
Synthesis of Novel Triazine-Based Chalcones and 8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepines as Potential Leads in the Search of Anticancer, Antibacterial and Antifungal Agents

 ,
,  , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results and Discussion
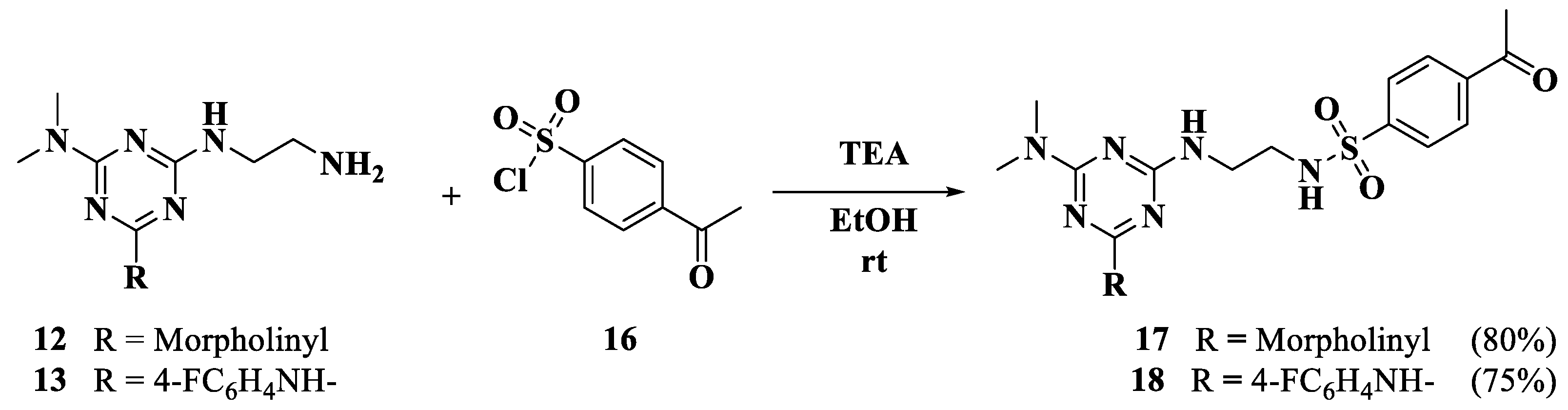
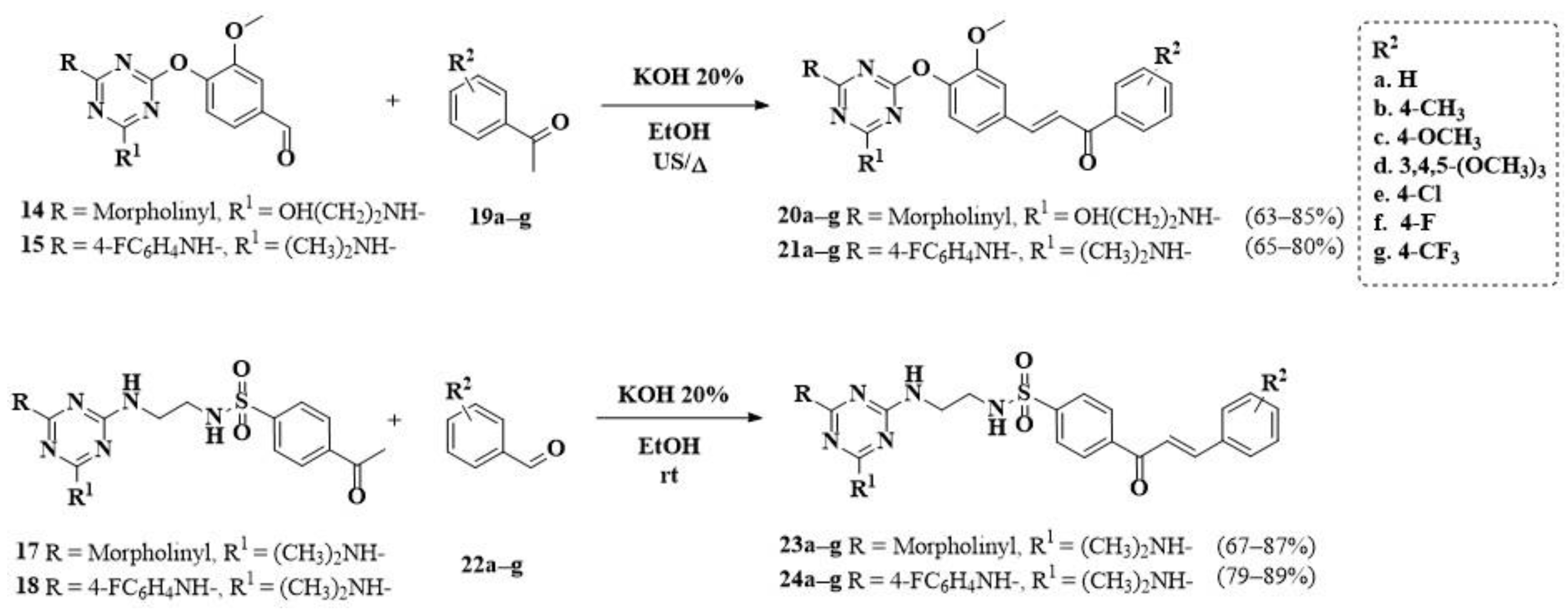
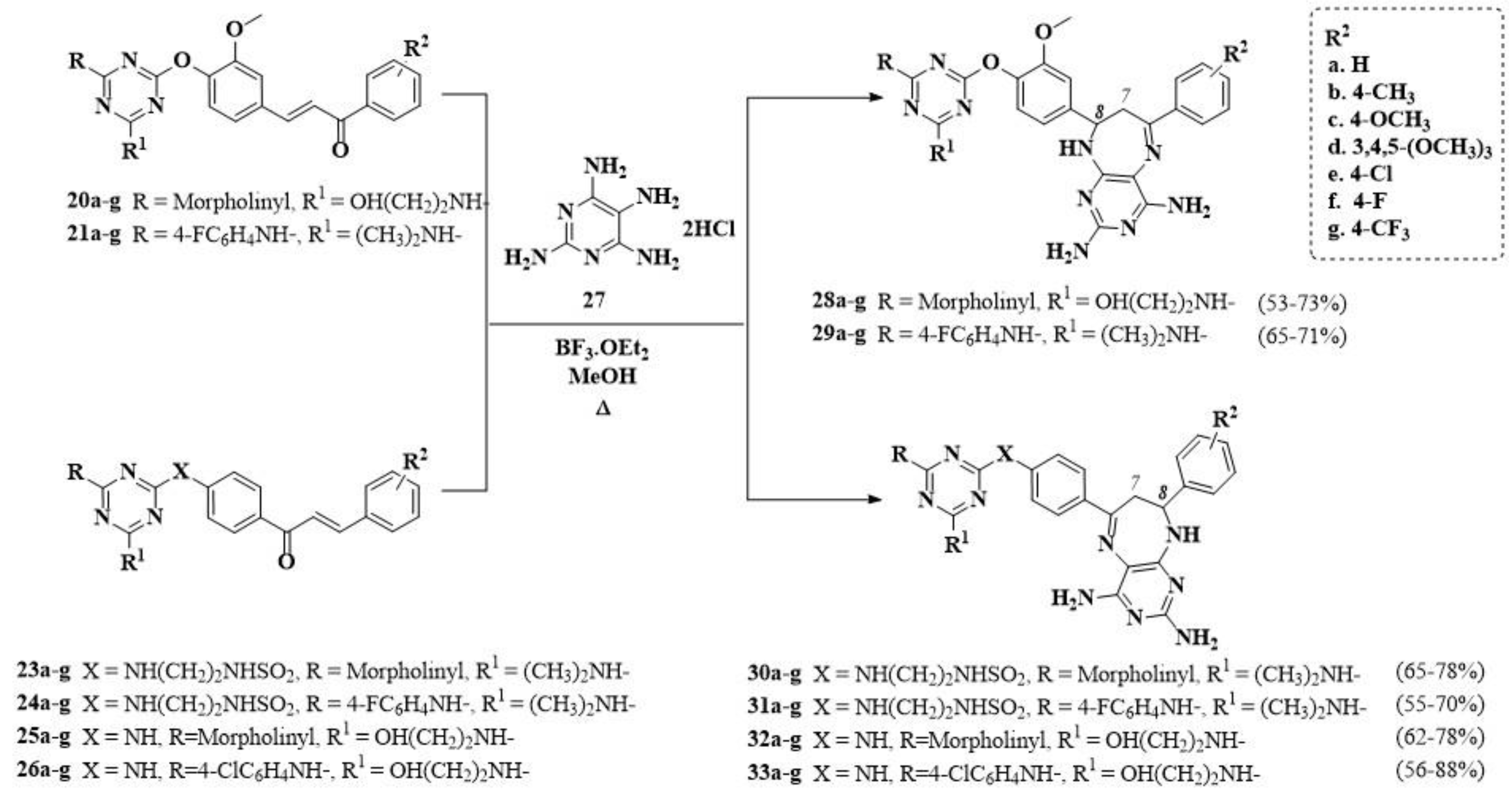
2.1. Chemistry
2.2. Biological Activity Studies
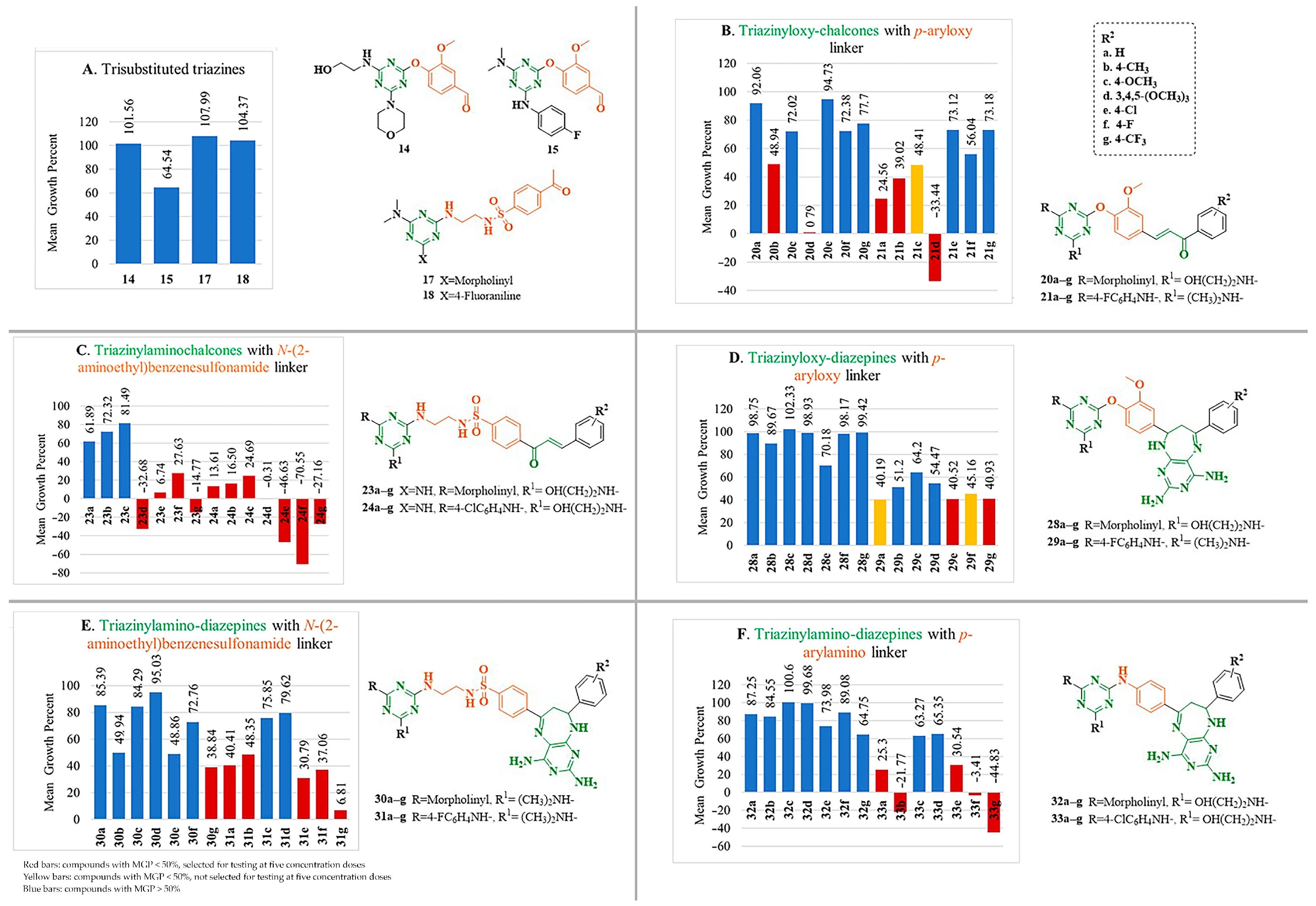
2.2.1. Anticancer Activity
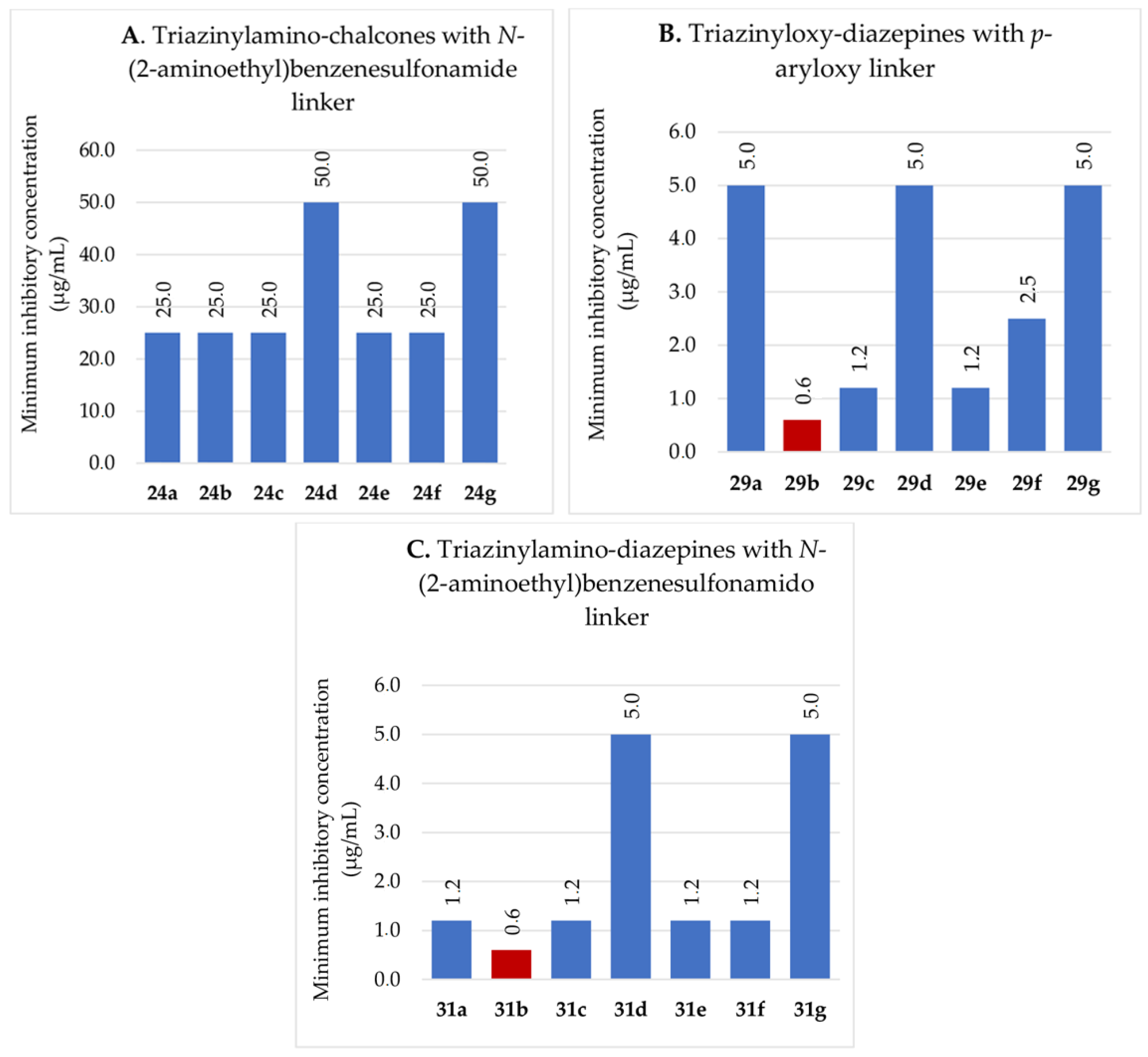
2.2.2. Antibacterial Activity
Antitubercular Activity
2.2.3. Antifungal Activity
2.2.4. In Silico Physicochemical Parameter Predictions
2.2.5. Hemolytic Activity
2.2.6. Toxicity Studies
3. Materials and Methods
3.1. Chemistry
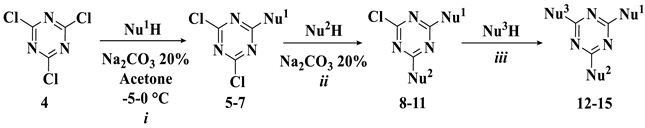
3.1.1. General Procedure for the Synthesis of Monosubstituted Triazines (5–7)
3.1.2. General Procedure for the Synthesis of Disubstituted Triazines (8–11)
3.1.3. Procedure for the Synthesis of Trisubstituted Triazine (12)
3.1.4. Procedure for the Synthesis of Trisubstituted Triazine (13)
3.1.5. Procedure for the Synthesis of Trisubstituted Triazine (14)
3.1.6. Procedure for the Synthesis of Trisubstituted Triazine (15)
3.1.7. General Procedure for the Synthesis of Trisubstituted Triazines (17,18)
3.1.8. General Procedure for the Synthesis of Chalcones (23,24a–g)
3.1.9. General Procedure for the Synthesis of Chalcones (20a–g)
3.1.10. General Procedure for the Synthesis of Chalcones (21a–g)
3.1.11. General Procedure for the Synthesis of the 8,9-dihydro-7H-pyrimido[4,5-b][1,4]Diazepines (28–33)a–g
3.1.12. Anticancer Activity
3.1.13. Antibacterial Activity
3.1.14. Antifungal Activity
3.1.15. Hemolytic Activity
3.1.16. Toxicity Studies In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Cancer Today. Available online: https://gco.iarc.fr/today/home (accessed on 17 June 2023).
