
Computational Modeling of the Interactions between DPP IV and Hemorphins


Abstract
1. Introduction
2. Results
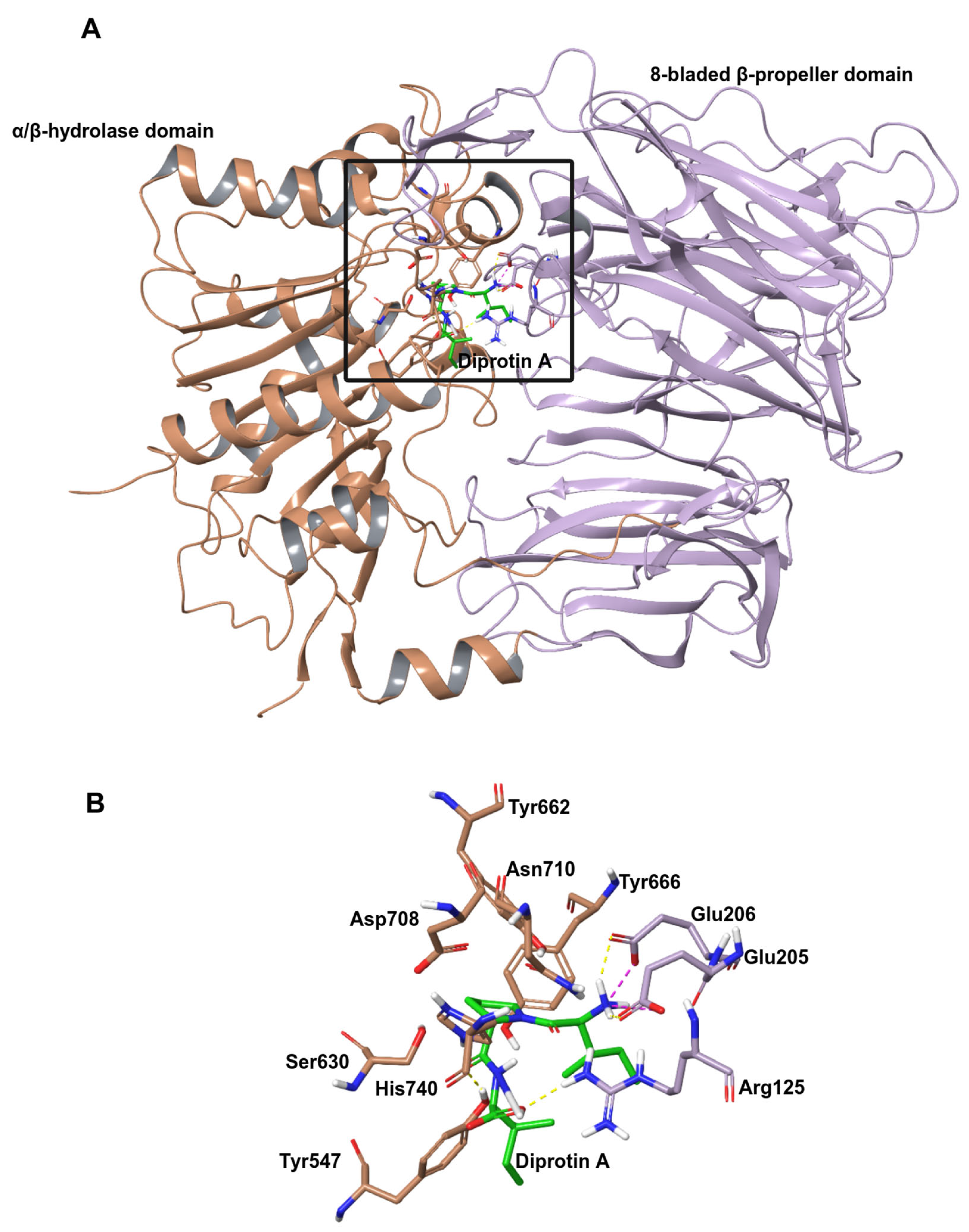
2.1. Molecular Docking
2.1.1. Docking of Hemorphins to DPP IV
2.1.2. Docking of Camel Hemorphins to DPP IV
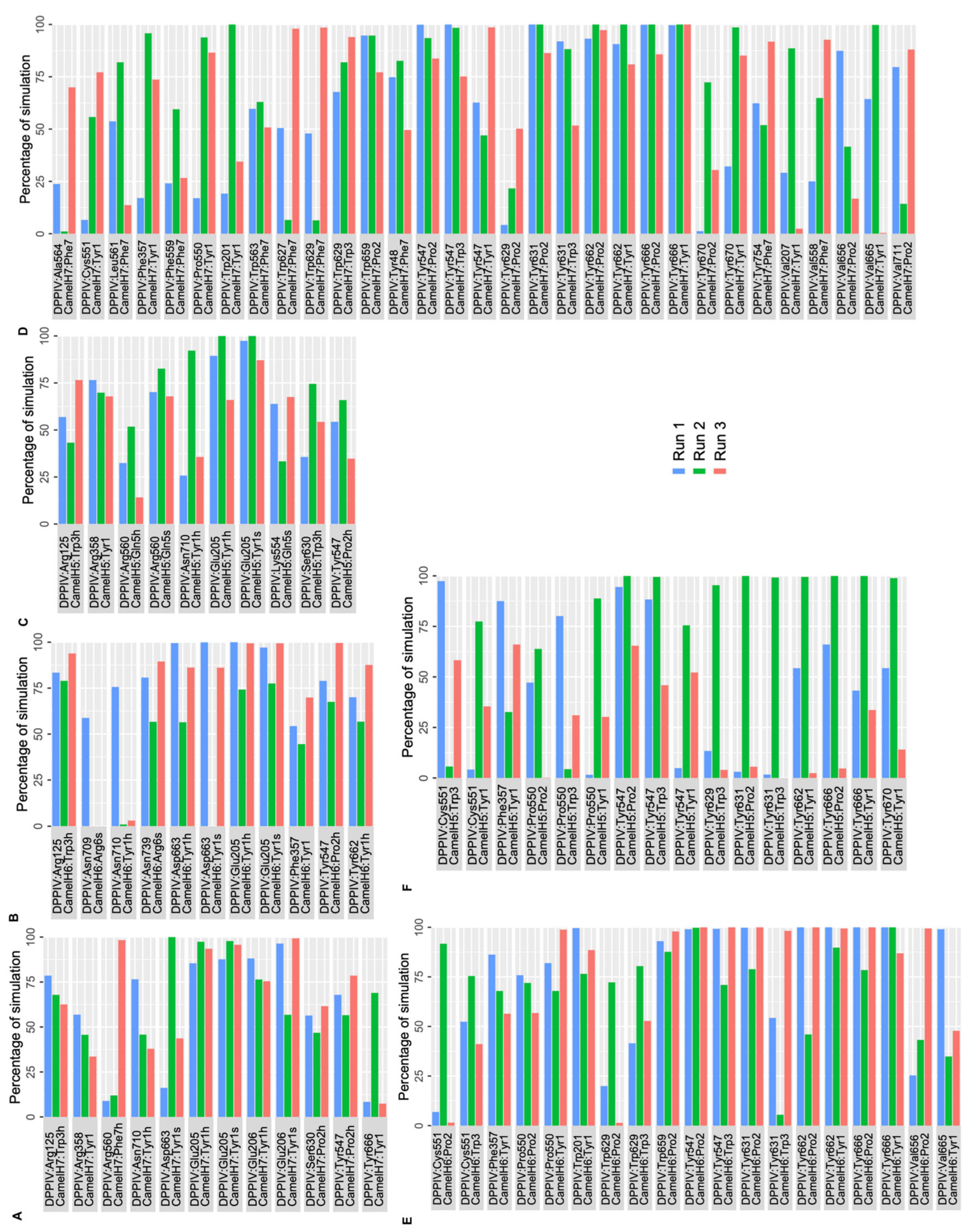
2.2. Molecular Dynamics Simulations
2.2.1. MD Simulations of DPP IV-Hemorphin Complexes
2.2.2. MD Simulations of DPP IV-Camel Hemorphin Complexes
3. Discussion
4. Materials and Methods
4.1. Protein Preparation and Structure Assessment
4.2. Active Site Identification and Grid Generation
4.3. Peptide Docking and Binding Free Energy Calculations
4.4. Protein–Peptide Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shaikh, S.; Lee, E.-J.; Ahmad, K.; Ahmad, S.-S.; Lim, J.-H.; Choi, I. A Comprehensive Review and Perspective on Natural Sources as Dipeptidyl Peptidase-4 Inhibitors for Management of Diabetes. Pharmaceuticals 2021, 14, 591. [Google Scholar] [CrossRef]
- Deacon, C.F. Dipeptidyl Peptidase 4 Inhibitors in the Treatment of Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2020, 16, 642–653. [Google Scholar] [CrossRef]
- Berger, J.P.; SinhaRoy, R.; Pocai, A.; Kelly, T.M.; Scapin, G.; Gao, Y.; Pryor, K.A.D.; Wu, J.K.; Eiermann, G.J.; Xu, S.S.; et al. A Comparative Study of the Binding Properties, Dipeptidyl Peptidase-4 (DPP -4) Inhibitory Activity and Glucose-lowering Efficacy of the DPP -4 Inhibitors Alogliptin, Linagliptin, Saxagliptin, Sitagliptin and Vildagliptin in Mice. Endocrinol. Diabet. Metabol. 2018, 1, e00002. [Google Scholar] [CrossRef]
- Wu, W.-L.; Hao, J.; Domalski, M.; Burnett, D.A.; Pissarnitski, D.; Zhao, Z.; Stamford, A.; Scapin, G.; Gao, Y.-D.; Soriano, A.; et al. Discovery of Novel Tricyclic Heterocycles as Potent and Selective DPP-4 Inhibitors for the Treatment of Type 2 Diabetes. ACS Med. Chem. Lett. 2016, 7, 498–501. [Google Scholar] [CrossRef]
