
Protein Kinase D Plays a Crucial Role in Maintaining Cardiac Homeostasis by Regulating Post-Translational Modifications of Myofilament Proteins

 ,
,  ,
,  and
and
Abstract
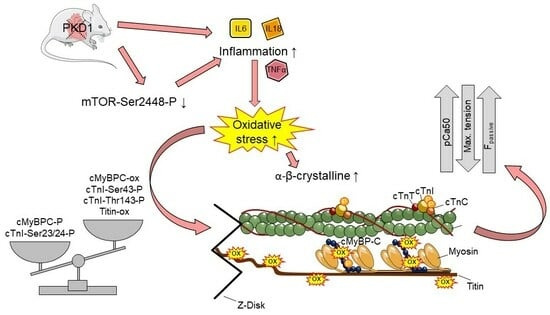
1. Introduction
2. Results
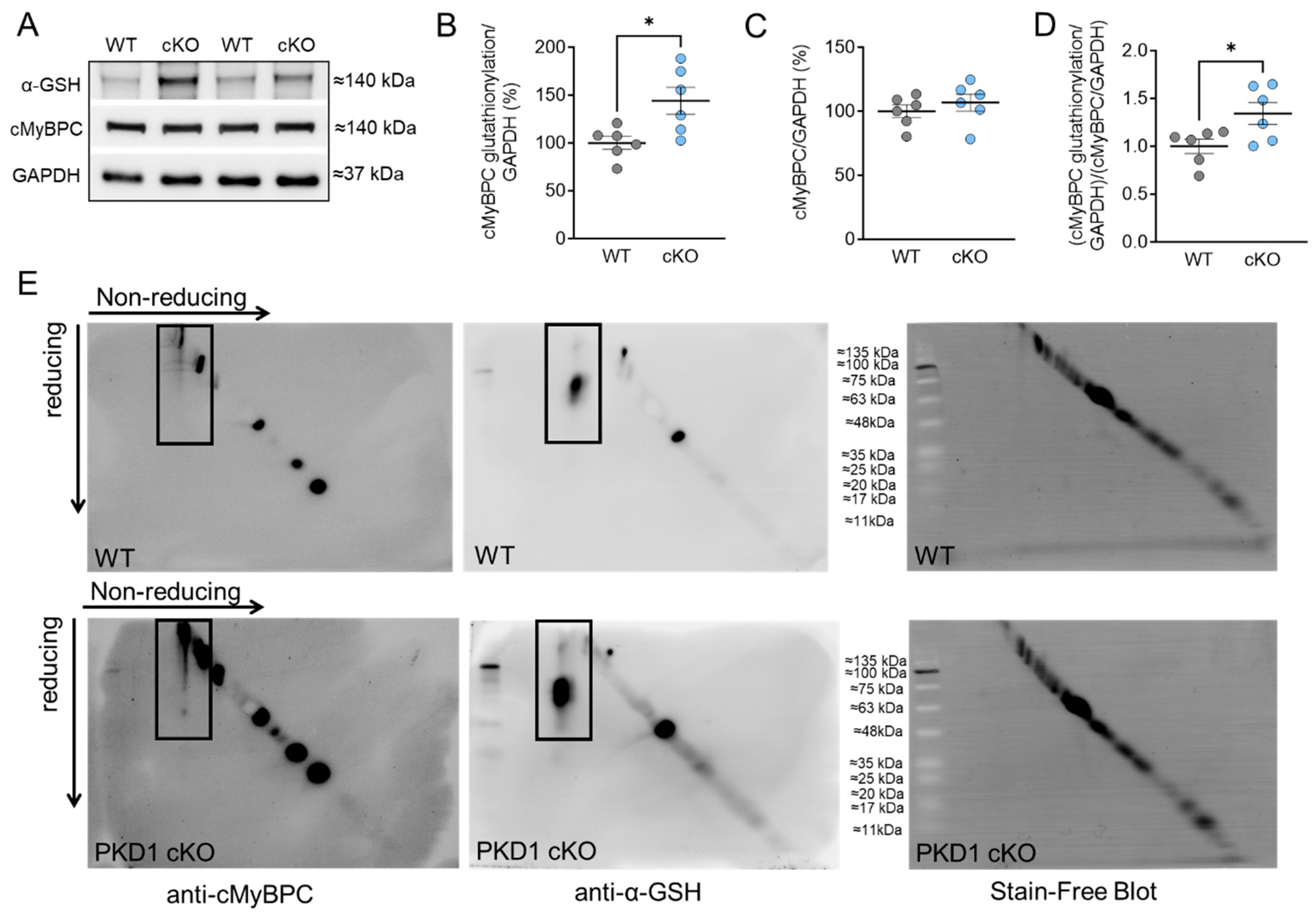
