
Inhibition of PRMT5/MEP50 Arginine Methyltransferase Activity Causes Cancer Vulnerability in NDRG2low Adult T-Cell Leukemia/Lymphoma

Abstract
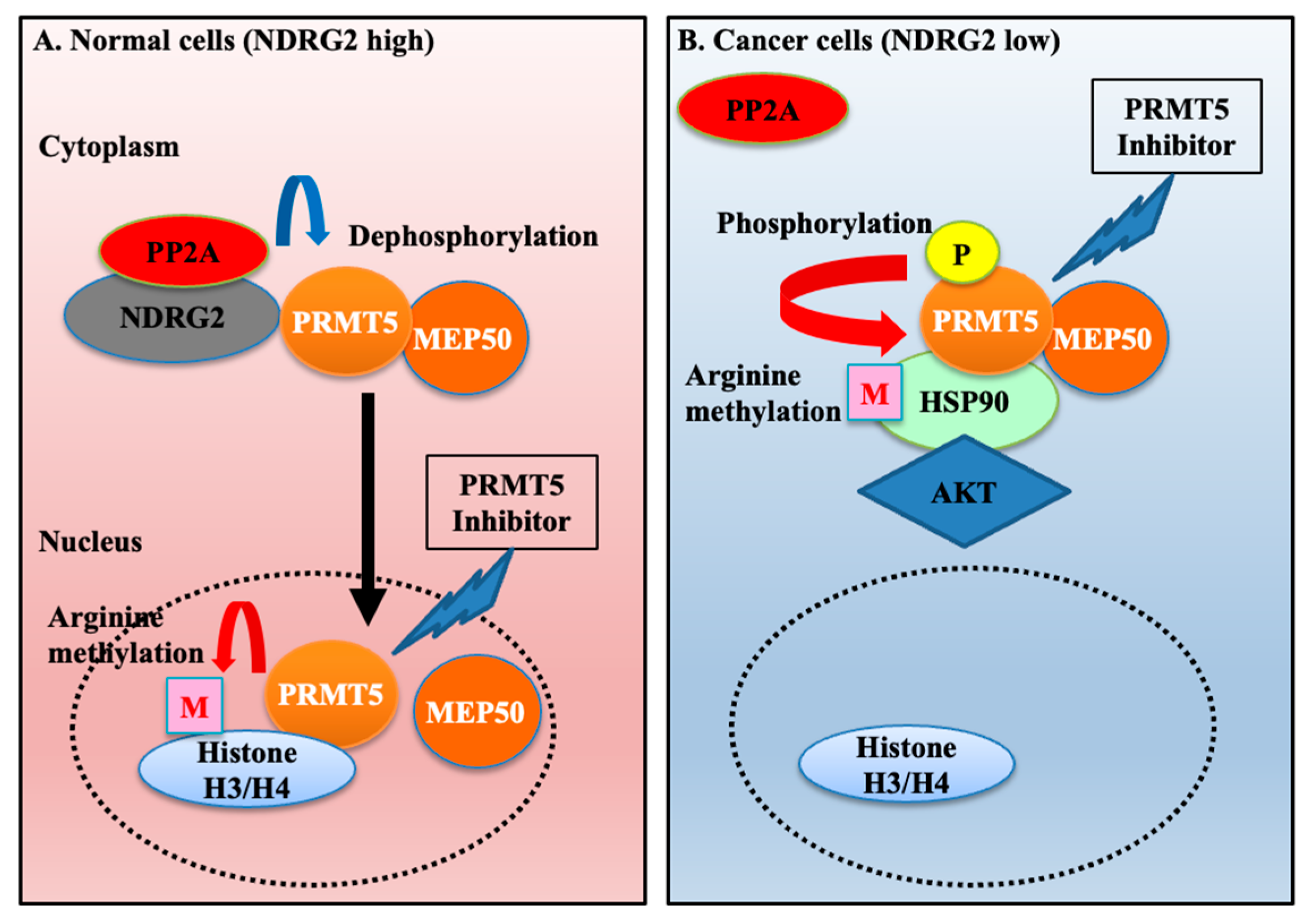
1. Introduction
2. Results
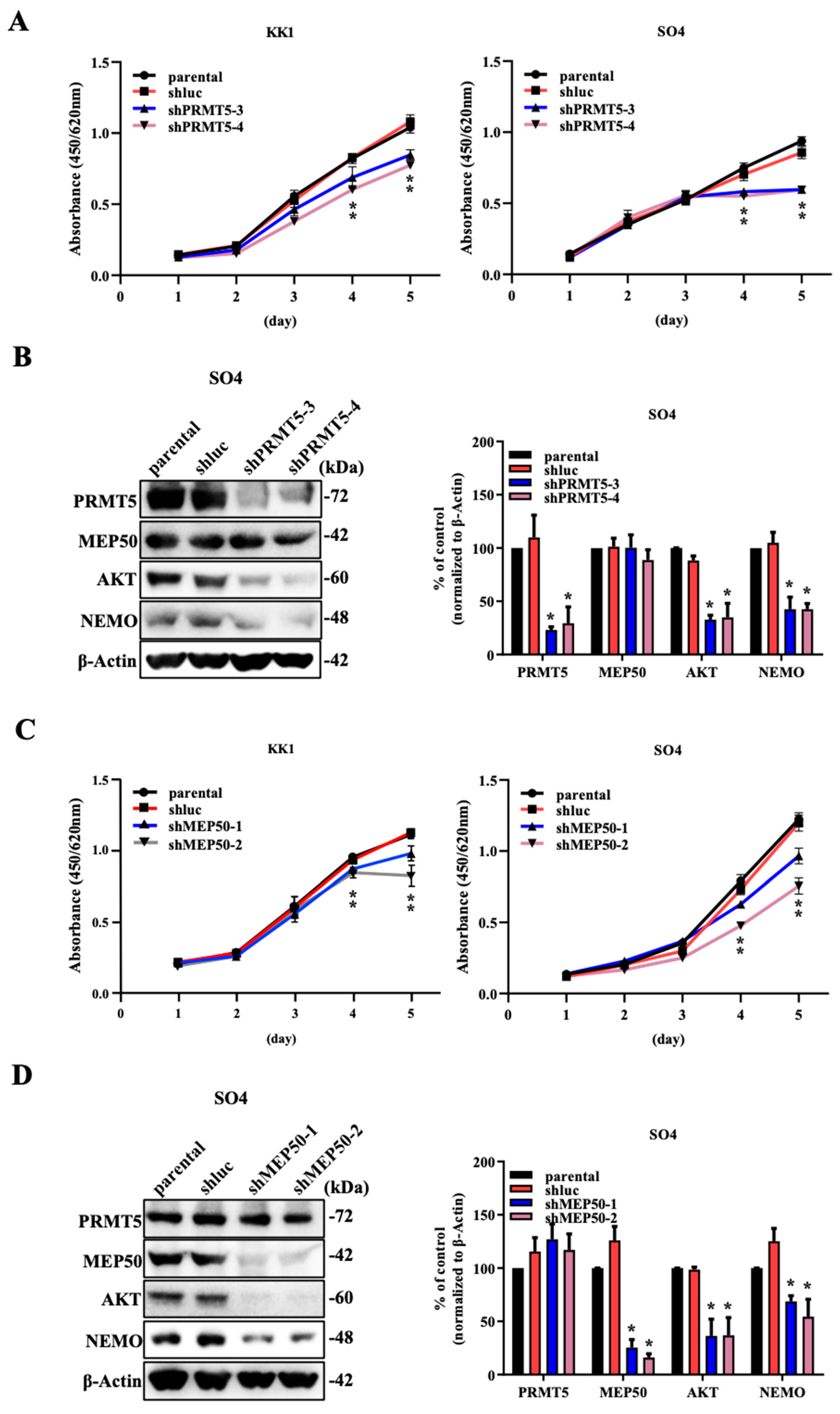
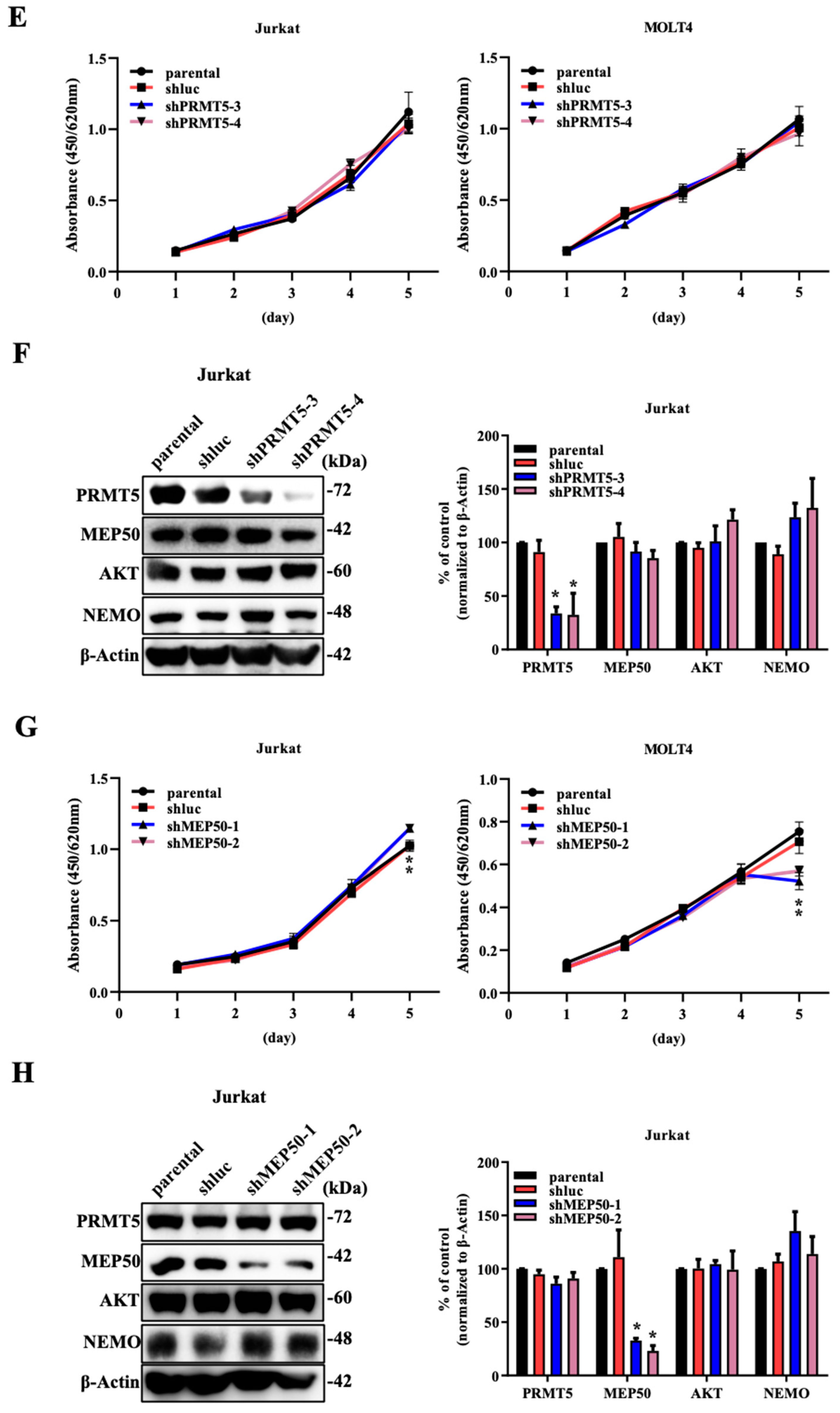
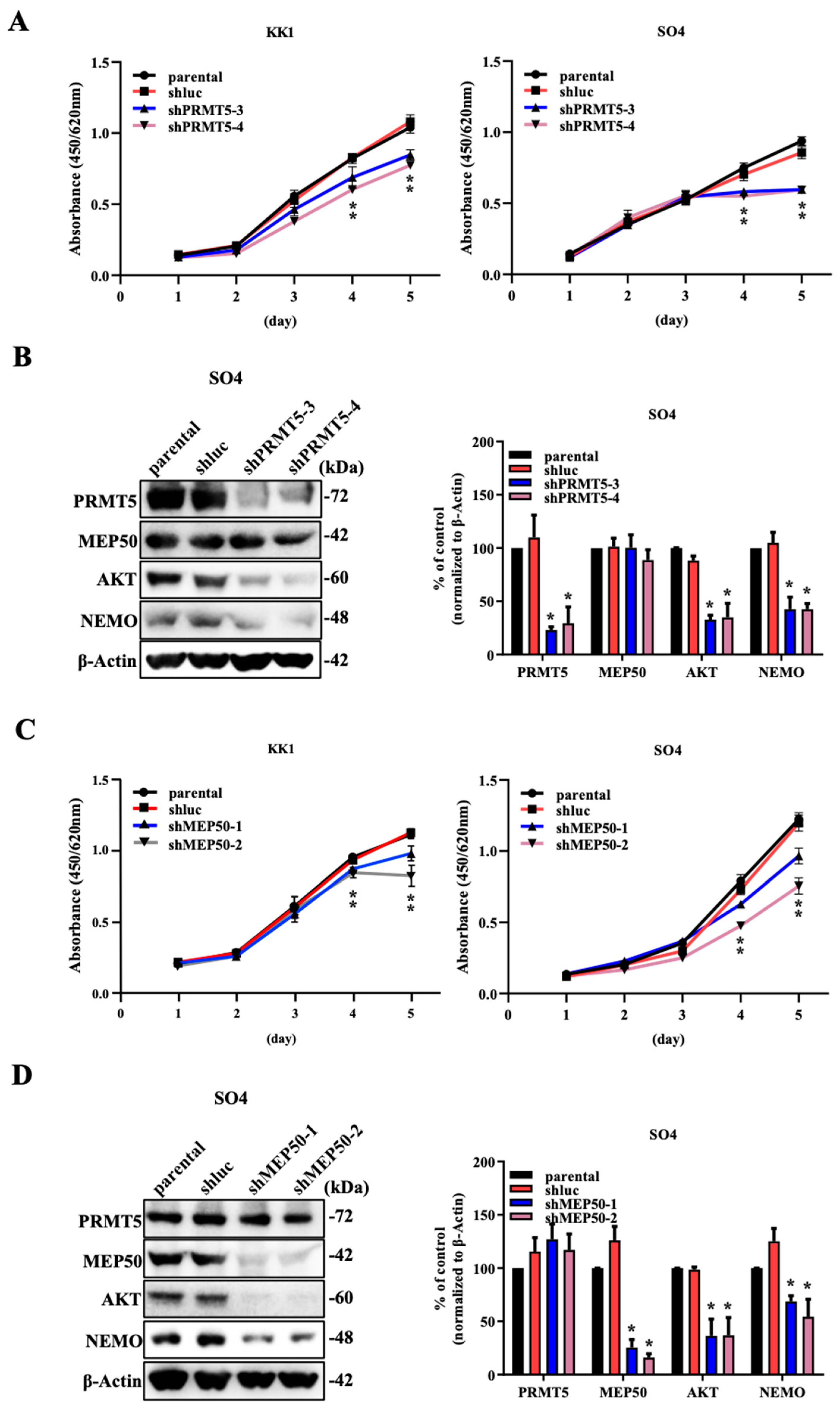
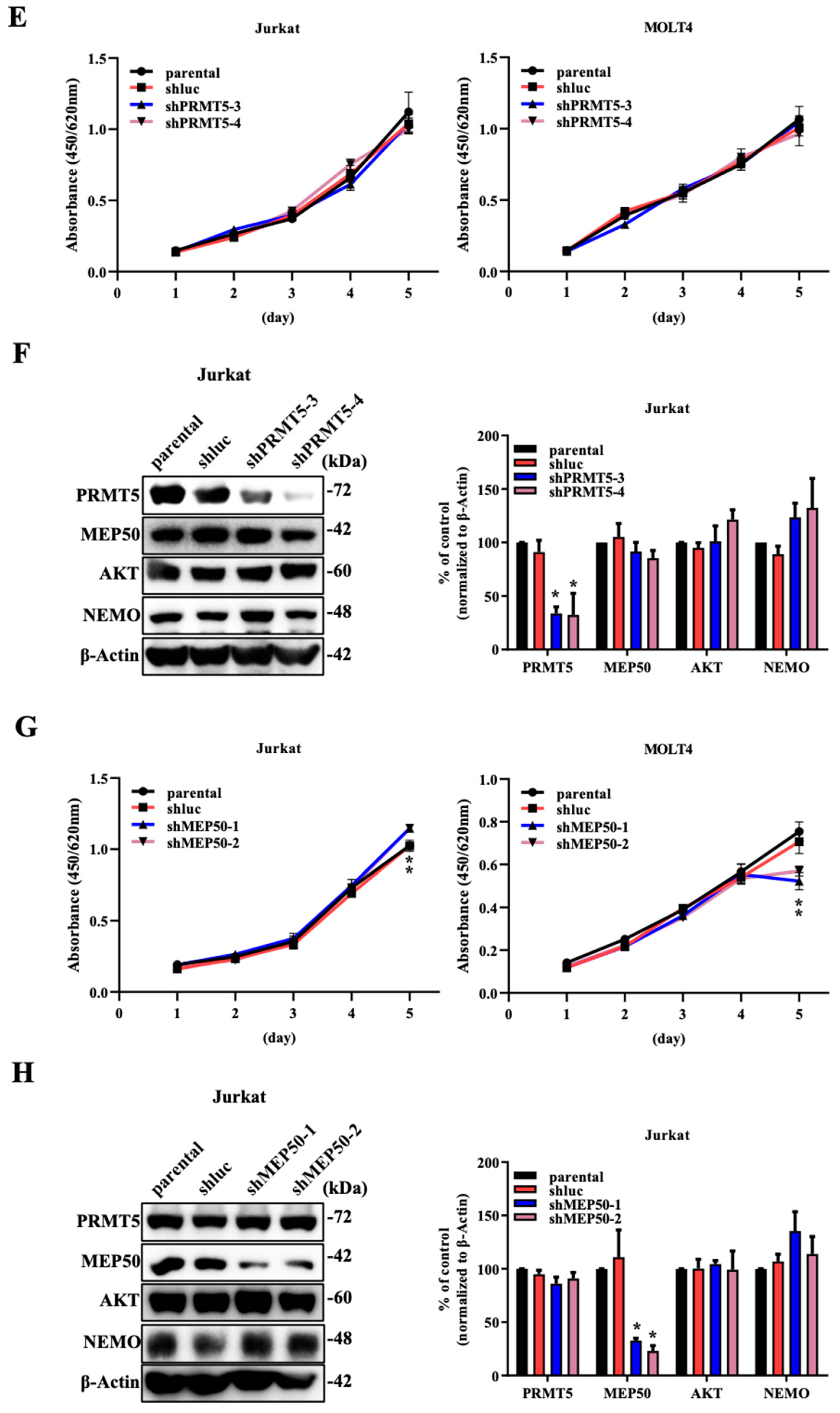
2.1. The Knockdown of PRMT5/MEP50 Expression Results in the Inhibition of Cell Proliferation through the Degradation of Client Proteins in ATL and Various Cancer Cells with Low NDRG2 Expression
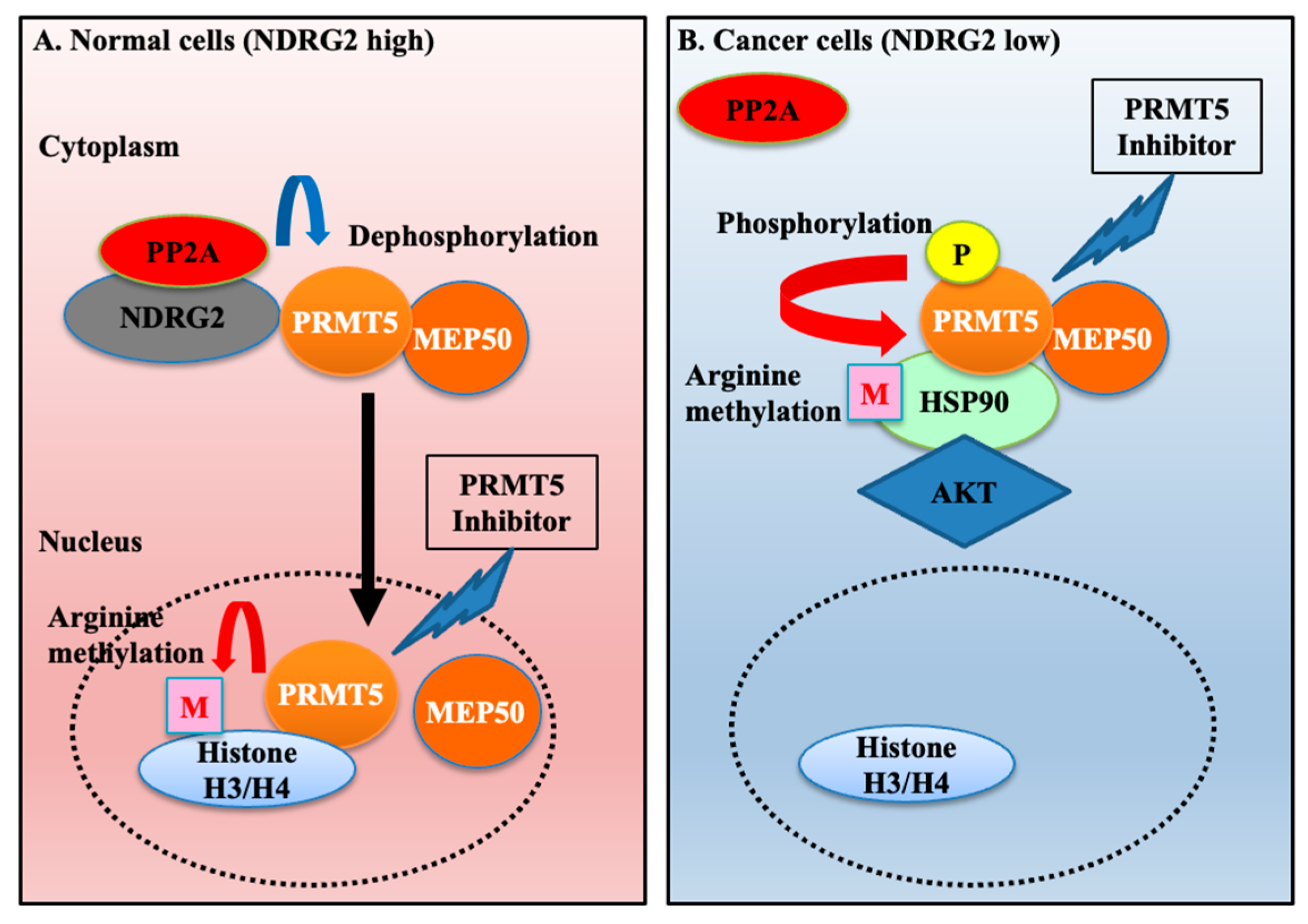
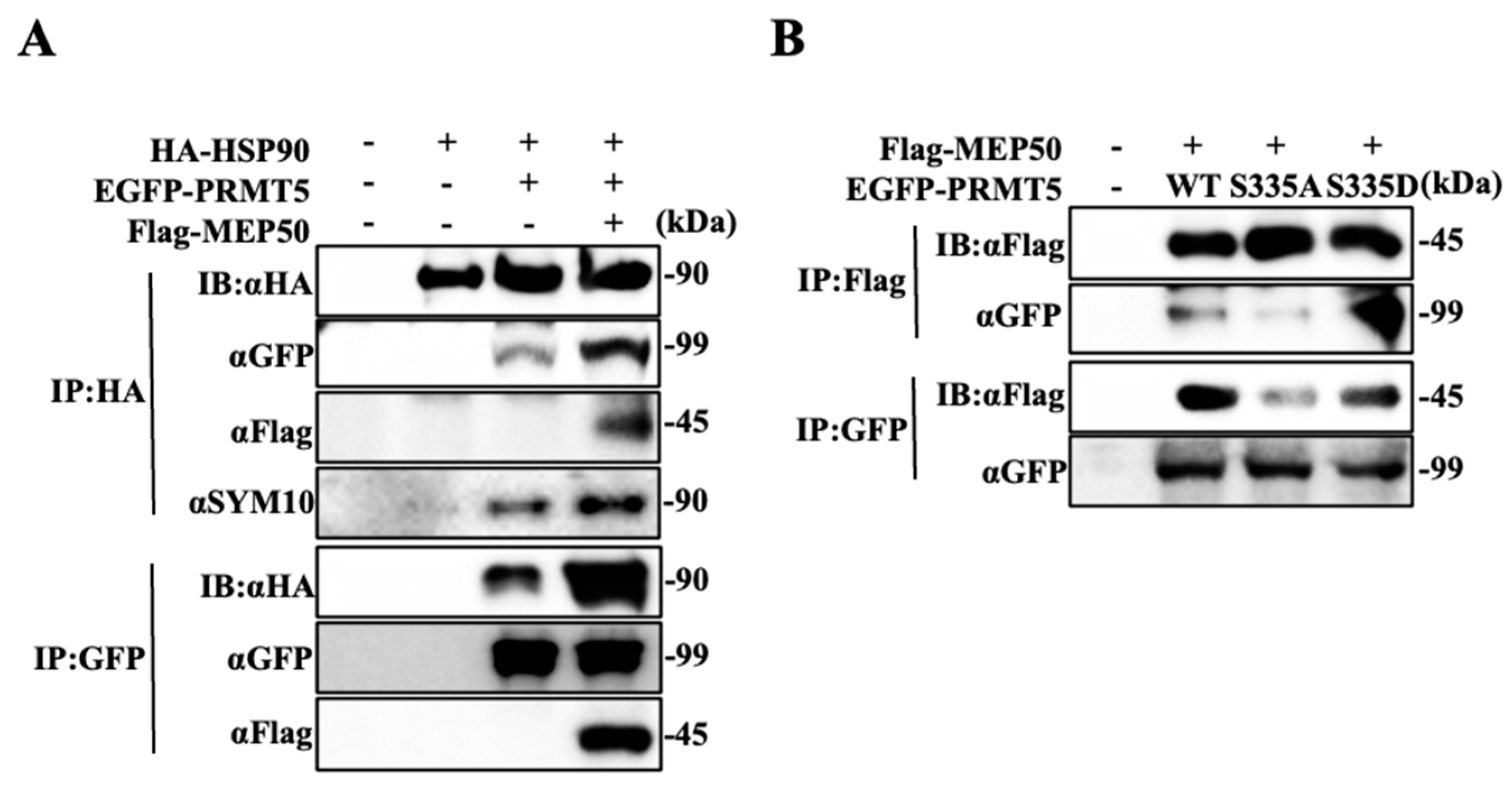
2.2. Hyperphosphorylated PRMT5 Binds to MEP50 and Promotes HSP90 Arginine Methylation
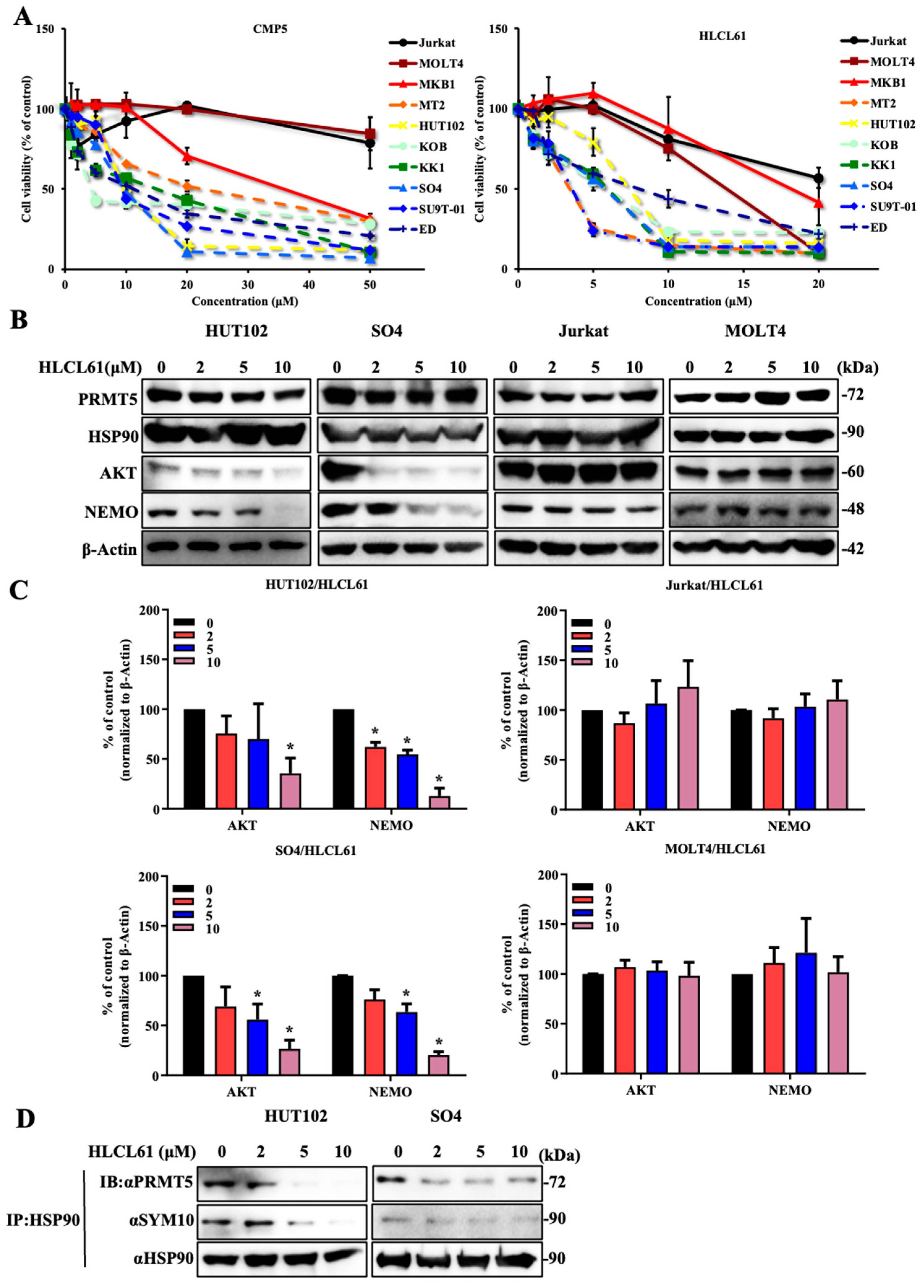
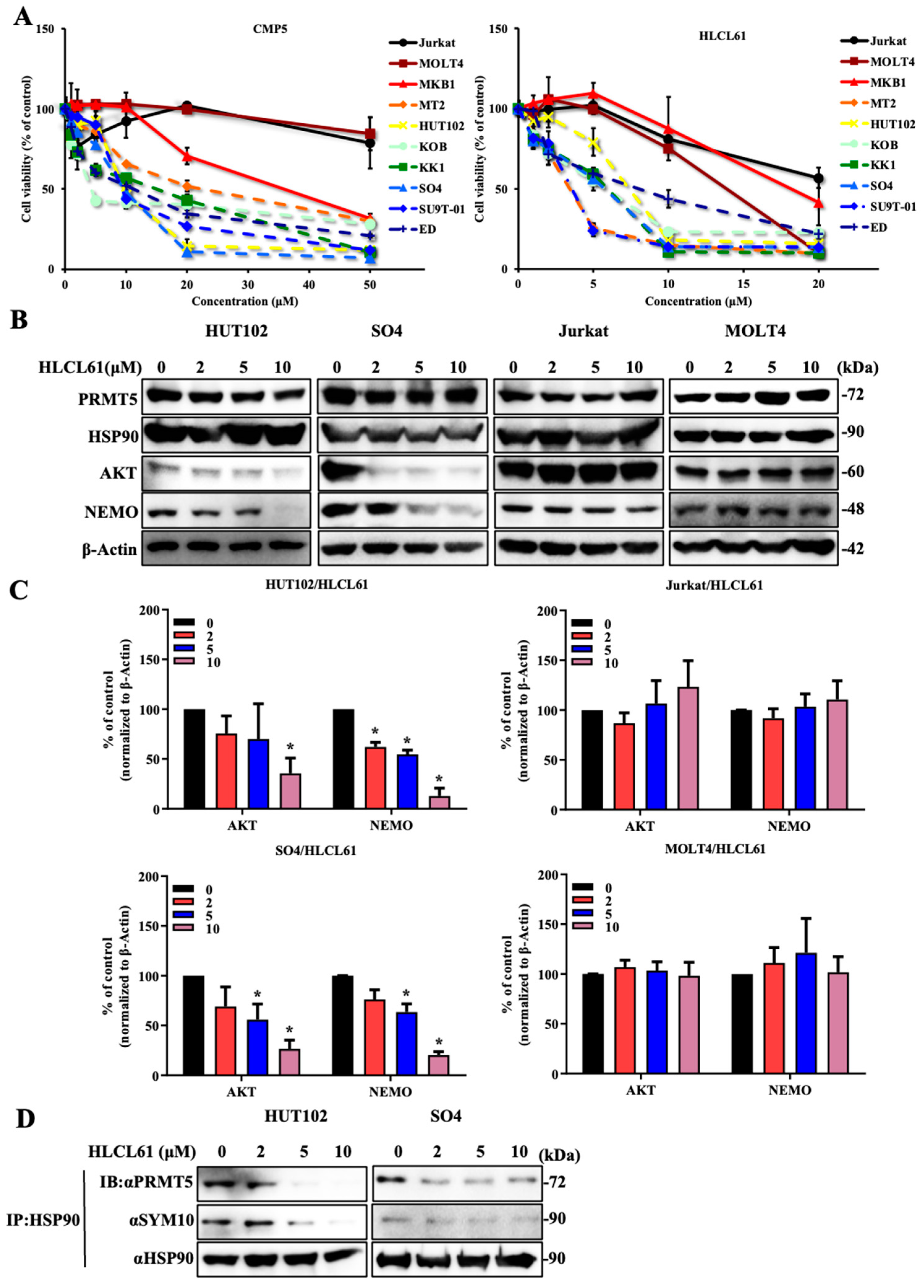
2.3. NDRG2low ATL Cells Are Sensitive to PRMT5 Inhibitors
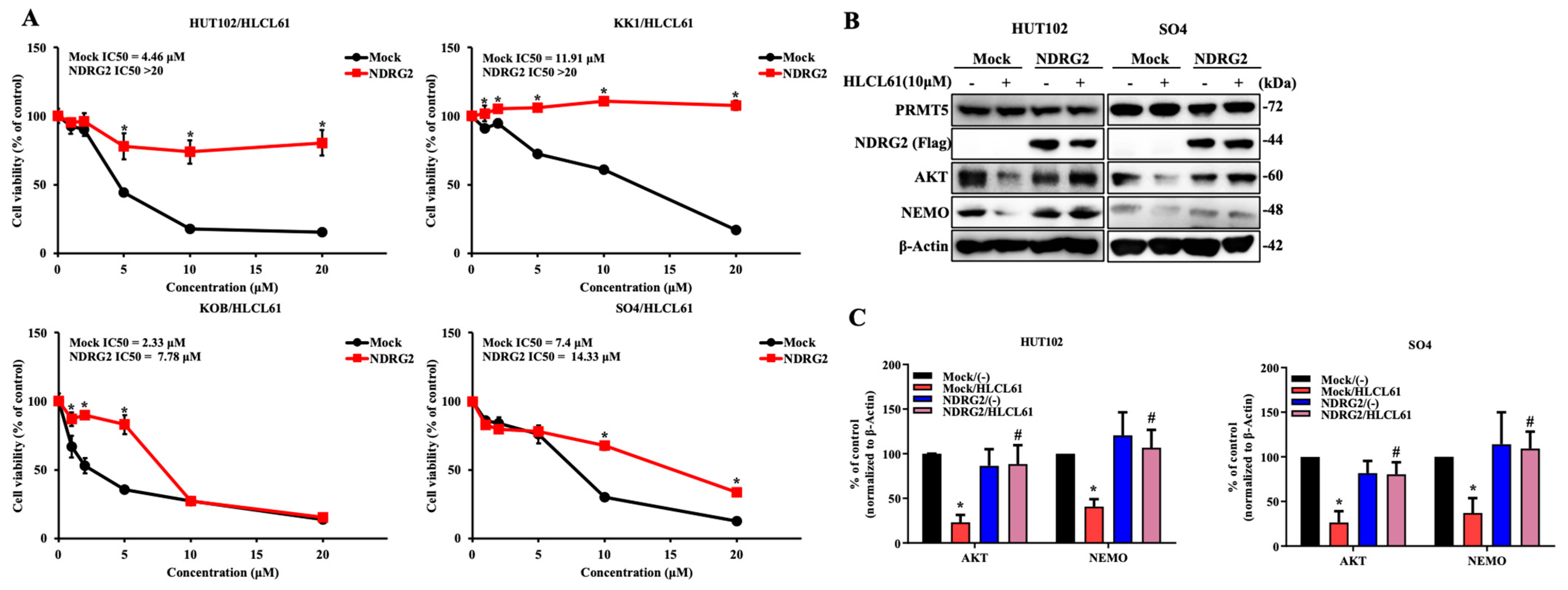
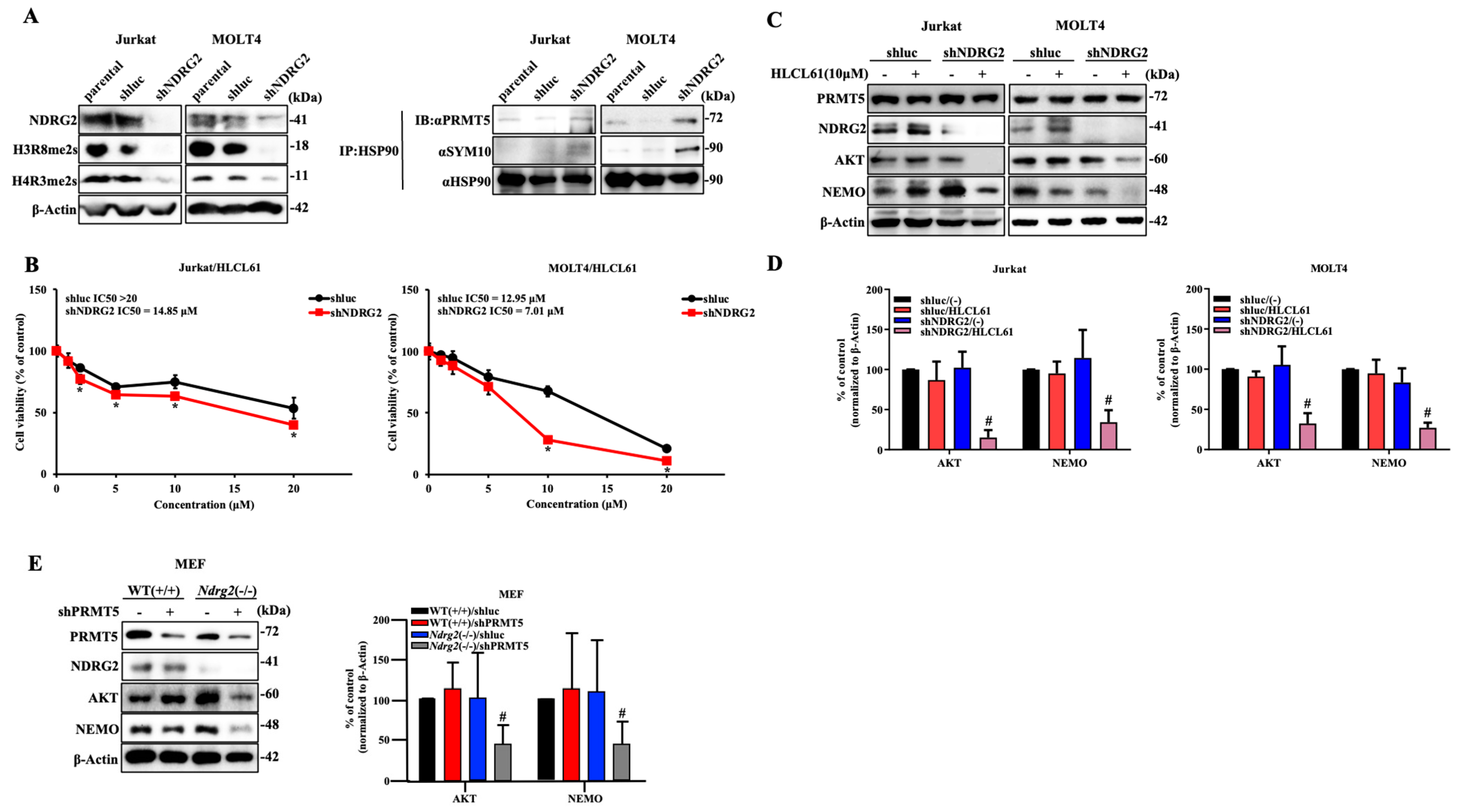
2.4. Knockdown of NDRG2 Expression in T-ALL Cells Enhances Sensitivity to PRMT5 Inhibitors
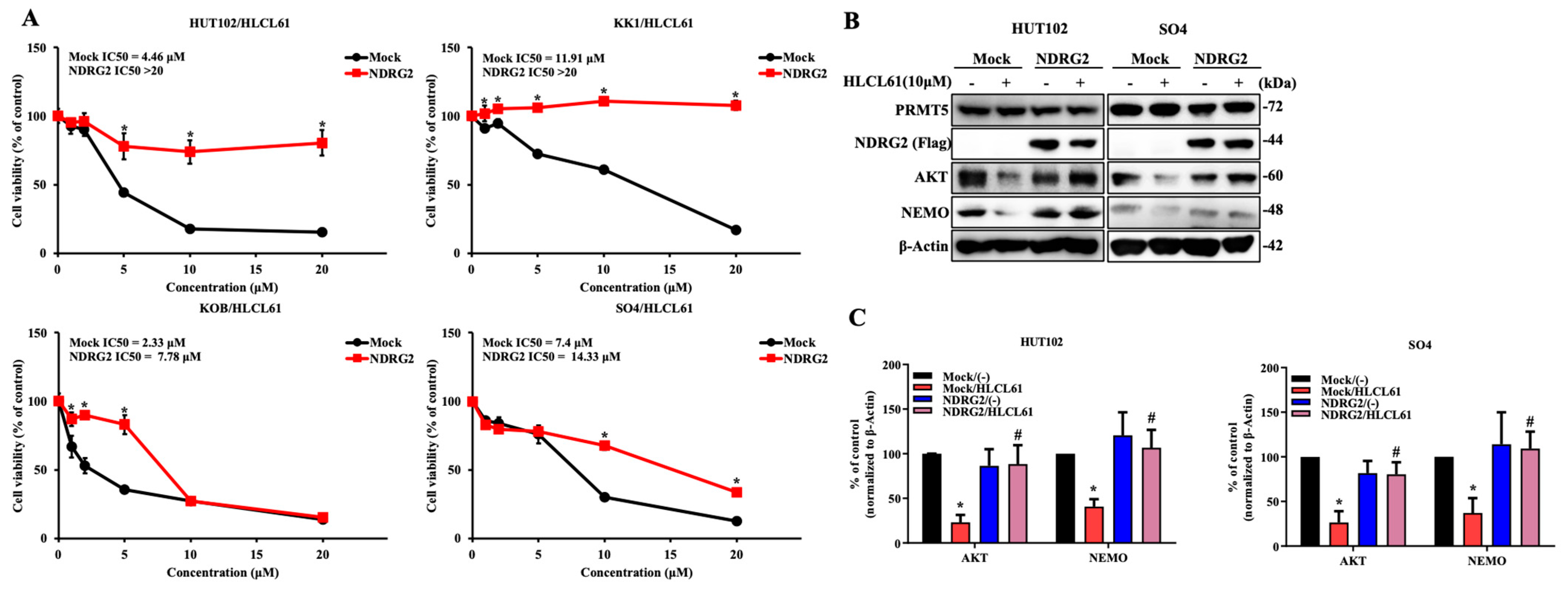
2.5. The Enhanced Expression of NDRG2 Attenuates the Antitumour Effects of PRMT5 Inhibitors in ATL and Solid Cancer Cells
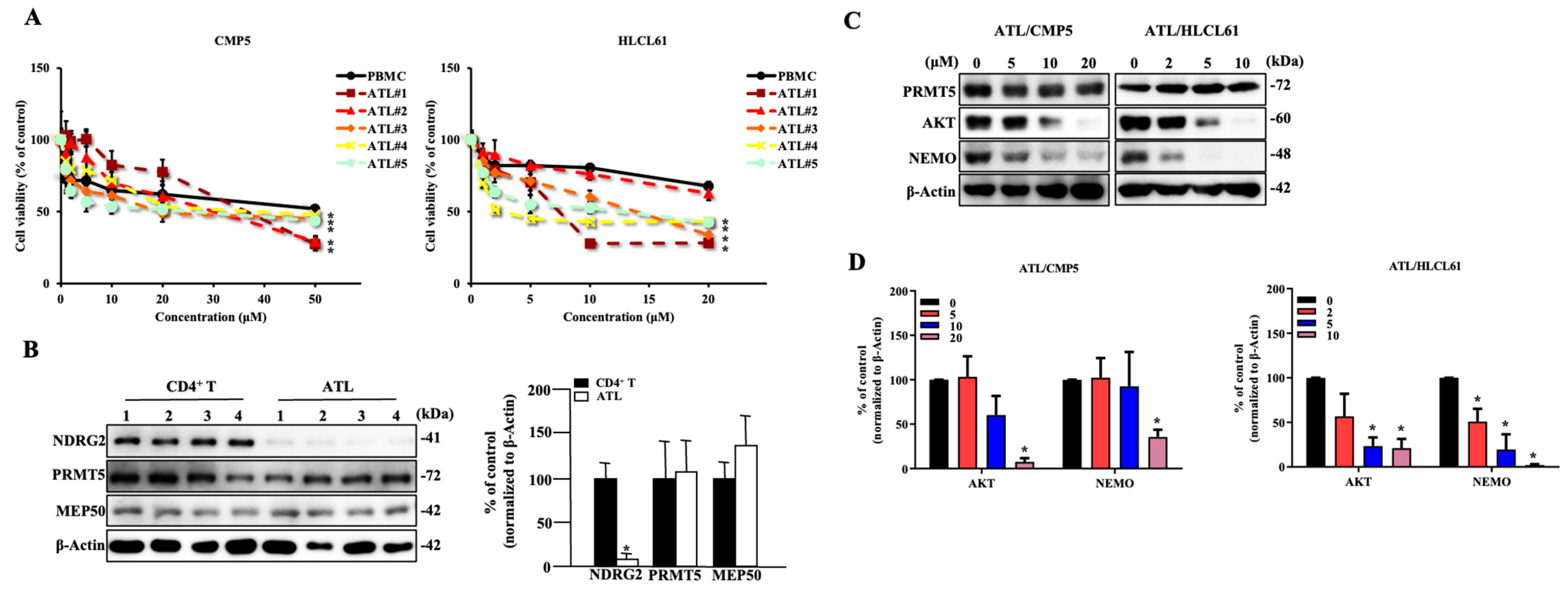
2.6. PRMT5 Inhibitors Are Effective against NDRG2low ATL Patient Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Plasmids
4.4. Patient Samples
4.5. Establishment of Stable Knockdown in Cancer Cell Line
4.6. Cell Proliferation Assay and Calculation of IC50
4.7. Western Blot and Immunoprecipitation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iwanaga, M. Epidemiology of HTLV-1 Infection and ATL in Japan: An Update. Front. Microbiol. 2020, 11, 1124. [Google Scholar] [CrossRef]