- Namoto, K.; Sirockin, F.; Ostermann, N.; Gessier, F.; Flohr, S.; Sedrani, R.; Gerhartz, B.; Trappe, J.; Hassiepen, U.; Duttaroy, A.; et al. Discovery of C-(1-Aryl-Cyclohexyl)-Methylamines as Selective, Orally Available Inhibitors of Dipeptidyl Peptidase IV. Bioorganic Med. Chem. Lett. 2014, 24, 731–736. [Google Scholar] [CrossRef]
- Hiramatsu, H.; Kyono, K.; Yamamoto, A.; Saeki, K.; Shima, H.; Sugiyama, S.; Inaka, K.; Shimizu, R. Crystal Structures of Human Dipeptidyl Peptidase IV in Its Apo and Diprotin B-Complexed Forms. Acta Biochim. Biophys. Sin. 2007, 39, 335–343. [Google Scholar] [CrossRef]
- Nabeno, M.; Akahoshi, F.; Kishida, H.; Miyaguchi, I.; Tanaka, Y.; Ishii, S.; Kadowaki, T. A Comparative Study of the Binding Modes of Recently Launched Dipeptidyl Peptidase IV Inhibitors in the Active Site. Biochem. Biophys. Res. Commun. 2013, 434, 191–196. [Google Scholar] [CrossRef]
- Aertgeerts, K. Crystal Structure of Human Dipeptidyl Peptidase IV in Complex with a Decapeptide Reveals Details on Substrate Specificity and Tetrahedral Intermediate Formation. Protein Sci. 2004, 13, 412–421. [Google Scholar] [CrossRef]
- Thoma, R.; Löffler, B.; Stihle, M.; Huber, W.; Ruf, A.; Hennig, M. Structural Basis of Proline-Specific Exopeptidase Activity as Observed in Human Dipeptidyl Peptidase-IV. Structure 2003, 11, 947–959. [Google Scholar] [CrossRef]
- Weihofen, W.A.; Liu, J.; Reutter, W.; Saenger, W.; Fan, H. Crystal Structures of HIV-1 Tat-Derived Nonapeptides Tat-(1–9) and Trp2-Tat-(1–9) Bound to the Active Site of Dipeptidyl-Peptidase IV (CD26). J. Biol. Chem. 2005, 280, 14911–14917. [Google Scholar] [CrossRef]
- Arulmozhiraja, S.; Matsuo, N.; Ishitsubo, E.; Okazaki, S.; Shimano, H.; Tokiwa, H. Comparative Binding Analysis of Dipeptidyl Peptidase IV (DPP-4) with Antidiabetic Drugs—An Ab Initio Fragment Molecular Orbital Study. PLoS ONE 2016, 11, e0166275. [Google Scholar] [CrossRef]
- Saini, K.; Sharma, S.; Khan, Y. DPP-4 Inhibitors for Treating T2DM—Hype or Hope? An Analysis Based on the Current Literature. Front. Mol. Biosci. 2023, 10, 1130625. [Google Scholar] [CrossRef]