- World Cancer Day 2022: Close the Care Gap—PAHO/WHO|Pan American Health Organization. Available online: https://www.paho.org/en/campaigns/world-cancer-day-2022-close-care-gap (accessed on 17 June 2023).
- No Time to Wait: Securing the Future from Drug-Resistant Infections. Available online: https://www.who.int/publications/i/item/no-time-to-wait-securing-the-future-from-drug-resistant-infections (accessed on 17 June 2023).
- Bhadoriya, U.; Kumar Jain, D. Fused Heterocycles As a Potent Biological Agents; Recent Advancement. Int. J. Pharm. Sci. Res. 2016, 7, 1874–1880. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.; Jonnalagadda, S. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Sprague, D.J.; Getschman, A.E.; Fenske, T.G.; Volkman, B.F.; Smith, B.C. Trisubstituted 1,3,5-Triazines: The First Ligands of the SY12-Binding Pocket on Chemokine CXCL12. ACS Med. Chem. Lett. 2021, 12, 1773–1782. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Paul, K. Synthesis, in Vitro Antitumor Activity, Dihydrofolate Reductase Inhibition, DNA Intercalation and Structure-Activity Relationship Studies of 1,3,5-Triazine Analogues. Bioorg. Med. Chem. Lett. 2016, 26, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Hashem, H.; Amr, A.E.G.; Nossier, E.; Anwar, M.; Azmy, E. New Benzimidazole-, 1,2,4-Triazole-, and 1,3,5-Triazine-Based Derivatives as Potential EGFR WT and EGFR T790M Inhibitors: Microwave-Assisted Synthesis, Anticancer Evaluation, and Molecular Docking Study. ACS Omega 2022, 7, 7155–7171. [Google Scholar] [CrossRef]
- Havránková, E.; Čalkovská, N.; Padrtová, T.; Csöllei, J.; Opatřilová, R.; Pazdera, P. Antioxidative Activity of 1,3,5-Triazine Analogues Incorporating Aminobenzene Sulfonamide, Aminoalcohol/Phenol, Piperazine, Chalcone, or Stilbene Motifs. Molecules 2020, 25, 1787. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Reddy, R.; Subba, V.; Gopireddy, R.; Poola, S.; Krishna, S.; Chintha, V. Ethyl-4-(Aryl)-6-Methyl-2-(Oxo/Thio)-3,4-Dihydro-1H-Pyrimidine-5-Carboxylates: Silica Supported Bismuth (III) Triflate Catalyzed Synthesis and Antioxidant Activity. Bioorg. Chem. 2022, 129, 106205. [Google Scholar] [CrossRef]
- Fader, L.; Bethell, R.; Bonneau, P.; Bös, M.; Bousquet, Y.; Cordingley, M.; Coulombe, R.; Deroy, P.; Faucher, A.M.; Gagnon, A.; et al. Discovery of a 1,5-Dihydrobenzo[b][1,4]Diazepine-2,4-Dione Series of Inhibitors of HIV-1 Capsid Assembly. Bioorg. Med. Chem. Lett. 2011, 21, 398–404. [Google Scholar] [CrossRef]
- Ludovici, D.; De Corte, B.; Kukla, M.; Ye, H.; Ho, C.; Lichtenstein, M.; Kavash, R.; Andries, K.; De Béthune, M.P.; Azijn, H.; et al. Evolution of Anti-HIV Drug Candidates. Part 2: Diaryltriazine (DATA) Analogues. Bioorg. Med. Chem. Lett. 2001, 11, 2235–2239. [Google Scholar] [CrossRef]
- Okazaki, S.; Mizuhara, T.; Shimura, K.; Murayama, H.; Ohno, H. Identification of Anti-HIV Agents with a Novel Benzo[4,5]Isothiazolo[2,3-a]Pyrimidine Scaffold. Bioorg. Med. Chem. 2015, 23, 1447–1452. [Google Scholar] [CrossRef]
- El-Subbagh, H.; Hassan, G.S.; El-Azab, A.S.; Abdel-Aziz, A.; Kadi, A.A.; Al-Obaid, A.; Al-Shabanah, O.; Sayed-Ahmed, M. Synthesis and Anticonvulsant Activity of Some New Thiazolo[3,2-a][1,3] Diazepine, Benzo[d]Thiazolo[5,2-a][12,6]Diazepine and Benzo[d]Oxazolo[5,2-a][12, 6]Diazepine Analogues. Eur. J. Med. Chem. 2011, 46, 5567–5572. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Jawaid Akhtar, M.; Raj, K.; Singh, S.; Sharma, P.; Kalra, S.; Chawla, P.; Kumar, B. Design, Synthesis and Evaluation of Piperazine Clubbed 1,2,4-Triazine Derivatives as Potent Anticonvulsant Agents. J. Mol. Struct. 2022, 1257, 132587. [Google Scholar] [CrossRef]
- Sethuvasan, S.; Sugumar, P.; Ponnuswamy, M.N.; Ponnuswamy, S. Synthesis, Spectral Characterization and Conformational Assignment of N-Formyl-2,7-Diaryl-1,4-Diazepan-5-Ones as Potent Antibacterial Agents and Type I DHQase Inhibitors. J. Mol. Struct. 2021, 1236, 130293. [Google Scholar] [CrossRef]
- Tan, Y.; Li, D.; Li, F.; Fawad, M.; Fang, B.; Zhou, C. Pyrimidine-Conjugated Fluoroquinolones as New Potential Broad-Spectrum Antibacterial Agents. Bioorg. Med. Chem. Lett. 2022, 73, 128885. [Google Scholar] [CrossRef]
- Gupta, S.; Paul, K. Membrane-Active Substituted Triazines Antibacterial Agents against Staphylococcus Aureus with Potential for Low Drug Resistance and Broad Activity. Eur. J. Med. Chem. 2023, 258, 115551. [Google Scholar] [CrossRef] [PubMed]
- Gour, J.; Gatadi, S.; Pooladanda, V.; Ghouse, S.M.; Malasala, S.; Madhavi, Y.V.; Godugu, C.; Nanduri, S. Facile Synthesis of 1,2,3-Triazole-Fused Indolo- and Pyrrolo[1,4]Diazepines, DNA-Binding and Evaluation of Their Anticancer Activity. Bioorg. Chem. 2019, 93, 103306. [Google Scholar] [CrossRef] [PubMed]
- Ali, W.; Garbo, S.; Kincses, A.; Nové, M.; Spengler, G.; Di Bello, E.; Honkisz-Orzechowska, E.; Karcz, T.; Szymańska, E.; Żesławska, E.; et al. Seleno-vs. Thioether Triazine Derivatives in Search for New Anticancer Agents Overcoming Multidrug Resistance in Lymphoma. Eur. J. Med. Chem. 2022, 243, 114761. [Google Scholar] [CrossRef]
- Timaniya, J.B.; Parikh, P.H.; Patel, M.J.; Dave, G.; Patel, K.P. Design, Synthesis and Evaluation of Novel Substituted Fused Pyrido Diazepine and Pyrimido Piperazine Derivatives: In Vitro Cytotoxicity Study over Various Cancer Cell Lines. Results Chem. 2023, 5, 100707. [Google Scholar] [CrossRef]
- Dai, X.; Xue, L.; Ji, S.; Zhou, Y.; Gao, Y.; Zheng, Y. Triazole-Fused Pyrimidines in Target-Based Anticancer Drug Discovery. Eur. J. Med. Chem. 2023, 249, 115101. [Google Scholar] [CrossRef]
- Venkatraj, M.; Ariën, K.; Heeres, J.; Joossens, J.; Messagie, J.; Michiels, J.; Van Der Veken, P.; Vanham, G.; Lewi, P.; Augustyns, K. Synthesis, Evaluation and Structure-Activity Relationships of Triazine Dimers as Novel Antiviral Agents. Bioorg. Med. Chem. Lett. 2012, 22, 7174–7178. [Google Scholar] [CrossRef] [PubMed]