- Einsiedel, L.; Pham, H.; Talukder, M.R.R.; Liddle, J.; Taylor, K.; Wilson, K.; Jersmann, H.; Gessain, A.; Woodman, R.; Kaldor, J. Pulmonary Disease Is Associated With Human T-Cell Leukemia Virus Type 1c Infection: A Cross-sectional Survey in Remote Aboriginal Communities. Clin. Infect. Dis. 2020, 73, e1498–e1506. [Google Scholar] [CrossRef]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Cook, L.B.; Phillips, A.A. How I treat adult T-cell leukemia/lymphoma. Blood 2021, 137, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Nakahata, S.; Fujii, M.; Iha, H.; Shimoda, K.; Morishita, K. The regulation of NDRG2 expression during ATLL development after HTLV-1 infection. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 2633–2646. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ichikawa, T.; Nakahata, S.; Kondo, Y.; Tagawa, Y.; Yamamoto, K.; Nagai, K.; Baba, T.; Yamaguchi, R.; Futakuchi, M.; et al. Loss of NDRG2 Expression Confers Oral Squamous Cell Carcinoma with Enhanced Metastatic Potential. Cancer Res. 2017, 77, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Lim, S.; Kim, K.D. The Function of N-Myc Downstream-Regulated Gene 2 (NDRG2) as a Negative Regulator in Tumor Cell Metastasis. Int. J. Mol. Sci. 2022, 23, 9365. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, S.; Ichikawa, T.; Maneesaay, P.; Saito, Y.; Nagai, K.; Tamura, T.; Manachai, N.; Yamakawa, N.; Hamasaki, M.; Kitabayashi, I.; et al. Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat. Commun. 2014, 5, 3393. [Google Scholar] [CrossRef]
- Ichikawa, T.; Nakahata, S.; Fujii, M.; Iha, H.; Morishita, K. Loss of NDRG2 enhanced activation of the NF-κB pathway by PTEN and NIK phosphorylation for ATL and other cancer development. Sci. Rep. 2015, 5, 12841. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Shanab, O.; Nakahata, S.; Shimosaki, S.; Manachai, N.; Ono, M.; Iha, H.; Shimoda, K.; Morishita, K. Novel PRMT5-mediated arginine methylations of HSP90A are essential for maintenance of HSP90A function in NDRG2low ATL and various cancer cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1867, 118615. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.-V.; Wang, B.; Jiang, G.; Wei, H.; Sun, M.; Prabhu, L.; Martin, M.; Safa, A.; Sun, S.; Liu, Y.; et al. Regulation of a PRMT5/NF-κB Axis by Phosphorylation of PRMT5 at Serine 15 in Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 3684. [Google Scholar] [CrossRef] [PubMed]
- Sipos, A.; Iván, J.; Bécsi, B.; Darula, Z.; Tamás, I.; Horváth, D.; Medzihradszky, K.F.; Erdődi, F.; Lontay, B. Myosin phosphatase and RhoA-activated kinase modulate arginine methylation by the regulation of protein arginine methyltransferase 5 in hepatocellular carcinoma cells. Sci. Rep. 2017, 7, 40590. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cheng, G.; Hamard, P.-J.; Greenblatt, S.; Wang, L.; Man, N.; Perna, F.; Xu, H.; Tadi, M.; Luciani, L.; et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Investig. 2015, 125, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, K.M.; Blomquist, C.; Acharya, N.; Li, R.; Ranaghan, M.J.; O’keefe, M.; Rodriguez, D.J.; Young, M.J.; Kesar, D.; Pal, D.; et al. Molecular basis for substrate recruitment to the PRMT5 methylosome. Mol. Cell 2021, 81, 3481–3495.e7. [Google Scholar] [CrossRef] [PubMed]
- Guderian, G.; Peter, C.; Wiesner, J.; Sickmann, A.; Schulze-Osthoff, K.; Fischer, U.; Grimmler, M. RioK1, a New Interactor of Protein Arginine Methyltransferase 5 (PRMT5), Competes with pICln for Binding and Modulates PRMT5 Complex Composition and Substrate Specificity. J. Biol. Chem. 2011, 286, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; El Messaoudi, S.; Rodier, G.; Le Cam, A.; Sardet, C.; Fabbrizio, E. The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep. 2008, 9, 452–458. [Google Scholar] [CrossRef]