- Ojeda-Montes, M.J.; Gimeno, A.; Tomas-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. Activity and Selectivity Cliffs for DPP-IV Inhibitors: Lessons We Can Learn from SAR Studies and Their Application to Virtual Screening. Med. Res. Rev. 2018, 38, 1874–1915. [Google Scholar] [CrossRef]
- Rasmussen, H.B.; Branner, S.; Wiberg, F.C.; Wagtmann, N. Crystal Structure of Human Dipeptidyl Peptidase IV/CD26 in Complex with a Substrate Analog. Nat. Struct. Biol. 2003, 10, 19–25. [Google Scholar] [CrossRef]
- Madsen, C.T.; Refsgaard, J.C.; Teufel, F.G.; Kjærulff, S.K.; Wang, Z.; Meng, G.; Jessen, C.; Heljo, P.; Jiang, Q.; Zhao, X.; et al. Combining Mass Spectrometry and Machine Learning to Discover Bioactive Peptides. Nat. Commun. 2022, 13, 6235. [Google Scholar] [CrossRef]
- Chang, L.; Mondal, A.; Perez, A. Towards Rational Computational Peptide Design. Front. Bioinform. 2022, 2, 1046493. [Google Scholar] [CrossRef]
- Vanhee, P.; van der Sloot, A.M.; Verschueren, E.; Serrano, L.; Rousseau, F.; Schymkowitz, J. Computational Design of Peptide Ligands. Trends Biotechnol. 2011, 29, 231–239. [Google Scholar] [CrossRef]
- Mielczarek, P.; Hartman, K.; Drabik, A.; Hung, H.-Y.; Huang, E.Y.-K.; Gibula-Tarlowska, E.; Kotlinska, J.H.; Silberring, J. Hemorphins—From Discovery to Functions and Pharmacology. Molecules 2021, 26, 3879. [Google Scholar] [CrossRef]
- Nyberg, F.; Sanderson, K.; Glämsta, E.-L. The Hemorphins: A New Class of Opioid Peptides Derived from the Blood Protein Hemoglobin. Biopolymers 1997, 43, 147–156. [Google Scholar] [CrossRef]
- Ali, A.; Baby, B.; Soman, S.S.; Vijayan, R. Molecular Insights into the Interaction of Hemorphin and Its Targets. Sci. Rep. 2019, 9, 14747. [Google Scholar] [CrossRef]
- Ali, A.; Alzeyoudi, S.A.R.; Almutawa, S.A.; Alnajjar, A.N.; Vijayan, R. Molecular Basis of the Therapeutic Properties of Hemorphins. Pharmacol. Res. 2020, 158, 104855. [Google Scholar] [CrossRef]
- Ayoub, M.A.; Vijayan, R. Hemorphins Targeting G Protein-Coupled Receptors. Pharmaceuticals 2021, 14, 225. [Google Scholar] [CrossRef]