- Shahari, M.S.B.; Dolzhenko, A.V. A Closer Look at N2,6-Substituted 1,3,5-Triazine-2,4-Diamines: Advances in Synthesis and Biological Activities. Eur. J. Med. Chem. 2022, 241, 114645. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spanò, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. 1,3,5-Triazines: A Promising Scaffold for Anticancer Drugs Development. Eur. J. Med. Chem. 2017, 142, 523–549. [Google Scholar] [CrossRef]
- Vathanaruba, M.; Raja, S.J.; Princess, R.; Tharmaraj, P. Pharmacological and Molecular Docking Studies of New Copper (II) Complexes of N2-Phenyl-N4,N6-Di(Thiazol-2-Yl)-1,3,5-Triazine-2,4,6-Triamine. J. Mol. Struct. 2022, 1253, 132275. [Google Scholar] [CrossRef]
- Miroslav, S.; Bouillon, I.; Krchňák, V. Combinatorial Libraries of Bis-Heterocyclic Compounds with Skeletal Diversity. J. Comb. Chem. 2008, 10, 923–933. [Google Scholar] [CrossRef]
- Wu, W.L.; Wen, Z.Y.; Qian, J.J.; Zou, J.P.; Liu, S.M.; Yang, S.; Qin, T.; Yang, Q.; Liu, Y.H.; Liu, W.W.; et al. Design, Synthesis, Characterization and Evaluation of 1,3,5-Triazine-Benzimidazole Hybrids as Multifunctional Acetylcholinesterases Inhibitors. J. Mol. Struct. 2022, 1257, 132498. [Google Scholar] [CrossRef]
- Kucwaj-Brysz, K.; Ali, W.; Kurczab, R.; Sudoł-Tałaj, S.; Wilczyńska-Zawal, N.; Jastrzębska-Więsek, M.; Satała, G.; Mordyl, B.; Żesławska, E.; Agnieszka-Olejarz-Maciej; et al. An Exit beyond the Pharmacophore Model for 5-HT6R Agents—A New Strategy to Gain Dual 5-HT6/5-HT2A Action for Triazine Derivatives with Procognitive Potential. Bioorg. Chem. 2022, 121, 105695. [Google Scholar] [CrossRef]
- Green, K.; Pang, A.; Thamban, N.; Garzan, A.; Baughn, A.; Tsodikov, O.; Garneau-Tsodikova, S. Discovery and Optimization of 6-(1-Substituted Pyrrole-2-Yl)-s-Triazine Containing Compounds as Antibacterial Agents. ACS Infect. Dis. 2022, 8, 757–767. [Google Scholar] [CrossRef]
- El-Wakil, M.H.; Khattab, S.N.; El-Yazbi, A.F.; El-Nikhely, N.; Soffar, A.; Khalil, H.H. New Chalcone-Tethered 1,3,5-Triazines Potentiate the Anticancer Effect of Cisplatin against Human Lung Adenocarcinoma A549 Cells by Enhancing DNA Damage and Cell Apoptosis. Bioorg. Chem. 2020, 105, 104393. [Google Scholar] [CrossRef]
- Solankee, A.; Kapadia, K.; Ćirić, A.; Soković, M.; Doytchinova, I.; Geronikaki, A. Synthesis of Some New S-Triazine Based Chalcones and Their Derivatives as Potent Antimicrobial Agents. Eur. J. Med. Chem. 2010, 45, 510–518. [Google Scholar] [CrossRef]
- Subbarayal, R.D.; Dannana, G.S.; Vasudeva, R.A.; Venkata, S.M.B. Synthesis, Characterization and in Vitro Biological Evaluation of Some New 1,3,5-triazine-chalcone Hybrid Molecules as Mycobacterium Tuberculosis H37Rv Inhibitors. Eur. J. Chem. 2014, 5, 570–577. [Google Scholar] [CrossRef]
- Shor, R.E.; Dai, J.; Lee, S.Y.; Pisarsky, L.; Matei, I.; Lucotti, S.; Lyden, D.; Bissell, M.J.; Ghajar, C.M. The PI3K/MTOR Inhibitor Gedatolisib Eliminates Dormant Breast Cancer Cells in Organotypic Culture, but Fails to Prevent Metastasis in Preclinical Settings. Mol. Oncol. 2022, 16, 130–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, G.; Li, L.; Liu, Y.; Wang, F.; Song, X.; Mao, Y. New and Practical Synthesis of Gedatolisib. Org. Process Res. Dev. 2018, 22, 62–66. [Google Scholar] [CrossRef]
- Venkatesan, A.M.; Dehnhardt, C.M.; Delos Santos, E.D.; Chen, Z.; Dos Santos, O.D.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis(Morpholino-l,3,5-Triazine) Derivatives: Potent Adenosine 5′-Triphosphate Competitive Phosphatidylinositol-3-Kinase/Mammalian Target of Rapamycin Inhibitors: Discovery of Compound 26 (PKI-587), a Highly Efficacious Dual Inhibitor. J. Med. Chem. 2010, 53, 2636–2645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tortorella, M. Molecular Design of Dual Inhibitors of PI3K and Potential Molecular Target of Cancer for Its Treatment: A Review. Eur. J. Med. Chem. 2022, 228, 114039. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Song, S.; Wang, X.; Hao, J. Small-Molecule Inhibitors of Breast Cancer-Related Targets: Potential Therapeutic Agents for Breast Cancer. Eur. J. Med. Chem. 2021, 210, 112954. [Google Scholar] [CrossRef] [PubMed]
- Elkanzi, N.A.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef]
- Wong, E. The Role of Chalcones and Flavanones in Flavonoid Biosynthesis. Phytochemistry 1968, 7, 1751–1758. [Google Scholar] [CrossRef]
- Comert Onder, F.; Kahraman, N.; Bellur Atici, E.; Cagir, A.; Kandemir, H.; Tatar, G.; Taskin Tok, T.; Kara, G.; Karliga, B.; Durdagi, S.; et al. Target-Driven Design of a Coumarinyl Chalcone Scaffold Based Novel EF2 Kinase Inhibitor Suppresses Breast Cancer Growth in Vivo. ACS Pharmacol. Transl. Sci. 2021, 4, 926–940. [Google Scholar] [CrossRef]
- Dan, W.; Dai, J. Recent Developments of Chalcones as Potential Antibacterial Agents in Medicinal Chemistry. Eur. J. Med. Chem. 2020, 187, 111980. [Google Scholar] [CrossRef]
- Ould Lamara, K.; Makhloufi-Chebli, M.; Benazzouz-Touami, A.; Terrachet-Bouaziz, S.; Robert, A.; Machado-Rodrigues, C.; Behr, J.B. Synthesis, Biological Activities of Chalcones and Novel 4-Acetylpyridine Oximes, Molecular Docking of the Synthesized Products as Acetylcholinesterase Ligands. J. Mol. Struct. 2022, 1252, 132153. [Google Scholar] [CrossRef]
- Dorababu, A.; Vijayalaxmi, S.; Sanjeevamurthy, R.; Vidya, L.; Prasannakumar, R.; Raghavendra, M. Identification of Quinoline-Chalcones and Heterocyclic Chalcone-Appended Quinolines as Broad-Spectrum Pharmacological Agents. Bioorg. Chem. 2020, 105, 104419. [Google Scholar] [CrossRef]
- Ur Rashid, H.; Xu, Y.; Ahmad, N.; Muhammad, Y.; Wang, L. Promising Anti-Inflammatory Effects of Chalcones via Inhibition of Cyclooxygenase, Prostaglandin E 2, Inducible NO Synthase and Nuclear Factor Κb Activities. Bioorg. Chem. 2019, 87, 335–365. [Google Scholar] [CrossRef] [PubMed]
- Urmann, C.; Bieler, L.; Priglinger, E.; Aigner, L.; Couillard-Despres, S.; Riepl, H.M. Neuroregenerative Potential of Prenyl- And Pyranochalcones: A Structure-Activity Study. J. Nat. Prod. 2021, 84, 2675–2682. [Google Scholar] [CrossRef]
- National Center for Advancing Translational Sciences (NCATS) Metochalcone. Available online: https://drugs.ncats.io/substance/1754ZE4075 (accessed on 14 March 2022).