- Sun, L.; Wang, M.; Lv, Z.; Yang, N.; Liu, Y.; Bao, S.; Gong, W.; Xu, R.-M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. USA 2011, 108, 20538–20543. [Google Scholar] [CrossRef]
- Li, Y.; Chitnis, N.; Nakagawa, H.; Kita, Y.; Natsugoe, S.; Yang, Y.; Li, Z.; Wasik, M.; Klein-Szanto, A.J.; Rustgi, A.K.; et al. PRMT5 Is Required for Lymphomagenesis Triggered by Multiple Oncogenic Drivers. Cancer Discov. 2015, 5, 288–303. [Google Scholar] [CrossRef]
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2016, 36, 373–386. [Google Scholar] [CrossRef]
- Ibrahim, R.; Matsubara, D.; Osman, W.; Morikawa, T.; Goto, A.; Morita, S.; Ishikawa, S.; Aburatani, H.; Takai, D.; Nakajima, J.; et al. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial-mesenchymal transition. Hum. Pathol. 2014, 45, 1397–1405. [Google Scholar] [CrossRef]
- Yin, S.; Liu, L.; Brobbey, C.; Palanisamy, V.; Ball, L.E.; Olsen, S.K.; Ostrowski, M.C.; Gan, W. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun. 2021, 12, 3444. [Google Scholar] [CrossRef]
- Owens, J.L.; Beketova, E.; Liu, S.; Shen, Q.; Pawar, J.S.; Asberry, A.M.; Yang, J.; Deng, X.; Elzey, B.D.; Ratliff, T.L.; et al. Targeting Protein Arginine Methyltransferase 5 Suppresses Radiation-induced Neuroendocrine Differentiation and Sensitizes Prostate Cancer Cells to Radiation. Mol. Cancer Ther. 2022, 21, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-Associated PRMT5 Methylates Histone H3 Arginine 8 and Negatively Regulates Expression of ST7 and NM23 Tumor Suppressor Genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2015, 7, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ma, Y.; Hu, X.; Zheng, Y.; Chen, X. Targeting PRMT5/Akt signalling axis prevents human lung cancer cell growth. J. Cell. Mol. Med. 2018, 23, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Alinari, L.; Mahasenan, K.V.; Yan, F.; Karkhanis, V.; Chung, J.