- Jobe, A.; Antony, P.; Altabbal, S.; Al Dhaheri, Y.; Vijayan, R. Interaction of Hemorphins with ACE Homologs. Sci. Rep. 2023, 13, 3743. [Google Scholar] [CrossRef]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM Database of Bioactive Peptides: Current Opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef]
- Daliri, E.; Oh, D.; Lee, B. Bioactive Peptides. Foods 2017, 6, 32. [Google Scholar] [CrossRef]
- Cohen, M.; Fruitier-Arnaudin, I.; Piot, J.M. Hemorphins: Substrates and/or Inhibitors of Dipeptidyl Peptidase IV. Biochimie 2004, 86, 31–37. [Google Scholar] [CrossRef]
- Vidal-Limon, A.; Aguilar-Toalá, J.E.; Liceaga, A.M. Integration of Molecular Docking Analysis and Molecular Dynamics Simulations for Studying Food Proteins and Bioactive Peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef]
- Biswas, S.; Mahmud, S.; Mita, M.A.; Afrose, S.; Hasan, M.R.; Sultana Shimu, M.S.; Saleh, M.A.; Mostafa-Hedeab, G.; Alqarni, M.; Obaidullah, A.J.; et al. Molecular Docking and Dynamics Studies to Explore Effective Inhibitory Peptides Against the Spike Receptor Binding Domain of SARS-CoV-2. Front. Mol. Biosci. 2022, 8, 791642. [Google Scholar] [CrossRef]
- Rahfeld, J.; Schierborn, M.; Hartrodt, B.; Neubert, K.; Heins, J. Are Diprotin A (Ile-Pro-Ile) and Diprotin B (Val-Pro-Leu) Inhibitors or Substrates of Dipeptidyl Peptidase IV? Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 1991, 1076, 314–316. [Google Scholar] [CrossRef]
- Umezawa, H.; Aoyagi, T.; Ogawa, K.; Naganawa, H.; Hamada, M.; Takeuchi, T. Diprotins A and B, Inhibitors of Dipeptidyl Aminopeptidase IV, Produced by Bacteria. J. Antibiot. 1984, 37, 422–425. [Google Scholar] [CrossRef]
- Pantaleão, S.; Philot, E.; De Resende-Lara, P.; Lima, A.; Perahia, D.; Miteva, M.; Scott, A.; Honorio, K. Structural Dynamics of DPP-4 and Its Influence on the Projection of Bioactive Ligands. Molecules 2018, 23, 490. [Google Scholar] [CrossRef]
- Liu, R.; Cheng, J.; Wu, H. Discovery of Food-Derived Dipeptidyl Peptidase IV Inhibitory Peptides: A Review. Int. J. Mol. Sci. 2019, 20, 463. [Google Scholar] [CrossRef]