- Higuchi, K.; Watanabe, T.; Tanigawa, T.; Tominaga, K.; Fujiwara, Y.; Arakawa, T. Sofalcone, a Gastroprotective Drug, Promotes Gastric Ulcer Healing Following Eradication Therapy for Helicobacter Pylori: A Randomized Controlled Comparative Trial with Cimetidine, an H2-Receptor Antagonist. J. Gastroenterol. Hepatol. 2010, 25, 155–160. [Google Scholar] [CrossRef]
- Moreno, L.M.; Quiroga, J.; Abonia, R.; Lauria, A.; Martorana, A.; Insuasty, H.; Insuasty, B. Synthesis, Biological Evaluation, and: In Silico Studies of Novel Chalcone: In Pyrazoline-Based 1,3,5-Triazines as Potential Anticancer Agents. RSC Adv. 2020, 10, 34114–34129. [Google Scholar] [CrossRef]
- Longley, D.; Harkin, D.; Johnston, P. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Rose, M.G.; Farrell, M.P.; Schmitz, J.C. Thymidylate Synthase: A Critical Target for Cancer Chemotherapy. Clin. Colorectal Cancer 2002, 1, 220–229. [Google Scholar] [CrossRef]
- Mohammed, K.; Elbeily, E.; El-Taweel, F.; Fadda, A. Synthesis, Characterization, and Antioxidant Evaluation of Some Novel Pyrazolo[3,4-c][1,2]Diazepine and Pyrazolo[3,4-c]Pyrazole Derivatives. J. Heterocycl. Chem. 2019, 56, 493–500. [Google Scholar] [CrossRef]
- Ramírez, J.; Svetaz, L.; Quiroga, J.; Abonia, R.; Raimondi, M.; Zacchino, S.; Insuasty, B. Synthesis of Novel Thiazole-Based 8,9-Dihydro-7H-Pyrimido[4,5-b] [1,4]Diazepines as Potential Antitumor and Antifungal Agents. Eur. J. Med. Chem. 2015, 92, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.R.; Fröde, T.S.; Nardi, G.M.; Vita, N.; Reeb, R.; Ferrara, P.; Ribeiro-do-Valle, R.M.; Farges, R.C. Anti-Inflammatory Effects of Peripheral Benzodiazepine Receptor Ligands in Two Mouse Models of Inflammation. Eur. J. Pharmacol. 2000, 408, 199–211. [Google Scholar] [CrossRef]
- Insuasty, B.; Ramírez, J.; Becerra, D.; Echeverry, C.; Quiroga, J.; Abonia, R.; Robledo, S.M.; Vélez, I.D.; Upegui, Y.; Muñoz, J.A.; et al. An Efficient Synthesis of New Caffeine-Based Chalcones, Pyrazolines and Pyrazolo[3,4-b][1,4]Diazepines as Potential Antimalarial, Antitrypanosomal and Antileishmanial Agents. Eur. J. Med. Chem. 2015, 93, 401–413. [Google Scholar] [CrossRef]
- Insuasty, B.; Orozco, F.; Lizarazo, C.; Quiroga, J.; Abonia, R.; Hursthouse, M.; Nogueras, M.; Cobo, J. Synthesis of New Indeno[1,2-e]Pyrimido[4,5-b][1,4]Diazepine-5,11-Diones as Potential Antitumor Agents. Bioorg. Med. Chem. 2008, 16, 8492–8500. [Google Scholar] [CrossRef]
- Insuasty, B.; Orozco, F.; Quiroga, J.; Abonia, R.; Nogueras, M.; Cobo, J. Microwave Induced Synthesis of Novel 8,9-Dihydro-7H-Pyrimido[4,5-b][1,4]Diazepines as Potential Antitumor Agents. Eur. J. Med. Chem. 2008, 43, 1955–1962. [Google Scholar] [CrossRef]
- Insuasty, B.; García, A.; Quiroga, J.; Abonia, R.; Nogueras, M.; Cobo, J. Synthesis of Novel 6,6a,7,8-Tetrahydro-5H-Naphtho[1,2-e]Pyrimido[4,5-b][1,4]Diazepines under Microwave Irradiation as Potential Anti-Tumor Agents. Eur. J. Med. Chem. 2010, 45, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shen, Q.; Gao, L.; Tong, L.; Li, J.; Chen, Y.; Lu, W. Pyrazolo[4,3-b]Pyrimido[4,5-e][1,4]Diazepine Derivatives as New Multi-Targeted Inhibitors of Aurora A/B and KDR. Eur. J. Med. Chem. 2018, 158, 428–441. [Google Scholar] [CrossRef] [PubMed]
- El Haimer, M.; Palkó, M.; Haukka, M.; Gajdács, M.; Zupkó, I.; Fülöp, F. Synthesis and biological evaluation of the new ring system benzo [f] pyrimido [1, 2-d][1, 2, 3] triazolo [1, 5-a][1, 4] diazepine and its cycloalkane and cycloalkene condensed analogues. RSC Adv. 2021, 11, 6952–6957. [Google Scholar] [CrossRef]
- Gracias, V.; Ji, Z.; Akritopoulou-Zanze, I.; Abad-Zapatero, C.; Huth, J.R.; Song, D.; Hajduk, P.J.; Johnson, E.F.; Glaser, K.B.; Marcotte, P.A.; et al. Scaffold Oriented Synthesis. Part 2: Design, Synthesis and Biological Evaluation of Pyrimido-Diazepines as Receptor Tyrosine Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2691–2695. [Google Scholar] [CrossRef]
- Deng, X.; Elkins, J.M.; Zhang, J.; Yang, Q.; Erazo, T.; Gomez, N.; Choi, H.G.; Wang, J.; Dzamko, N.; Lee, J.D.; et al. Structural Determinants for ERK5 (MAPK7) and Leucine Rich Repeat Kinase 2 Activities of Benzo[e]Pyrimido-[5,4-b]Diazepine-6(11H)-Ones. Eur. J. Med. Chem. 2013, 70, 758–767. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E.J.; Fraga, C.A.M. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent Advances (2015–2016) in Anticancer Hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, O.; Shapi, M.; Tuszynski, J.A. A Review of the Recent Developments of Molecular Hybrids Targeting Tubulin Polymerization. Int. J. Mol. Sci. 2022, 23, 4001. [Google Scholar] [CrossRef] [PubMed]
- Osman, S.M.; Alasmary, F.A.; Kenawy, E.R.; Aly, E.S.A.; Khattab, S.N.; El-Faham, A. Synthesis, Characterization and Comparative Thermal Degradation Kinetics of s-Triazine Based Polymers. J. Polym. Res. 2021, 28, 304. [Google Scholar] [CrossRef]
- Venkata Krishna Reddy, M.; Vasu Govardhana Reddy, P.; Suresh Reddy, C. PEPPSI-SONO-SP2: A New Highly Efficient Ligand-Free Catalyst System for the Synthesis of Tri-Substituted Triazine Derivatives: Via Suzuki-Miyaura and Sonogashira Coupling Reactions under a Green Approach. New J. Chem. 2016, 40, 5135–5142. [Google Scholar] [CrossRef]
- Anamika, S.; Ghabbour, H.; Khan, S.T.; De la Torre, B.G.; Albericio, F.; El-Faham, A. Novel Pyrazolyl-s-Triazine Derivatives, Molecular Structure Ans Antimicrobial Activity. J. Mol. Struct. 2017, 1145, 244–253. [Google Scholar] [CrossRef]
- Padilla-Salinas, R.; Sun, L.; Anderson, R.; Yang, X.; Zhang, S.; Chen, Z.J.; Yin, H. Discovery of Small-Molecule Cyclic GMP-AMP Synthase Inhibitors. J. Org. Chem. 2020, 85, 1579–1600. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Paul, K. Triazine-Benzimidazole Conjugates: Synthesis, Spectroscopic and Molecular Modelling Studies for Interaction with Calf Thymus DNA. RSC Adv. 2016, 6, 14741–14750. [Google Scholar] [CrossRef]
- Rosenau, T.; Renfrew, A.H.M.; Adelwöhrer, C.; Potthast, A.; Kosma, P. Cellulosics Modified with Slow-Release Reagents. Part I. Synthesis of Triazine-Anchored Reagents for Slow Release of Active Substances from Cellulosic Materials. Polymer 2005, 46, 1453–1458. [Google Scholar] [CrossRef]
- Adhikari, N.; Choudhury, A.A.K.; Shakya, A.; Ghosh, S.K.; Patgiri, S.J.; Singh, U.P.; Bhat, H.R. Design and Development of Novel N-(4-Aminobenzoyl)- l-Glutamic Acid Conjugated 1,3,5-Triazine Derivatives as Pf-DHFR Inhibitor: An in-Silico and in-Vitro Study. J. Biochem. Mol. Toxicol. 2023, 37, e23290. [Google Scholar] [CrossRef]
- Moreno, L.; Quiroga, J.; Abonia, R.; Ramírez-Prada, J.; Insuasty, B. Synthesis of New 1,3,5-Triazine-Based 2-Pyrazolines as Potential Anticancer Agents. Molecules 2018, 23, 1956. [Google Scholar] [CrossRef]
- NCI-60 Screening Methodology|NCI-60 Human Tumor Cell Lines Screen|Discovery & Development Services|Developmental Therapeutics Program (DTP). Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 26 September 2023).