-H.; Smith, E.M.; Quinion, C.; Smith, P.L.; Kim, L.; Patton, J.T.; et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015, 125, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Tarighat, S.S.; Santhanam, R.; Frankhouser, D.; Radomska, H.S.; Lai, H.; Anghelina, M.; Wang, H.; Huang, X.; Alinari, L.; Walker, A.; et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: Gene activation and repression via histone arginine methylation. Leukemia 2016, 30, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Fan, C.; Jiang, P.; Ma, Z.; Yan, X.; Di, S.; Jiang, S.; Li, T.; Cheng, Y.; Yang, Y. Emerging role of N-myc downstream-regulated gene 2 (NDRG2) in cancer. Oncotarget 2015, 7, 209–223. [Google Scholar] [CrossRef]
- Hu, W.; Yang, Y.; Fan, C.; Ma, Z.; Deng, C.; Li, T.; Lv, J.; Yao, W.; Gao, J. Clinical and pathological significance of N-Myc downstream-regulated gene 2 (NDRG2) in diverse human cancers. Apoptosis 2016, 21, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Lim, S.; Kim, K.D. N-myc Downstream-Regulated Gene 2 (NDRG2) Function as a Positive Regulator of Apoptosis: A New Insight into NDRG2 as a Tumor Suppressor. Cells 2021, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Topatana, W.; Juengpanich, S.; Cao, J.; Hu, J.; Zhang, B.; Ma, D.; Cai, X.; Chen, M. Development of synthetic lethality in cancer: Molecular and cellular classification. Signal Transduct. Target. Ther. 2020, 5, 241. [Google Scholar] [CrossRef]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Cai, S.; Li, S.; Ryan, C.E.; Guo, Z.; Schaiff, W.T.; Lin, L.; Hoog, J.; Goiffon, R.J.; Prat, A.; et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J. Clin. Investig. 2012, 122, 1541–1552. [Google Scholar] [CrossRef]