- Engel, M.; Hoffmann, T.; Wagner, L.; Wermann, M.; Heiser, U.; Kiefersauer, R.; Huber, R.; Bode, W.; Demuth, H.-U.; Brandstetter, H. The Crystal Structure of Dipeptidyl Peptidase IV (CD26) Reveals Its Functional Regulation and Enzymatic Mechanism. Proc. Natl. Acad. Sci. USA 2003, 100, 5063–5068. [Google Scholar] [CrossRef]
- Engel, M.; Hoffmann, T.; Manhart, S.; Heiser, U.; Chambre, S.; Huber, R.; Demuth, H.-U.; Bode, W. Rigidity and Flexibility of Dipeptidyl Peptidase IV: Crystal Structures of and Docking Experiments with DPIV. J. Mol. Biol. 2006, 355, 768–783. [Google Scholar] [CrossRef]
- Chien, C.-H.; Huang, L.-H.; Chou, C.-Y.; Chen, Y.-S.; Han, Y.-S.; Chang, G.-G.; Liang, P.-H.; Chen, X. One Site Mutation Disrupts Dimer Formation in Human DPP-IV Proteins. J. Biol. Chem. 2004, 279, 52338–52345. [Google Scholar] [CrossRef]
- Kuhn, B.; Hennig, M.; Mattei, P. Molecular Recognition of Ligands in Dipeptidyl Peptidase IV. Curr. Top. Med. Chem. 2007, 7, 609–620. [Google Scholar] [CrossRef]
- Zettl, H.; Schubert-Zsilavecz, M.; Steinhilber, D. Medicinal Chemistry of Incretin Mimetics and DPP-4 Inhibitors. ChemMedChem 2010, 5, 179–185. [Google Scholar] [CrossRef]
- Pei, Z.; Li, X.; Von Geldern, T.W.; Longenecker, K.; Pireh, D.; Stewart, K.D.; Backes, B.J.; Lai, C.; Lubben, T.H.; Ballaron, S.J.; et al. Discovery and Structure−Activity Relationships of Piperidinone- and Piperidine-Constrained Phenethylamines as Novel, Potent, and Selective Dipeptidyl Peptidase IV Inhibitors. J. Med. Chem. 2007, 50, 1983–1987. [Google Scholar] [CrossRef]
- Sakashita, H.; Akahoshi, F.; Yoshida, T.; Kitajima, H.; Hayashi, Y.; Ishii, S.; Takashina, Y.; Tsutsumiuchi, R.; Ono, S. Lead Optimization of [(S)-γ-(Arylamino)Prolyl]Thiazolidine Focused on γ-Substituent: Indoline Compounds as Potent DPP-IV Inhibitors. Bioorg. Med. Chem. 2007, 15, 641–655. [Google Scholar] [CrossRef]
- Araki, M.; Kanegawa, N.; Iwata, H.; Sagae, Y.; Ito, K.; Masuda, K.; Okuno, Y. Hydrophobic Interactions at Subsite S1′ of Human Dipeptidyl Peptidase IV Contribute Significantly to the Inhibitory Effect of Tripeptides. Heliyon 2020, 6, e04227. [Google Scholar] [CrossRef]
