECs) | Endothelial dysfunction |
| F. Bock [38] | 2013 | Animal (mice) | Diabetes-related nephropathy |
| First Author | Year | Study Population | Pathophysiological Condition |
| J. A. Larke [58] | 2023 | Healthy adults | Gastrointestinal inflammation |
| Y. Ikubo [59] | 2022 | Patients with CTEPH | Pulmonary hypertension |
| R. L. Walker [60] | 2021 | Framingham Heart Study cohort | Cardiometabolic diseases |
| T. V. Rohm [61] | 2021 | Obese and non-obese adults | Obesity |
| X. Wang [62] | 2020 | Patients with ESRD | Renal disease |
| Y. Wan [63] | 2020 | Adults in different BMI categories | Cardiometabolic diseases |
| F. Piñero [64] | 2019 | Patients with cirrhosis | Liver cancer |
| J. Shapiro [65] | 2019 | Patients with psoriasis | Autoimmune diseases |
| M. Trøseid [66] | 2015 | Patients with chronic heart failure | Cardiovascular diseases |
| M. Rossi [67] | 2014 | Patients with CKD | Renal disease |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da C. Pinaffi-Langley, A.C.; Melia, E.; Hays, F.A. Exploring the Gut–Mitochondrial Axis: p66Shc Adapter Protein and Its Implications for Metabolic Disorders. Int. J. Mol. Sci. 2024, 25, 3656. https://doi.org/10.3390/ijms25073656
da C. Pinaffi-Langley AC, Melia E, Hays FA. Exploring the Gut–Mitochondrial Axis: p66Shc Adapter Protein and Its Implications for Metabolic Disorders. International Journal of Molecular Sciences. 2024; 25(7):3656. https://doi.org/10.3390/ijms25073656
Chicago/Turabian Styleda C. Pinaffi-Langley, Ana Clara, Elizabeth Melia, and Franklin A. Hays. 2024. "Exploring the Gut–Mitochondrial Axis: p66Shc Adapter Protein and Its Implications for Metabolic Disorders" International Journal of Molecular Sciences 25, no. 7: 3656. https://doi.org/10.3390/ijms25073656
APA Styleda C. Pinaffi-Langley, A. C., Melia, E., & Hays, F. A. (2024). Exploring the Gut–Mitochondrial Axis: p66Shc Adapter Protein and Its Implications for Metabolic Disorders. International Journal of Molecular Sciences, 25(7), 3656. https://doi.org/10.3390/ijms25073656