- Grever, M.; Schepartz, S.; Chabner, B. The National Cancer Institute: Cancer Drug Discovery and Development Program. Semin. Oncol. 1992, 19, 622–638. [Google Scholar] [PubMed]
- Weinstein, J.N.; Myers, T.G.; O’Connor, P.M.; Friend, S.H.; Fornace, A.J.; Kohn, K.W.; Fojo, T.; Bates, S.E.; Rubinstein, L.V.; Anderson, N.L.; et al. An Information-Intensive Approach to the Molecular Pharmacology of Cancer. Science 1997, 275, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Verbitskiy, E.; Baskakova, S.; Belyaev, D.; Vakhrusheva, D.; Eremeeva, N.; Rusinov, G.; Charushin, V. Renaissance of 4-(5-Nitrofuran-2-Yl)-5-Arylamino Substituted Pyrimidines: Microwave-Assisted Synthesis and Antitubercular Activity. Mendeleev Commun. 2021, 31, 210–212. [Google Scholar] [CrossRef]
- Salmerón, P.; Viñado, B.; El Ouazzani, R.; Hernández, M.; Barbera, M.J.; Alberny, M.; Jané, M.; Larrosa, N.; Pumarola, T.; Hoyos-Mallecot, Y.; et al. Antimicrobial Susceptibility of Neisseria Gonorrhoeae in Barcelona during a Five-Year Period, 2013 to 2017. Euro Surveill 2020, 25, 1900576. [Google Scholar] [CrossRef] [PubMed]
- Unemo, M.; Shafer, W.M. Antimicrobial Resistance in Neisseria Gonorrhoeae in the 21st Century: Past, Evolution, and Future. Clin. Microbiol. Rev. 2014, 27, 587–613. [Google Scholar] [CrossRef]
- Unemo, M.; Shafer, W.M. Antibiotic Resistance in Neisseria Gonorrhoeae: Origin, Evolution, and Lessons Learned for the Future. Ann. N. Y. Acad. Sci. 2011, 1230, 1230. [Google Scholar] [CrossRef]
- Insuasty, B.; García, A.; Bueno, J.; Quiroga, J.; Abonia, R.; Ortiz, A. Antimycobacterial Activity of Pyrimido[4,5-b]Diazepine Derivatives. Arch. Pharm.—Chem. Life Sci. 2012, 345, 739–744. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Liu, Y.; Harshbarger, W.; Huang, L.; Wang, J.; Deng, X.; Capuzzi, S.J.; Muratov, E.N.; Tropsha, A.; Muthuswamy, S.; et al. Synthesis and Structure−activity Relationships of DCLK1 Kinase Inhibitors Based on a 5,11-Dihydro-6H-benzo[e]Pyrimido[5,4-b][1,4]Diazepin-6-One Scaffold. J. Med. Chem. 2020, 63, 10088. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. M27-A3. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard, 3rd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008; ISBN 1-56238-666-2. [Google Scholar]
- Clinical and Laboratory Standards Institute. M38-Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi, 3rd ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017; Volume 37, ISBN 1-56238-831-2. [Google Scholar]
- Borman, A.M.; Muller, J.; Walsh-Quantick, J.; Szekely, A.; Patterson, Z.; Palmer, M.D.; Fraser, M.; Johnson, E.M. MIC Distributions for Amphotericin B, Fluconazole, Itraconazole, Voriconazole, Flucytosine and Anidulafungin and 35 Uncommon Pathogenic Yeast Species from the UK Determined Using the CLSI Broth Microdilution Method. J. Antimicrob. Chemother. 2020, 75, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Conceição, K.; Konno, K.; Richardson, M.; Antoniazzi, M.M.; Jared, C.; Daffre, S.; Camargo, A.C.M.; Pimenta, D.C. Isolation and Biochemical Characterization of Peptides Presenting Antimicrobial Activity from the Skin of Phyllomedusa Hypochondrialis. Peptides 2006, 27, 3092–3099. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Leber, A. Clinical Microbiology: Procedures Handbook, 4th ed.; Wiley: Washington, DC, USA, 2016; ISBN 9781683670766. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). M100. Performance Standards for Antimicrobial Susceptibility Testing, 27th ed.; Clinical and Laboratory Standards Institute: Pittsburgh, PA, USA, 2023; ISBN 9781684401048. [Google Scholar]
- Schön, T.; Werngren, J.; Machado, D.; Borroni, E.; Wijkander, M.; Lina, G.; Mouton, J.; Matuschek, E.; Kahlmeter, G.; Giske, C.; et al. Antimicrobial Susceptibility Testing of Mycobacterium Tuberculosis Complex Isolates—The EUCAST Broth Microdilution Reference Method for MIC Determination. Clin. Microbiol. Infect. 2020, 26, 1488–1492. [Google Scholar] [CrossRef]
- Piatek, M.; Sheehan, G.; Kavanagh, K. Galleria Mellonella: The Versatile Host for Drug Discovery, in Vivo Toxicity Testing and Characterising Host-Pathogen Interactions. Antibiotics 2021, 10, 1545. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Monosubstituted precursors 5–7 | |||
| Compound | Nu1H | i time, equivalents | Yield (%) |
| 5 [68,69,70] | Morpholine | 2 h, 4:Nu1H (1:1) | 88 |
| 6 [71,72] | 4-Fluoroaniline | 20 min, 4:Nu1H (1:1) | 97 |
| 7 | 4-Hydroxy-3-methoxybenzaldehyde | 4 h, 4:Nu1H (2.7:1) | 96 |
| Disubstituted precursors 8–11 | |||
| Compound | Nu2H | ii temperature, time, equivalents | |
| 8 [74] | Dimethylamine | rt, 4 h, 5:Nu2H (1:1) | 90 |
| 9 [71] | Dimethylamine | rt, 4 h, 6:Nu2H (1:1.5) | 77 |
| 10 | 4-Hydroxy-3-methoxybenzaldehyde | rt, 3 h, 6:Nu2H (1:1) | 91 |
| 11 [73] | Morpholine | −5–0 °C, 7 h, 7:Nu2H (1.2:1) | 88 |
| Trisubstituted precursors 12–15 | |||
| Compound | Nu3H | iii solvent, temperature, time, equivalents | |
| 12 | Ethylenediamine | Solvent free, rt, 18 h, 8:Nu3H (1:16) | 78 |
| 13 | Ethylenediamine | Solvent free, reflux, 4 h, 9:Nu3H (1:16) | 73 |
| 14 | Dimethylamine | Dioxane, rt, 1 h, 10:Nu3H (1:1) | 83 |
| 15 | Ethanolamine | Dioxane, rt, 24 h, 11:Nu3H (1:1.5) | 86 |
| Compound | Panel Name | Most Sensitive Cell Line | GI50 a (μM) | LC50 b (μM) | Range GI50 d (μM) | GI50 5-FU (μM) (NS 18893) e |
|---|---|---|---|---|---|---|
| 20b | Melanoma | LOX IMVI | 0.43 | 4.09 | 0.43–100 | 0.25 |
| Leukemia | SR | 1.06 | >100 | 0.02 | ||
| Renal cancer | UO-31 | 1.49 | - | 1.43 | ||
| Breast cancer | MCF7 | 1.50 | >100 | 0.08 | ||
| 20d | Melanoma | LOX IMVI | 0.64 | 4.99 | 0.64–4.72 | 0.25 |
| Leukemia | SR | 0.78 | >100 | 0.02 | ||
| Colon cancer | HCT-116 | 1.53 | 6.78 | 0.23 | ||
| Leukemia | HL-60(TB) | 1.54 | >100 | 2.30 | ||
| 21a | Melanoma | LOX IMVI | 1.60 | 6.23 | 1.60–48.60 | 0.25 |
| Colon cancer | SW-620 | 1.81 | 8.70 | 0.93 | ||
| Non-small cell lung cancer | HOP-92 | 2.08 | >100 | 77.98 | ||