- McCabe, N.; Hanna, C.; Walker, S.M.; Gonda, D.; Li, J.; Wikstrom, K.; Savage, K.I.; Butterworth, K.T.; Chen, C.; Harkin, D.P.; et al. Mechanistic Rationale to Target PTEN-Deficient Tumor Cells with Inhibitors of the DNA Damage Response Kinase ATM. Cancer Res. 2015, 75, 2159–2165. [Google Scholar] [CrossRef]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef]
- Ernzen, K.; Melvin, C.; Yu, L.; Phelps, C.; Niewiesk, S.; Green, P.L.; Panfil, A.R. The PRMT5 inhibitor EPZ015666 is effective against HTLV-1-transformed T-cell lines in vitro and in vivo. Front. Microbiol. 2023, 14, 1101544. [Google Scholar] [CrossRef]
- Panfil, A.R.; Al-Saleem, J.; Howard, C.M.; Mates, J.M.; Kwiek, J.J.; Baiocchi, R.A.; Green, P.L. PRMT5 Is Upregulated in HTLV-1-Mediated T-Cell Transformation and Selective Inhibition Alters Viral Gene Expression and Infected Cell Survival. Viruses 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Asberry, A.M.; Cai, X.; Deng, X.; Santiago, U.; Liu, S.; Sims, H.S.; Liang, W.; Xu, X.; Wan, J.; Jiang, W.; et al. Discovery and Biological Characterization of PRMT5:MEP50 Protein–Protein Interaction Inhibitors. J. Med. Chem. 2022, 65, 13793–13812. [Google Scholar] [CrossRef] [PubMed]
- Krzyzanowski, A.; Esser, L.M.; Willaume, A.; Prudent, R.; Peter, C.; Hart, P.; Waldmann, H. Development of Macrocyclic PRMT5–Adaptor Protein Interaction Inhibitors. J. Med. Chem. 2022, 65, 15300–15311. [Google Scholar] [CrossRef] [PubMed]
- McKinney, D.C.; McMillan, B.J.; Ranaghan, M.J.; Moroco, J.A.; Brousseau, M.; Mullin-Bernstein, Z.; O’keefe, M.; McCarren, P.; Mesleh, M.F.; Mulvaney, K.M.; et al. Discovery of a First-in-Class Inhibitor of the PRMT5–Substrate Adaptor Interaction. J. Med. Chem. 2021, 64, 11148–11168. [Google Scholar] [CrossRef]