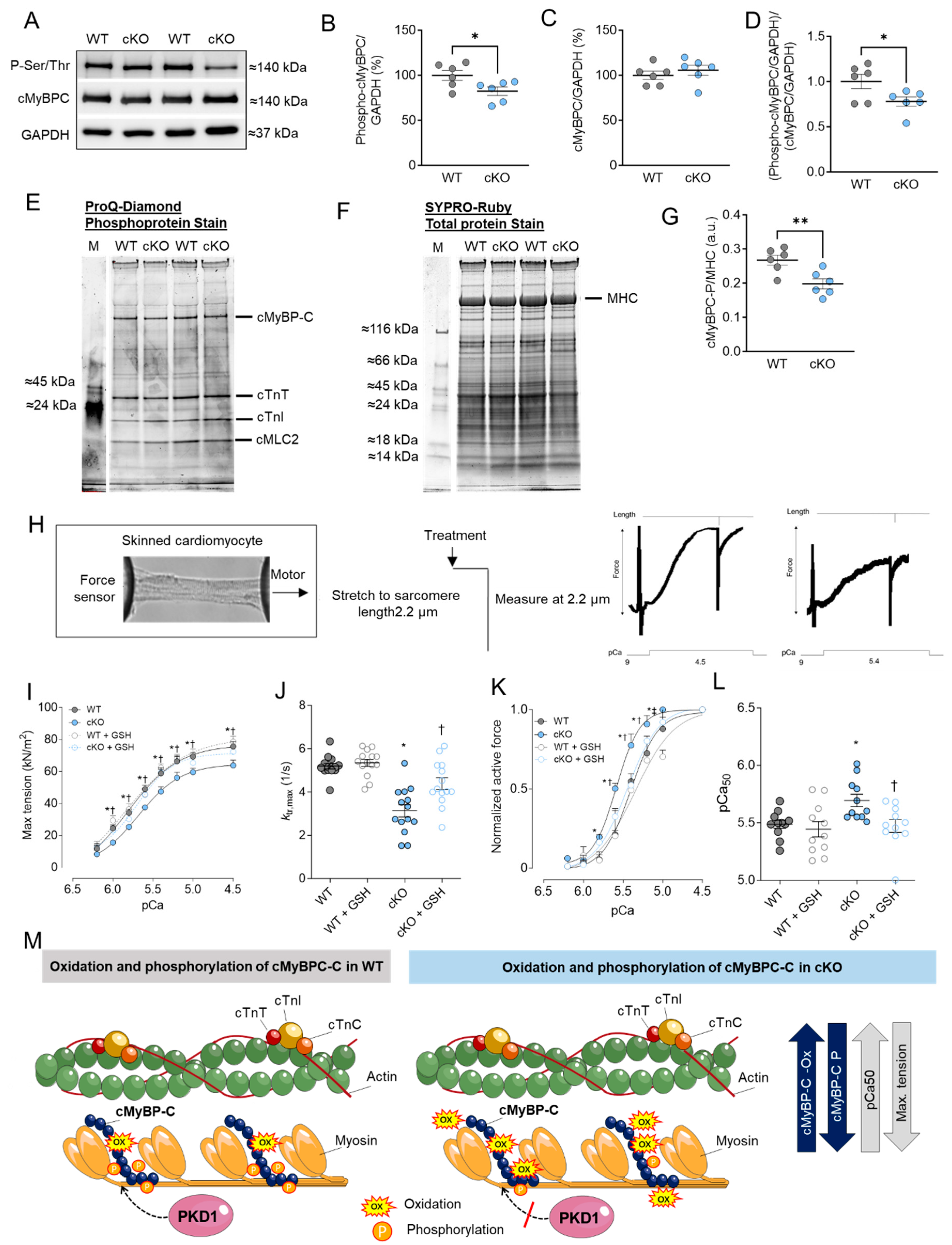
2.1. Altered Maximum Ca2+-Activated Tension and Ca2+ Sensitivity Are Associated with Changes in