| Leukemia | MOLT-4 | 2.56 | >100 | 0.35 | ||
| 21b | Melanoma | LOX IMVI | 1.84 | 6.69 | 1.84–51.90 | 0.25 |
| Colon cancer | HCT-116 | 2.28 | 24.40 | 0.23 | ||
| Colon cancer | SW-620 | 2.36 | 20.10 | 0.93 | ||
| Leukemia | MOLT-4 | 2.80 | >100 | 0.35 | ||
| 21d | Leukemia | MOLT-4 | 0.47 | >100 | 0.47–8.42 | 0.35 |
| Melanoma | LOX IMVI | 0.48 | 4.11 | 0.25 | ||
| CNS cancer | U251 | 0.50 | 9.59 | 0.91 | ||
| Leukemia | SR | 0.70 | >100 | 0.02 | ||
| 23a | Colon cancer | HCT-116 | 1.82 | 7.10 | 1.82–20.20 | 0.23 |
| Melanoma | LOX IMVI | 1.97 | 8.68 | 0.25 | ||
| Colon cancer | KM12 | 2.00 | 8.93 | 0.21 | ||
| Leukemia | K-562 | 2.19 | >100 | 3.98 | ||
| 23d | Renal cancer | RXF 393 | 1.22 | 8.63 | 1.22–14.30 | 2.61 |
| Leukemia | RPMI-8226 | 1.39 | >100 | 0.04 | ||
| Leukemia | K-562 | 1.46 | >100 | 3.58 | ||
| CNS cancer | U251 | 1.54 | 5.54 | 0.91 | ||
| 23e | Melanoma | LOX IMVI | 1.71 | 6.13 | 1.71–17.90 | 0.25 |
| Colon cancer | HCT-15 | 2.07 | 9.93 | 0.11 | ||
| Breast cancer | MCF7 | 2.37 | 34.60 | 0.08 | ||
| Leukemia | SR | 2.52 | >100 | 0.02 | ||
| 23f | Melanoma | LOX IMVI | 1.63 | 6.19 | 1.63–16.10 | 0.25 |
| Leukemia | RPMI-8226 | 1.91 | >100 | 0.04 | ||
| Colon cancer | HCT-15 | 2.15 | 13.70 | 0.11 | ||
| Ovarian cancer | IGROV1 | 2.27 | 25.40 | 1.22 | ||
| 23g | Melanoma | LOX IMVI | 1.68 | 5.94 | 1.68–17.80 | 0.25 |
| Breast cancer | MCF7 | 1.68 | 6.63 | 0.08 | ||
| Leukemia | RPMI-8226 | 1.77 | >100 | 0.04 | ||
| Ovarian cancer | IGROV1 | 1.77 | 6.97 | 1.22 | ||
| 24a | Colon cancer | HCT-116 | 1.72 | 6.10 | 1.72–14.60 | 0.23 |
| Breast cancer | MCF7 | 1.99 | 8.23 | 0.08 | ||
| Colon cancer | HCT-15 | 2.04 | 7.25 | 0.11 | ||
| Colon cancer | HCC-2998 | 2.35 | 17.90 | 0.05 | ||
| 24b | Colon cancer | HCT-116 | 1.75 | 6.17 | 1.75–17.40 | 0.23 |
| Breast cancer | MCF7 | 2.01 | 25.20 | 0.08 | ||
| Colon cancer | HT29 | 2.38 | 16.20 | 0.18 | ||
| Leukemia | RPMI-8226 | 2.73 | >100 | 0.04 | ||
| 24c | Colon cancer | HT29 | 1.83 | 7.63 | 1.83–17.10 | 0.18 |
| Colon cancer | HCT-116 | 1.86 | 7.07 | 0.23 | ||
| Breast cancer | MCF7 | 1.93 | 46.10 | 0.08 | ||
| Non-small cell lung cancer | NCI-H522 | 2.55 | 67.10 | 7.28 | ||
| 24d | Renal cancer | RXF 393 | 1.45 | 6.74 | 1.45–21.80 | 2.61 |
| Colon cancer | HCC-2998 | 1.62 | 5.52 | 0.05 | ||
| CNS cancer | SF-539 | 1.71 | 5.62 | 0.06 | ||
| Melanoma | LOX IMVI | 1.71 | 5.93 | 0.25 | ||
| 24e | Colon cancer | HCT-116 | 1.66 | 3.11 | 1.66–12.80 | 0.23 |
| Colon cancer | HT29 | 2.15 | 5.72 | 0.18 | ||
| Breast cancer | MCF7 | 2.28 | 6.33 | 0.08 | ||
| Leukemia | RPMI-8226 | 2.47 | 7.11 | 0.04 | ||
| 24f | Colon cancer | HCT-116 | 1.60 | 3.01 | 1.60–8.39 | 0.23 |
| Melanoma | LOX IMVI | 1.66 | 3.07 | 0.25 | ||
| Renal cancer | SN12C | 1.70 | 3.36 | 0.50 | ||
| Ovarian cancer | OVCAR-3 | 1.74 | 3.24 | 0.02 | ||
| 24g | Colon cancer | HCT-116 | 1.68 | 3.13 | 1.68–17.40 | 0.23 |
| Breast cancer | MCF7 | 1.70 | 3.56 | 0.08 | ||
| Colon cancer | HCC-2998 | 1.77 | 3.61 | 0.05 | ||
| Melanoma | LOX IMVI | 1.86 | 3.84 | 0.25 | ||
| 29e | Renal cancer | RXF 393 | 2.46 | >100 | 2.46–100 | 2.61 |
| Non-small cell lung cancer | HOP-92 | 3.00 | >100 | 77.98 | ||
| CNS cancer | SNB-75 | 3.24 | >100 | 78.70 | ||
| Renal cancer | CAKI-1 | 3.38 | >100 | 0.07 | ||
| 29g | Renal cancer | RXF 393 | 1.22 | 34.10 | 1.22–100 | 2.61 |
| Renal cancer | CAKI-1 | 1.65 | >100 | 0.07 | ||
| Leukemia | MOLT-4 | 1.71 | >100 | 0.35 | ||
| Colon cancer | HCT-116 | 1.76 | 7.56 | 0.23 | ||
| 30g | Non-small cell lung cancer | HOP-92 | 7.48 | >100 | 7.48–100 | 77.98 |
| Renal cancer | RXF 393 | 10.30 | >100 | 2.61 | ||
| Leukemia | RPMI-8226 | 11.20 | >100 | 0.04 | ||
| Breast cancer | MDA-MB-468 | 13.70 | 69.50 | 6.61 | ||
| 31a | Non-small cell lung cancer | HOP-92 | 2.18 | 9.96 | 2.18–99.70 | 77.98 |
| Renal cancer | RXF 393 | 2.55 | 8.95 | 2.61 | ||
| Leukemia | MOLT-4 | 3.11 | >100 | 0.35 | ||
| Leukemia | RPMI-8226 | 3.32 | >100 | 0.04 | ||
| 31b | Melanoma | LOX IMVI | 2.09 | 5.11 | 2.09–78.80 | 0.25 |
| Leukemia | MOLT-4 | 2.20 | 7.53 | 0.35 | ||
| Renal cancer | RXF 393 | 2.27 | 9.12 | 2.61 | ||
| Colon cancer | HT29 | 2.27 | 4.65 | 0.18 | ||
| 31e | Melanoma | LOX IMVI | 1.77 | 3.55 | 1.77–72.70 | 0.25 |
| Breast cancer | MDA-MB-468 | 1.92 | 4.92 | 0.07 | ||
| Renal cancer | RXF 393 | 2.27 | 8.75 | 2.61 | ||
| Non-small cell lung cancer | HOP-92 | 2.53 | 13.20 | 77.98 | ||
| 31f | Breast cancer | MDA-MB-468 | 2.15 | 6.24 | 2.15–96.00 | 6.61 |
| Leukemia | MOLT-4 | 2.45 | 0.35 | |||
| Renal cancer | RXF 393 | 2.48 | 2.61 | |||
| CNS cancer | SNB-75 | 2.53 | 74.00 | 78.70 | ||
| 31g | CNS cancer | SNB-75 | 2.13 | 86.00 | 2.13–86.00 | 78.70 |
| Renal cancer | RXF 393 | 2.57 | 18.80 | 2.61 | ||
| Breast cancer | MDA-MB-468 | 2.67 | 11.70 | 6.61 | ||
| Leukemia | MOLT-4 | 2.74 | 86.00 | 0.35 | ||
| 33a | Leukemia | MOLT-4 | <0.01 | >100 | 0.01–17.70 | 0.35 |
| Leukemia | HL-60(TB) | 0.32 | >100 | 2.51 | ||
| Leukemia | SR | 0.55 | 0.02 | |||
| Leukemia | CCRF-CEM | 0.95 | >100 | 9.79 | ||
| 33b | Non-small cell lung cancer | HOP-92 | 1.30 | 6.30 | 1.30–16.70 | 77.98 |
| Leukemia | K-562 | 1.34 | 24.90 | 3.58 | ||
| Leukemia | MOLT-4 | 1.38 | >100 | 0.35 | ||
| Renal cancer | RXF 393 | 1.45 | 2.61 | |||
| 33e | Leukemia | K-562 | 0.71 | >100 | 0.71–14.80 | 3.58 |
| Leukemia | MOLT-4 | 0.79 | >100 | 0.35 | ||
| Leukemia | SR | 0.91 | >100 | 0.02 | ||
| Leukemia | CCRF-CEM | 1.14 | >100 | 9.79 | ||
| 33f | Leukemia | K-562 | 0.49 | >100 | 0.49–12.90 | 3.58 |
| Leukemia | MOLT-4 | 0.53 | >100 | 0.35 | ||
| Leukemia | SR | 0.67 | >100 | 0.02 | ||
| Leukemia | CCRF-CEM | 0.75 | >100 | 9.79 | ||
| 33g | Leukemia | K-562 | 0.64 | >100 | 0.64–2.55 | 3.58 |
| Leukemia | MOLT-4 | 0.89 | >100 | 0.35 | ||
| Colon cancer | HCT-116 | 1.05 | 5.54 | 0.23 | ||
| Renal cancer | RXF 393 | 1.14 | 5.48 | 2.61 |
| Compound | MIC (μg/mL) | Compound | MIC (μg/mL) | Compound | MIC (μg/mL) |
|---|---|---|---|---|---|
| 15 | ≥500 | 29c | 62.5 | 32b | 4 |