- Miyoshi, I.; Kubonishi, I.; Yoshimoto, S.; Akagi, T.; Ohtsuki, Y.; Shiraishi, Y.; Nagata, K.; Hinuma, Y. Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells. Nature 1981, 294, 770–771. [Google Scholar] [CrossRef]
- Yamada, Y.; Sugawara, K.; Hata, T.; Tsuruta, K.; Moriuchi, R.; Maeda, T.; Atogami, S.; Murata, K.; Fujimoto, K.; Kohno, T.; et al. Interleukin-15 (IL-15) Can Replace the IL-2 Signal in IL-2–Dependent Adult T-Cell Leukemia (ATL) Cell Lines: Expression of IL-15 Receptor α on ATL Cells. Blood 1998, 91, 4265–4272. [Google Scholar] [CrossRef]
- Maeda, T.; Yamada, Y.; Moriuchi, R.; Sugahara, K.; Tsuruda, K.; Joh, T.; Atogami, S.; Tsukasaki, K.; Tomonaga, M.; Kamihira, S. Fas Gene Mutation in the Progression of Adult T Cell Leukemia. J. Exp. Med. 1999, 189, 1063–1071. [Google Scholar] [CrossRef]
- Arima, N.; Molitor, J.A.; Smith, M.R.; Kim, J.H.; Daitoku, Y.; Greene, W.C. Human T-cell leukemia virus type I Tax induces expression of the Rel-related family of kappa B enhancer-binding proteins: Evidence for a pretranslational component of regulation. J. Virol. 1991, 65, 6892–6899. [Google Scholar] [CrossRef]
- Maeda, M.; Shimizu, A.; Ikuta, K.; Okamoto, H.; Kashihara, M.; Uchiyama, T.; Honjo, T.; Yodoi, J. Origin of human T-lymphotrophic virus I-positive T cell lines in adult T cell leukemia. Analysis of T cell receptor gene rearrangement. J. Exp. Med. 1985, 162, 2169–2174. [Google Scholar] [CrossRef]
- Lyles, R.H.; Poindexter, C.; Evans, A.; Brown, M.; Cooper, C.R. Nonlinear model-based estimates of IC50 for studies involving continuous therapeutic dose–response data. Contemp. Clin. Trials 2008, 29, 878–886. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | EPZ015866 | CMP5 | HLCL61 | Tax | NDRG2 |
|---|---|---|---|---|---|
| Jurkat | >100 | 71.7 | 22.72 | - | + |
| MOLT4 | >100 | 92.97 | 13.06 | - | + |
| MKB1 | >100 | 32.5 | 17.6 | - | + |