cMyBPC Oxidation and Phosphorylation in PKD1 cKO Cardiomyocytes
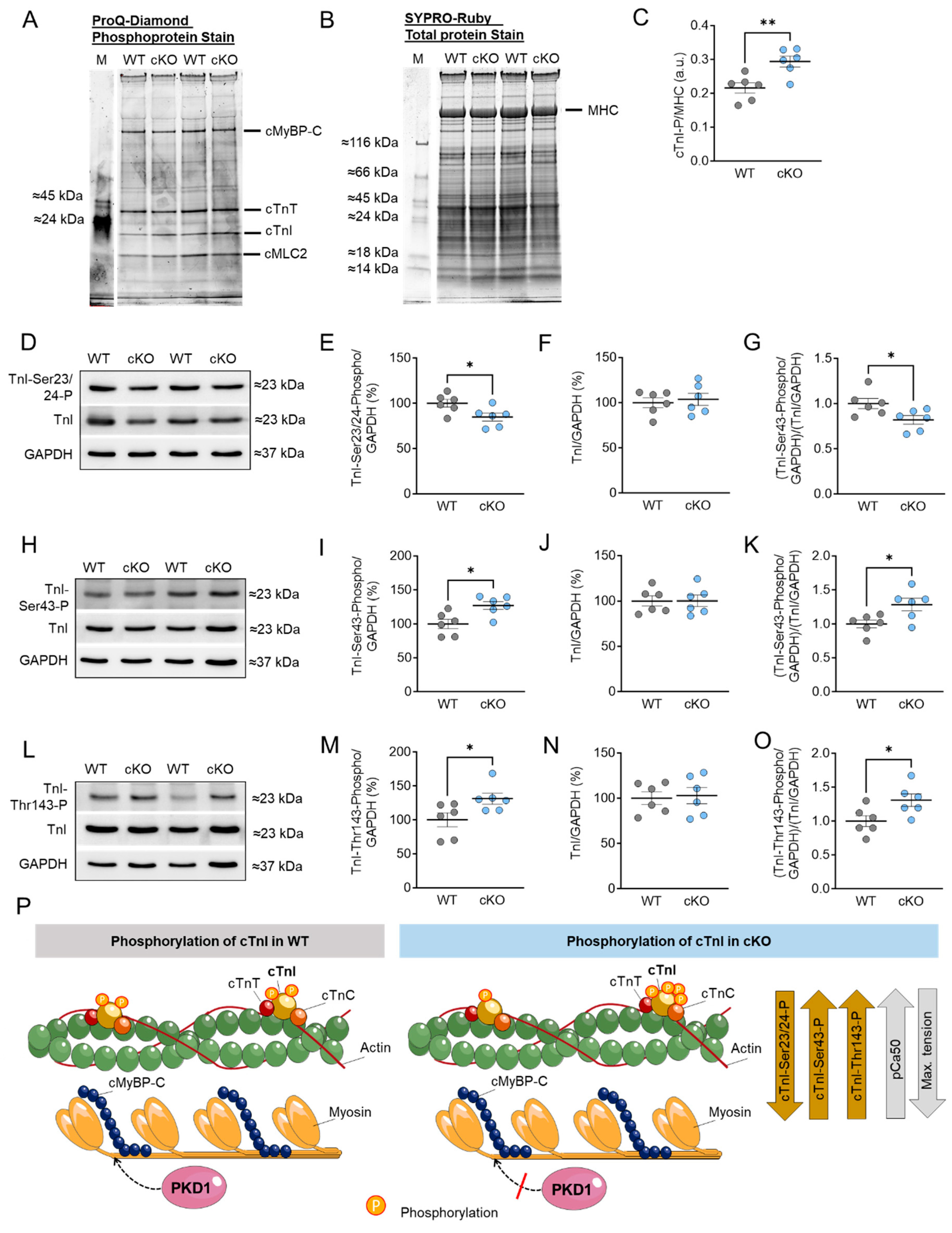
2.2. Altered Troponin I (TnI) Phosphorylation