- Bjelke, J.R.; Christensen, J.; Branner, S.; Wagtmann, N.; Olsen, C.; Kanstrup, A.B.; Rasmussen, H.B. Tyrosine 547 Constitutes an Essential Part of the Catalytic Mechanism of Dipeptidyl Peptidase IV. J. Biol. Chem. 2004, 279, 34691–34697. [Google Scholar] [CrossRef]
- Havale, S.H.; Pal, M. Medicinal Chemistry Approaches to the Inhibition of Dipeptidyl Peptidase-4 for the Treatment of Type 2 Diabetes. Bioorg. Med. Chem. 2009, 17, 1783–1802. [Google Scholar] [CrossRef]
- Li, C.; Shen, J.; Li, W.; Lu, C.; Liu, G.; Tang, Y. Possible Ligand Release Pathway of Dipeptidyl Peptidase IV Investigated by Molecular Dynamics Simulations: Ligand Release from DPP4. Proteins 2011, 79, 1800–1809. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; Dellafiora, L.; Paolella, S.; Galaverna, G.; Cozzini, P.; FitzGerald, R.J. In Silico Approaches Applied to the Study of Peptide Analogs of Ile-Pro-Ile in Relation to Their Dipeptidyl Peptidase IV Inhibitory Properties. Front. Endocrinol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Rodhi, A.M.; Yap, P.-G.; Abayomi, O.O.; Gan, C.-Y. A Review on the Types of Amino Acid at Ultimate, Penultimate and Antepenultimate Position in Some Dipeptidyl-Peptidase IV Inhibitory Peptides Based on Molecular Docking Analysis. Food Chem. Adv. 2023, 2, 100244. [Google Scholar] [CrossRef]
- Xu, F.; Yao, Y.; Xu, X.; Wang, M.; Pan, M.; Ji, S.; Wu, J.; Jiang, D.; Ju, X.; Wang, L. Identification and Quantification of DPP-IV-Inhibitory Peptides from Hydrolyzed-Rapeseed-Protein-Derived Napin with Analysis of the Interactions between Key Residues and Protein Domains. J. Agric. Food Chem. 2019, 67, 3679–3690. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, R.; Chen, X.; Zeng, Z.; Ma, H.; Chen, S. Dipeptidyl Peptidase IV-Inhibitory Peptides Derived from Silver Carp (Hypophthalmichthys Molitrix Val.) Proteins. J. Agric. Food Chem. 2016, 64, 831–839. [Google Scholar] [CrossRef]