| MT2 | >100 | 21.65 | 3.09 | + | - |
| HUT102 | >100 | 9.49 | 6.95 | + | - |
| KOB | >100 | 3.98 | 5.52 | + | - |
| SU9T-01 | >100 | 9.1 | 3.23 | + | - |
| KK1 | >100 | 14.07 | 5.7 | - | - |
| SO4 | >100 | 9.45 | 5.57 | - | - |
| ED | >100 | 10.81 | 7.58 | - | - |
| Cell Origin | CMP5 | HLCL61 |
|---|---|---|
| PBMC | 58.08 | 43.37 |
| ATL#1 | 33.12 | 6.97 |
| ATL#2 | 27.13 | 42.71 |
| ATL#3 | 18.81 | 28.46 |
| ATL#4 | 35 | 2.33 |
| ATL#5 | 23.94 | 12.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ichikawa, T.; Suekane, A.; Nakahata, S.; Iha, H.; Shimoda, K.; Murakami, T.; Morishita, K. Inhibition of PRMT5/MEP50 Arginine Methyltransferase Activity Causes Cancer Vulnerability in NDRG2low Adult T-Cell Leukemia/Lymphoma. Int. J. Mol. Sci. 2024, 25, 2842. https://doi.org/10.3390/ijms25052842
Ichikawa T, Suekane A, Nakahata S, Iha H, Shimoda K, Murakami T, Morishita K. Inhibition of PRMT5/MEP50 Arginine Methyltransferase Activity Causes Cancer Vulnerability in NDRG2low Adult T-Cell Leukemia/Lymphoma. International Journal of Molecular Sciences. 2024; 25(5):2842. https://doi.org/10.3390/ijms25052842
Chicago/Turabian StyleIchikawa, Tomonaga, Akira Suekane, Shingo Nakahata, Hidekatsu Iha, Kazuya Shimoda, Takashi Murakami, and Kazuhiro Morishita. 2024. "Inhibition of PRMT5/MEP50 Arginine Methyltransferase Activity Causes Cancer Vulnerability in NDRG2low Adult T-Cell Leukemia/Lymphoma" International Journal of Molecular Sciences 25, no. 5: 2842. https://doi.org/10.3390/ijms25052842
APA StyleIchikawa, T., Suekane, A., Nakahata, S., Iha, H., Shimoda, K., Murakami, T., & Morishita, K. (2024). Inhibition of PRMT5/MEP50 Arginine Methyltransferase Activity Causes Cancer Vulnerability in NDRG2low Adult T-Cell Leukemia/Lymphoma. International Journal of Molecular Sciences, 25(5), 2842. https://doi.org/10.3390/ijms25052842