Levels in PKD1 cKO Mice
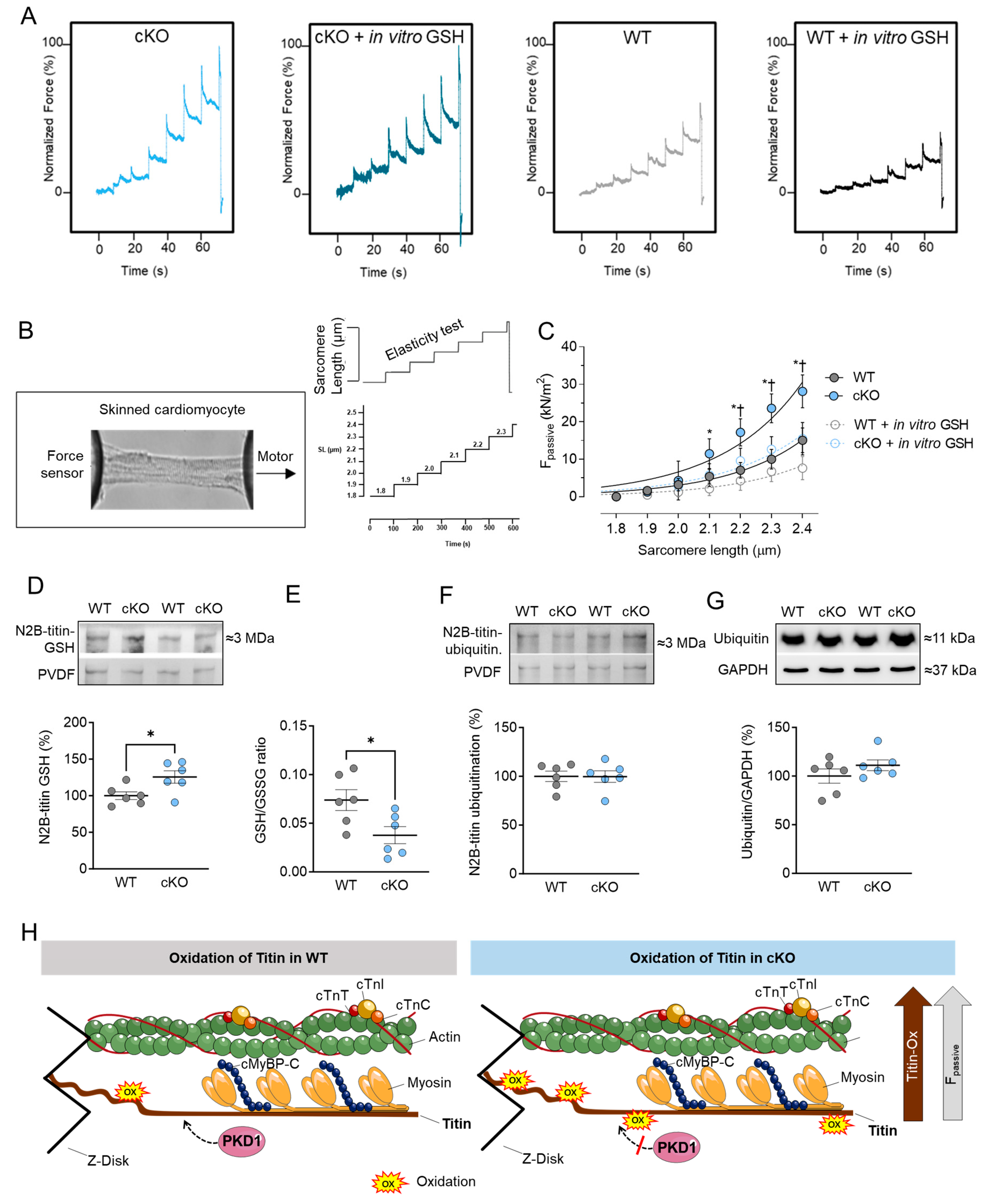
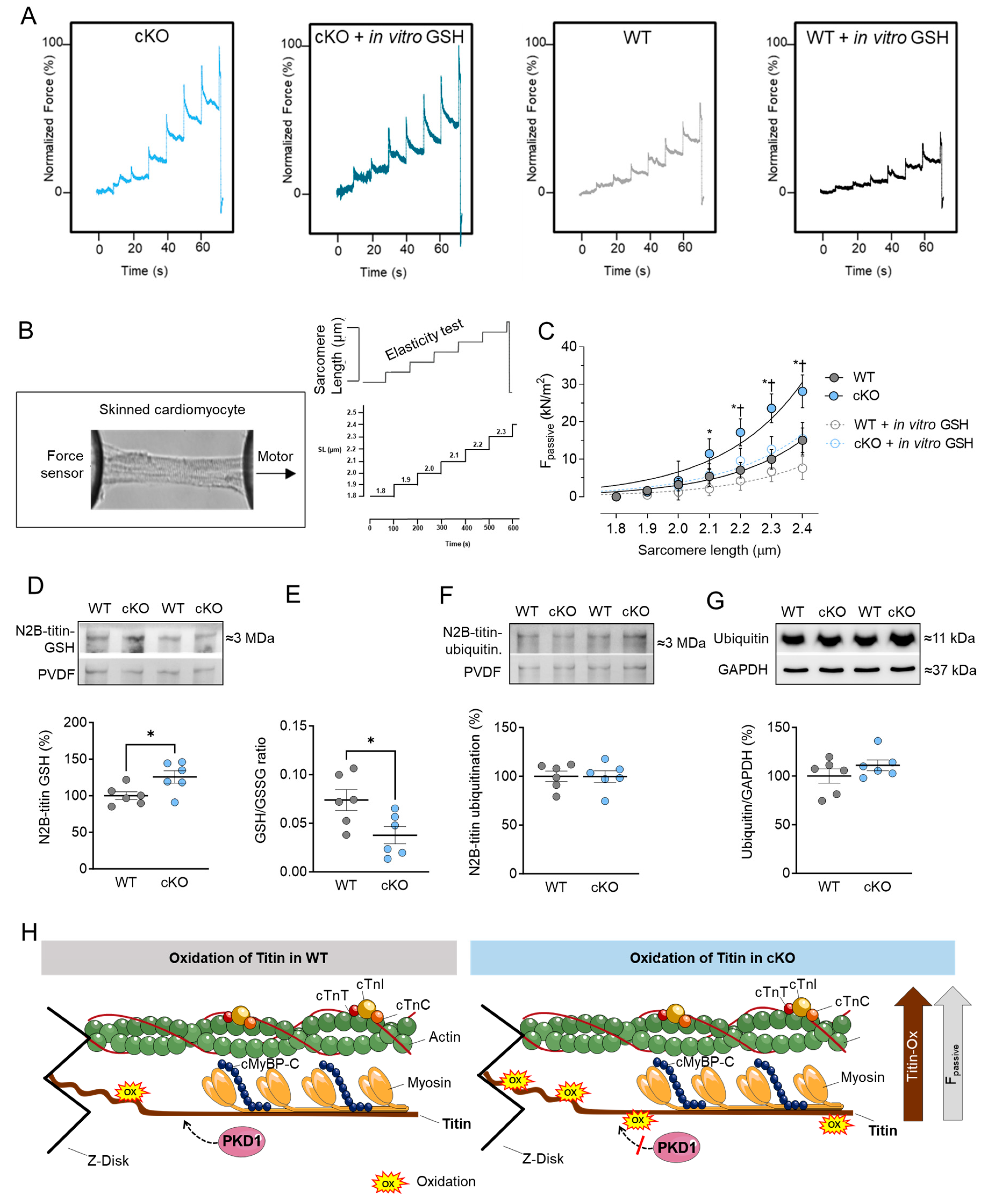
2.3. Increased Cardiomyocyte