- Brandt, W.; Lehmann, T.; Thondorf, I.; Born, I.; Schutkowski, M.; Rahfeld, J.-U.; Neubert, K.; Barth, A. A Model of the Active Site of Dipeptidyl Peptidase IV Predicted by Comparative Molecular Field Analysis and Molecular Modelling Simulations. Int. J. Pept. Protein Res. 1995, 46, 494–507. [Google Scholar] [CrossRef]
- Schrödinger Release 2022-4: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2022.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Metzler, W.J.; Yanchunas, J.; Weigelt, C.; Kish, K.; Klei, H.E.; Xie, D.; Zhang, Y.; Corbett, M.; Tamura, J.K.; He, B.; et al. Involvement of DPP-IV Catalytic Residues in Enzyme–Saxagliptin Complex Formation. Protein Sci. 2008, 17, 240–250. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Schrödinger Release 2020-4: Desmond Molecular Dynamics System; D.E. Shaw Research: New York, NY, USA, 2021.
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible Multiple Time Scale Molecular Dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A Fast SHAKE Algorithm to SolveDistance Constraint Equations forSmall Molecules in MolecularDynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
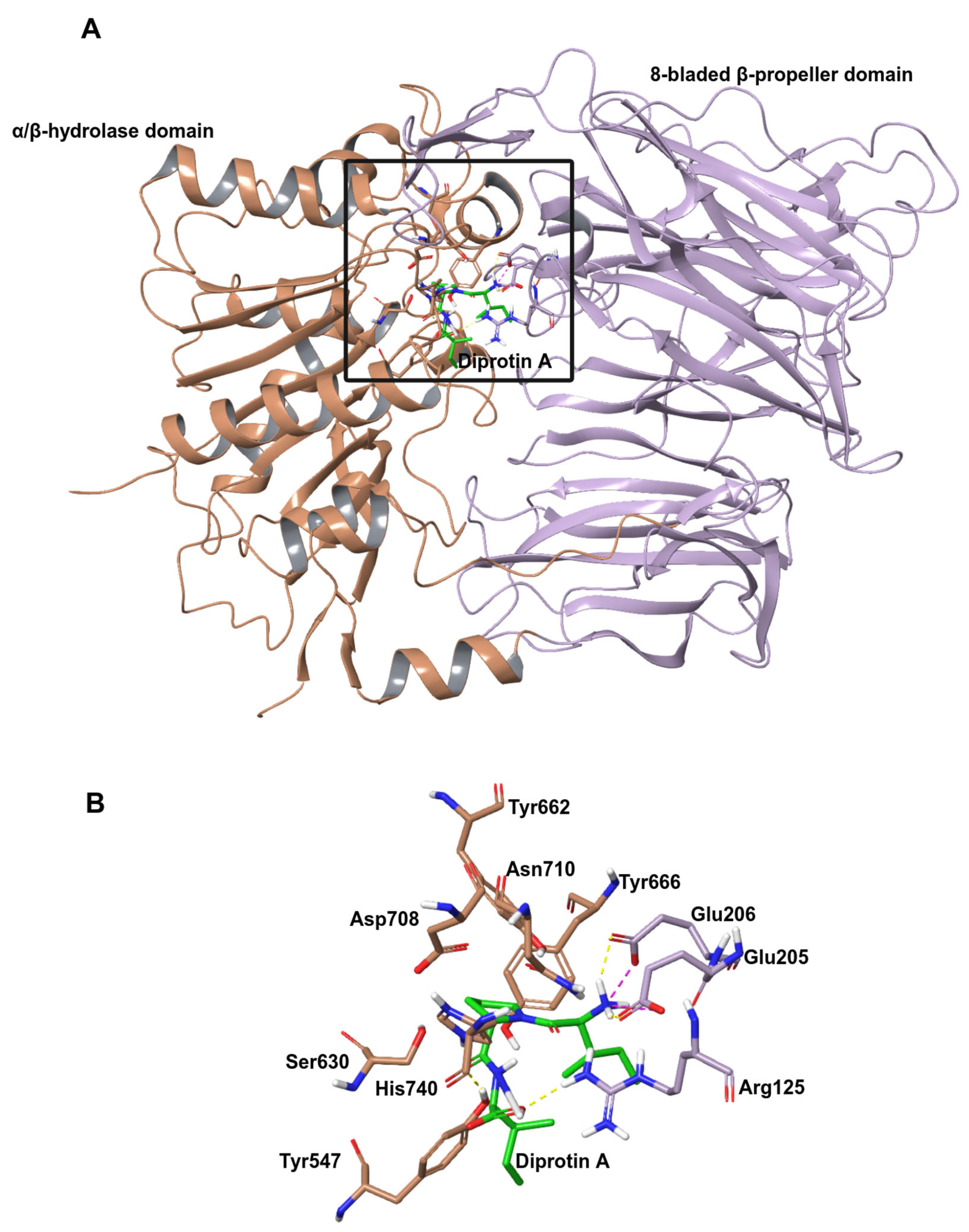

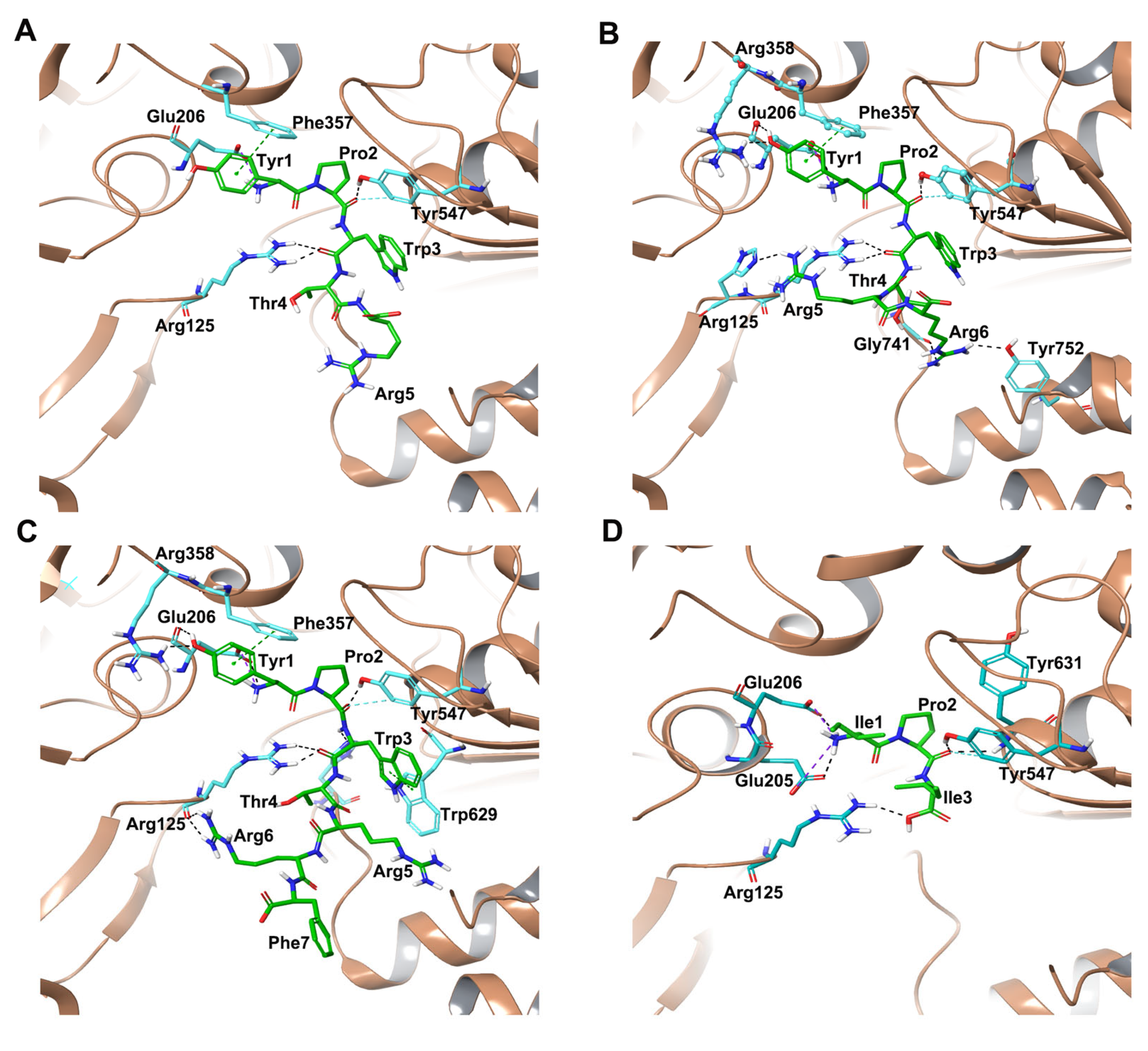

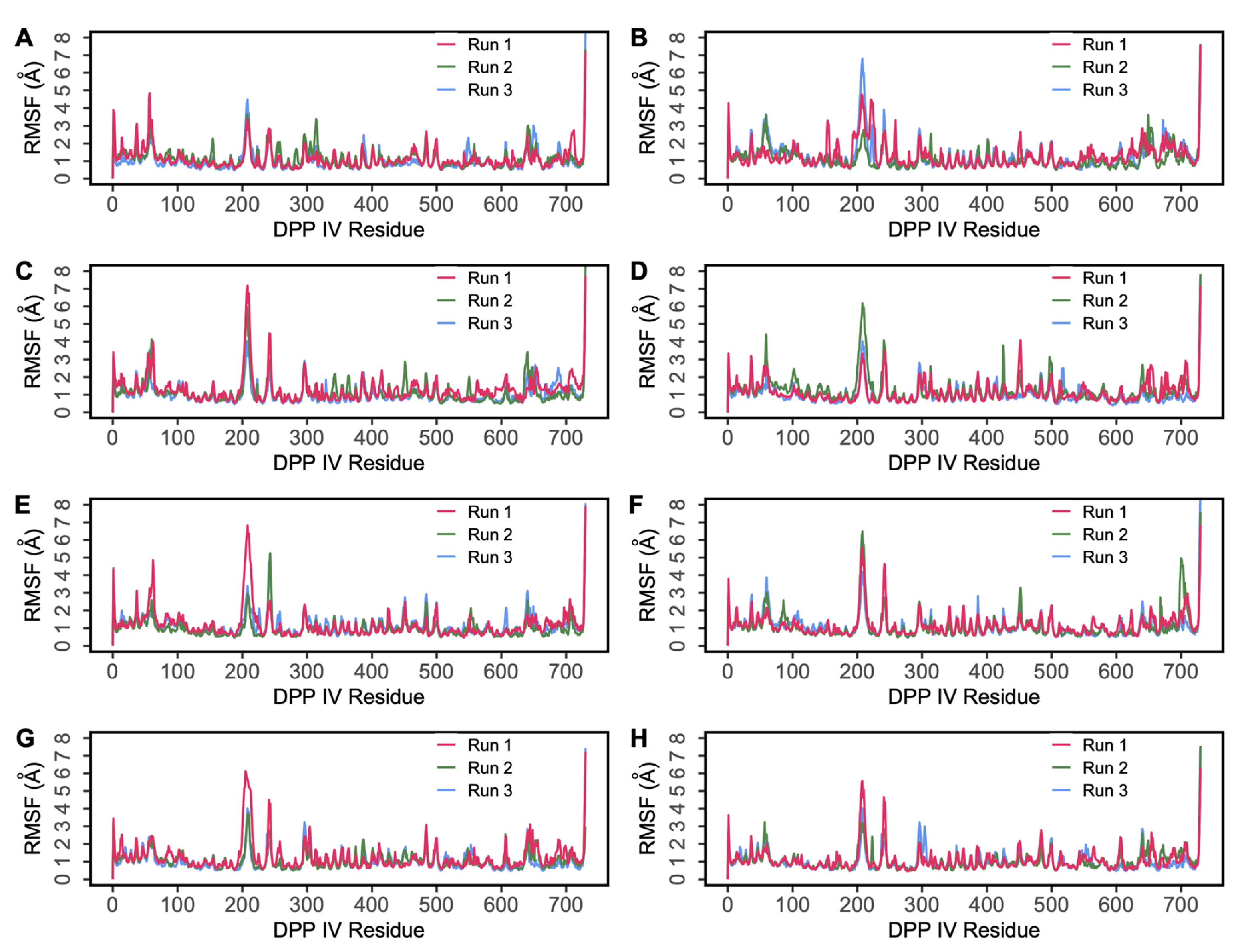
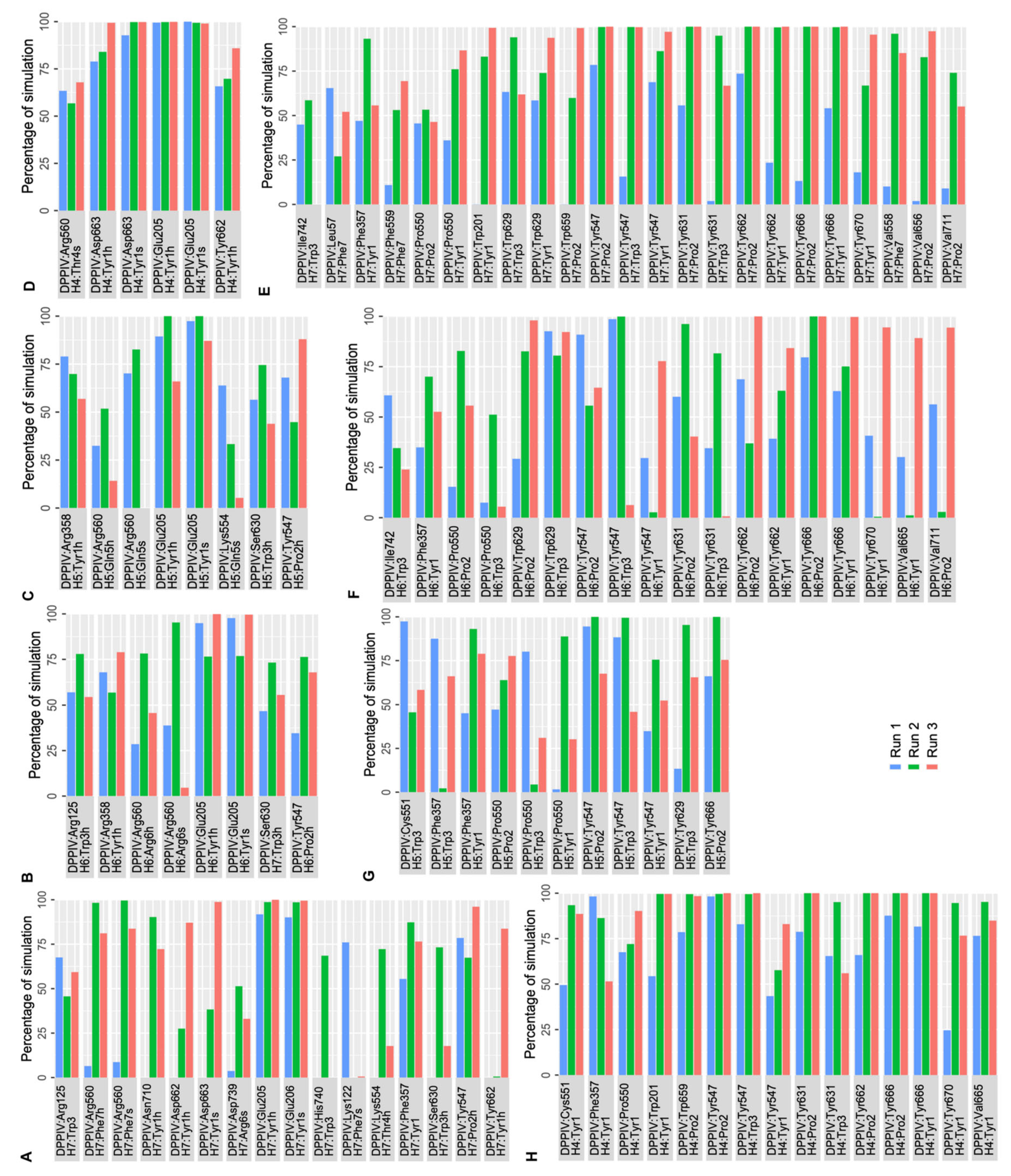

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Glide Score (GScore) Docking Score (kcal/mol) | MM-GBSA Binding Free Energy (kcal/mol) | Hydrogen Bonds | Hydrophobic Interactions | Salt Bridges | π–π Interactions |
|---|---|---|---|---|---|---|
| YPWTQRF (H7) | −11.44 | −119.64 | Glu206, Arg358, Arg125, Tyr547 Asp739 | Tyr48, Trp201, Val207, Phe357, Tyr631, Trp629, Val711, Tyr547, Tyr666, Tyr662, Trp659, Val656, Ile742, Ala743, Leu561, Leu49, Tyr752 | Glu206, Asp739 | Phe357 |
| YPWTQR (H6) | −10.53 | −92.92 | Tyr48, Arg 358, Glu206, Tyr547, Tyr 752, His748, Ala743 | Val207, Phe357, Tyr547, Tyr631, Trp629, Trp627, Tyr662, Trp659, Val656, Tyr666, Val711, Tyr752, Tyr48, Ala743 | Glu205 | Phe357 |