| 21f | 500 | 29d | 16.12 | 32d | 500 |
| 23d | >500 | 29e | ≥500 | 32e | 1000 |
| 24e | 500 | 29f | 0.5 | 32f | 2 |
| 28a | 4 | 29g | ≥500 | 32g | 4 |
| 28b | 4 | 30d | 16.12 | 33a | 1 |
| 28c | 0.5 | 31a | 8 | 33b | 0.5 |
| 28d | 2 | 31b | 8 | 33c | 2 |
| 28e | 8 | 31c | 8 | 33f | 1 |
| 28f | 8 | 31d | 500 | Penicillin a | 0.25 (0.25–1.0) |
| 28g | 8 | 31e | 0.5 | Tetracycline a | 1.00 (0.25–1.0) |
| 29a | 2 | 31f | 0.25 | ||
| 29b | ≥500 | 32a | 8 |
| Compounds | MIC b (μg/mL)/MFC (μg/mL) c | |||||||
|---|---|---|---|---|---|---|---|---|
| Ca | Cn | Mg | Tr | Tm | Afu | Ani | Afl | |
| 17 | >250 | >250 | >250 | 125 | >250 | >250 | >250 | >250 |
| 21d | >250 | >250 | >250 | 125 | >250 | >250 | 125 | >250 |
| 23e | 250 | 250 | >250 | 125 | >250 | >250 | >250 | >250 |
| 24c | >250 | >250 | >250 | 125 | >250 | >250 | >250 | >250 |
| 24e | >250 | >250 | >250 | >250 | 125 | >250 | >250 | >250 |
| 29c | >250 | >250 | 125 | >250 | >250 | >250 | >250 | >250 |
| 29e | >250 | >250 | >250 | 62.5 | >250 | >250 | >250 | >250 |
| 29f | >250 | >250 | 125 | >250 | >250 | >250 | >250 | >250 |
| 29g | >250 | >250 | 125 | >250 | >250 | >250 | >250 | >250 |
| 30d | >250 | >250 | >250 | 125 | >250 | >250 | >250 | >250 |
| 30g | 125 | 125 | >250 | 250 | >250 | >250 | >250 | >250 |
| 31g | >250 | >250 | >250 | 250 | 62.5 | 62.5 | >250 | >250 |
| Amphotericin B d | 0.5 | 0.25 | 0.125 | 0.075 | 0.075 | 0.50 | 0.50 | 0.50 |
| Terbinafine d | - | - | 0.04 | 0.01 | 0.025 | - | - | - |
| Fluconazole d | 0.03 | 0.25 | - | - | - | - | - | - |
| Itraconazole d | 0.5 | - | - | - | - | - | - | - |
| Compounds | 29b | 29e | 31b | 31f | 31g | 33a | Expected Value |
|---|---|---|---|---|---|---|---|
| MW | 621.67 | 697.79 | 642.09 | 675.64 | 751.76 | 609.08 | <500 |
| PSA | 174.61 | 222.73 | 174.61 | 174.61 | 222.73 | 197.20 | <140 |
| HBA | 9 | 10 | 9 | 12 | 13 | 7 | <10 |
| HBD | 4 | 6 | 4 | 4 | 6 | 7 | <5 |
| RB | 8 | 11 | 8 | 9 | 12 | 9 | <10 |
| Log P | 4.64 | 3.38 | 4.23 | 4.64 | 3.89 | 3.36 | 0–5 |
| Log S (ESOL) | −6.89 | −6.59 | −7.19 | −7.46 | −7.16 | −6.38 | >−6 |
| Lipinski violations | 2 | 3 | 2 | 2 | 3 | 3 | - |
| GI absorption | Low | Low | Low | Low | Low | Low | - |
| BBB permeant | No | No | No | No | No | No | - |
| Pgp substrate | No | No | No | No | No | No | - |
| CYP1A2 inhibitor | No | No | No | No | No | No | - |
| CYP2C19 inhibitor | No | No | No | No | No | No | - |
| CYP2C9 inhibitor | No | No | No | No | No | No | - |
| CYP2D6 inhibitor | No | No | No | No | No | No | - |
| CYP3A4 inhibitor | Yes | Yes | No | No | Yes | Yes | - |
| Synthetic Accessibility | 5.31 | 5.62 | 5.18 | 5.34 | 5.64 | 5.03 | <6 |
| Bioavailability score | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | >0.1 |
| Compound | % Hemolysis | Compound | % Hemolysis | Compound | % Hemolysis |
|---|---|---|---|---|---|
| 17 | 1.6 | 24g | 0.8 | 31a | 1 |
| 20b | 3.3 | 28a | 3 | 31b | 1.7 |
| 20d | 1.2 | 28b | 5 | 31c | 1.2 |
| 21a | 2 | 28c | 7 | 31d | 0.6 |
| 21b | 13 | 28d | 1 | 31e | 2.3 |
| 21d | 8 | 28e | 5 | 31f | 2.4 |
| 21f | 0.8 | 28f | 2 | 31g | 3 |
| 23a | 0.7 | 28g | 100 | 32a | 2 |
| 23b | 1.8 | 29a | 3 | 32b | 1 |
| 23c | 0 | 29c | 2.9 | 32f | 5 |
| 23d | 0.2 | 29d | 75 | 32g | 2 |
| 24a | 10.6 | 29f | 0 | 33a | 8 |
| 24b | 1.2 | 30a | 0.6 | 33b | 100 |
| 24c | 0.1 | 30b | 0.2 | 33c | 5 |
| 24d | 2.3 | 30d | 11 | 33e | 1.2 |
| 24e | 9.1 | 30e | 2.7 | 33f | 5 |
| 24f | 9.9 | 30g | 2.4 | 33g | 22 |
| Compound | 29b | 29e | 31b | 31f | 31g | 33a |
|---|---|---|---|---|---|---|
| Hepatotoxicity | Inactive 0.56 | Inactive 0.52 | Inactive 0.52 | Inactive 0.56 | Inactive 0.56 | Inactive 0.70 |
| Immunotoxicity | Inactive 0.52 | Active 0.99 | Active 0.99 | Active 0.73 | Active 0.84 | Active 0.57 |
| Carcinogenicity | Active 0.51 | Inactive 0.61 | Inactive 0.61 | Active 0.51 | Active 0.51 | Inactive 0.62 |
| Mutagenicity | Inactive 0.71 | Inactive 0.52 | Inactive 0.51 | Inactive 0.73 | Inactive 0.73 | Inactive 0.62 |
| Cytotoxicity | Inactive 0.50 | Inactive 0.56 | Inactive 0.55 | Active 0.50 | Active 0.50 | Active 0.54 |
| LD50(mg/Kg) | 3000 | 900 | 3000 | 3000 | 900 | 2958 |
| Toxicity class a | 5 | 4 | 5 | 5 | 4 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, L.M.; Quiroga, J.; Abonia, R.; Crespo, M.d.P.; Aranaga, C.; Martínez-Martínez, L.; Sortino, M.; Barreto, M.; Burbano, M.E.; Insuasty, B. Synthesis of Novel Triazine-Based Chalcones and 8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepines as Potential Leads in the Search of Anticancer, Antibacterial and Antifungal Agents. Int. J. Mol. Sci. 2024, 25, 3623. https://doi.org/10.3390/ijms25073623
Moreno LM, Quiroga J, Abonia R, Crespo MdP, Aranaga C, Martínez-Martínez L, Sortino M, Barreto M, Burbano ME, Insuasty B. Synthesis of Novel Triazine-Based Chalcones and 8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepines as Potential Leads in the Search of Anticancer, Antibacterial and Antifungal Agents. International Journal of Molecular Sciences. 2024; 25(7):3623. https://doi.org/10.3390/ijms25073623
Chicago/Turabian StyleMoreno, Leydi M., Jairo Quiroga, Rodrigo Abonia, María del P. Crespo, Carlos Aranaga, Luis Martínez-Martínez, Maximiliano Sortino, Mauricio Barreto, María E. Burbano, and Braulio Insuasty. 2024. "Synthesis of Novel Triazine-Based Chalcones and 8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepines as Potential Leads in the Search of Anticancer, Antibacterial and Antifungal Agents" International Journal of Molecular Sciences 25, no. 7: 3623. https://doi.org/10.3390/ijms25073623
APA StyleMoreno, L. M., Quiroga, J., Abonia, R., Crespo, M. d. P., Aranaga, C., Martínez-Martínez, L., Sortino, M., Barreto, M., Burbano, M. E., & Insuasty, B. (2024). Synthesis of Novel Triazine-Based Chalcones and 8,9-dihydro-7H-pyrimido[4,5-b][1,4]diazepines as Potential Leads in the Search of Anticancer, Antibacterial and Antifungal Agents. International Journal of Molecular Sciences, 25(7), 3623. https://doi.org/10.3390/ijms25073623