| YPWTQ (H5) | −10.76 | −99.12 | Glu206, Arg358, Tyr547, His740, Arg125, Gly741 | Val207, Trp201, Tyr547, Ile742, Ala743, Tyr752, Tyr48, Tyr662, Trp629, Trp659, Tyr631,Trp659, Val711, Val656, Tyr666, Phe357 | Glu206 | Phe357, Trp629 |
| YPWT (H4) | −7.6 | −84.98 | Arg358, Glu206, Glu205, Tyr547, Arg125, His740 | Trp201, Tyr547, Val207, Val656, Phe357, Trp659, Tyr666, Tyr662, Val711, Tyr631, Trp629 | Glu205 | Phe357 |
| YPWTRRF (Camel H7) | −10.54 | −113.28 | Arg358, Glu206, Tyr547, His740, Arg125 | Val207, Trp201, Tyr547, Trp563, Tyr752, Tyr48, Leu55, Leu57, Val656, Trp659, Tyr631, Trp659, Val711, Tyr666, Phe357, Tyr662 | Glu206 | Phe357 |
| YPWTRR (Camel H6) | −10.07 | −85.99 | Arg358, Glu206, Tyr547, His740, Arg125 | Val207, Trp201, Tyr547, Tyr120, Tyr128, Phe357, Ala743, Val656, Trp659, Tyr662 | Glu206 | Phe357 |
| YPWTR (Camel H5) | −9.42 | −102.88 | Arg125, Tyr547 | Trp201, Tyr547, Ile742, Ala743, Tyr752, Phe357, Tyr666, Val656, Tyr631, Trp659, Val711, Trp629, Tyr662 | Glu206 | Phe357 |
| Diprotin A (Positive control) | −8.05 | −83.00 | Arg125, Tyr547, Tyr631 | Phe357, Tyr662, Tyr666, Trp659, Val711, Tyr547, Tyr631, Trp629 | Glu 205, Glu 206 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antony, P.; Baby, B.; Jobe, A.; Vijayan, R. Computational Modeling of the Interactions between DPP IV and Hemorphins. Int. J. Mol. Sci. 2024, 25, 3059. https://doi.org/10.3390/ijms25053059
Antony P, Baby B, Jobe A, Vijayan R. Computational Modeling of the Interactions between DPP IV and Hemorphins. International Journal of Molecular Sciences. 2024; 25(5):3059. https://doi.org/10.3390/ijms25053059
Chicago/Turabian StyleAntony, Priya, Bincy Baby, Amie Jobe, and Ranjit Vijayan. 2024. "Computational Modeling of the Interactions between DPP IV and Hemorphins" International Journal of Molecular Sciences 25, no. 5: 3059. https://doi.org/10.3390/ijms25053059
APA StyleAntony, P., Baby, B., Jobe, A., & Vijayan, R. (2024). Computational Modeling of the Interactions between DPP IV and Hemorphins. International Journal of Molecular Sciences, 25(5), 3059. https://doi.org/10.3390/ijms25053059