Passive Stiffness (Fpassive) Is Accompanied by Increased S-Glutathionylation in PKD1 cKO Mice
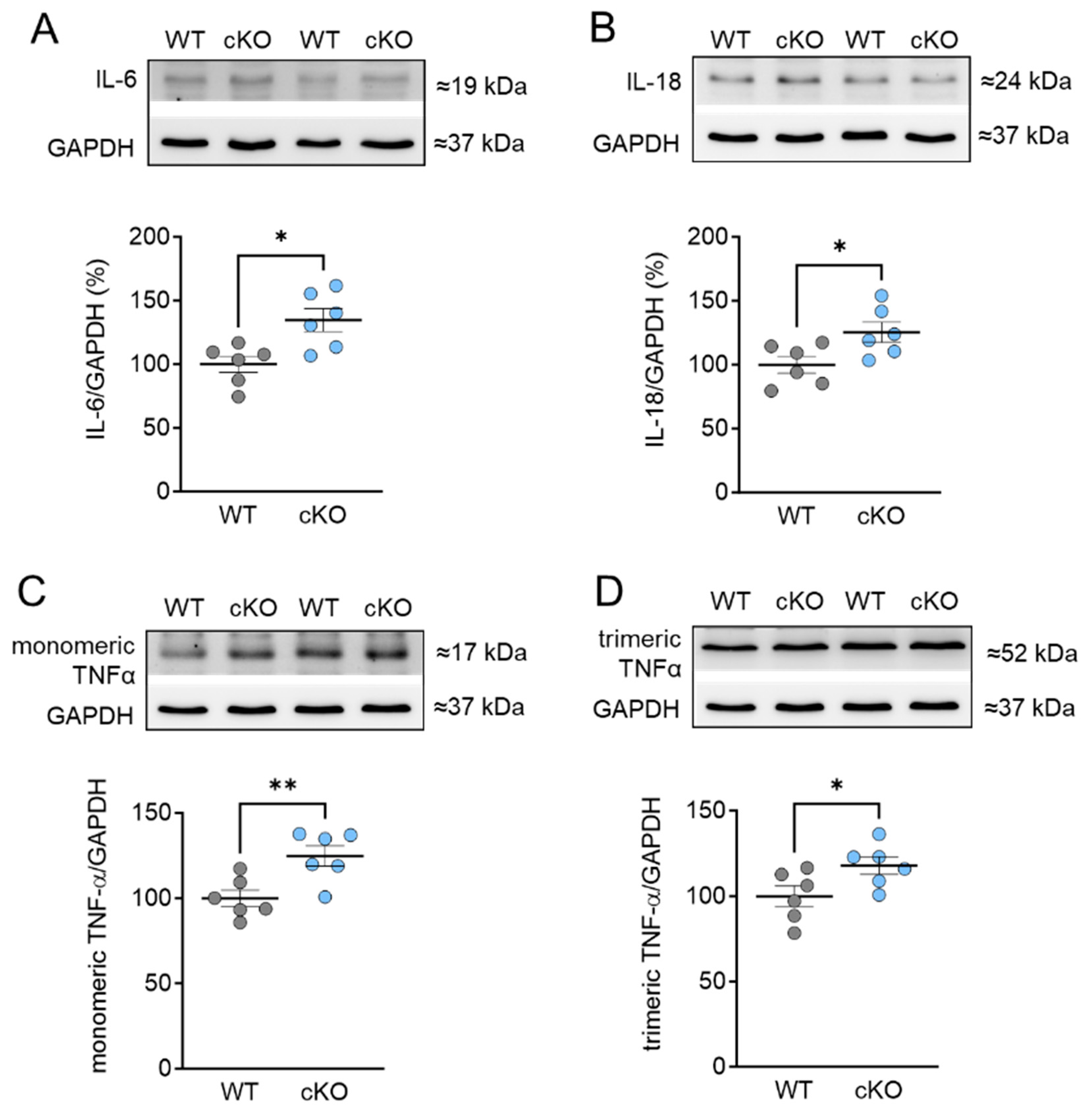
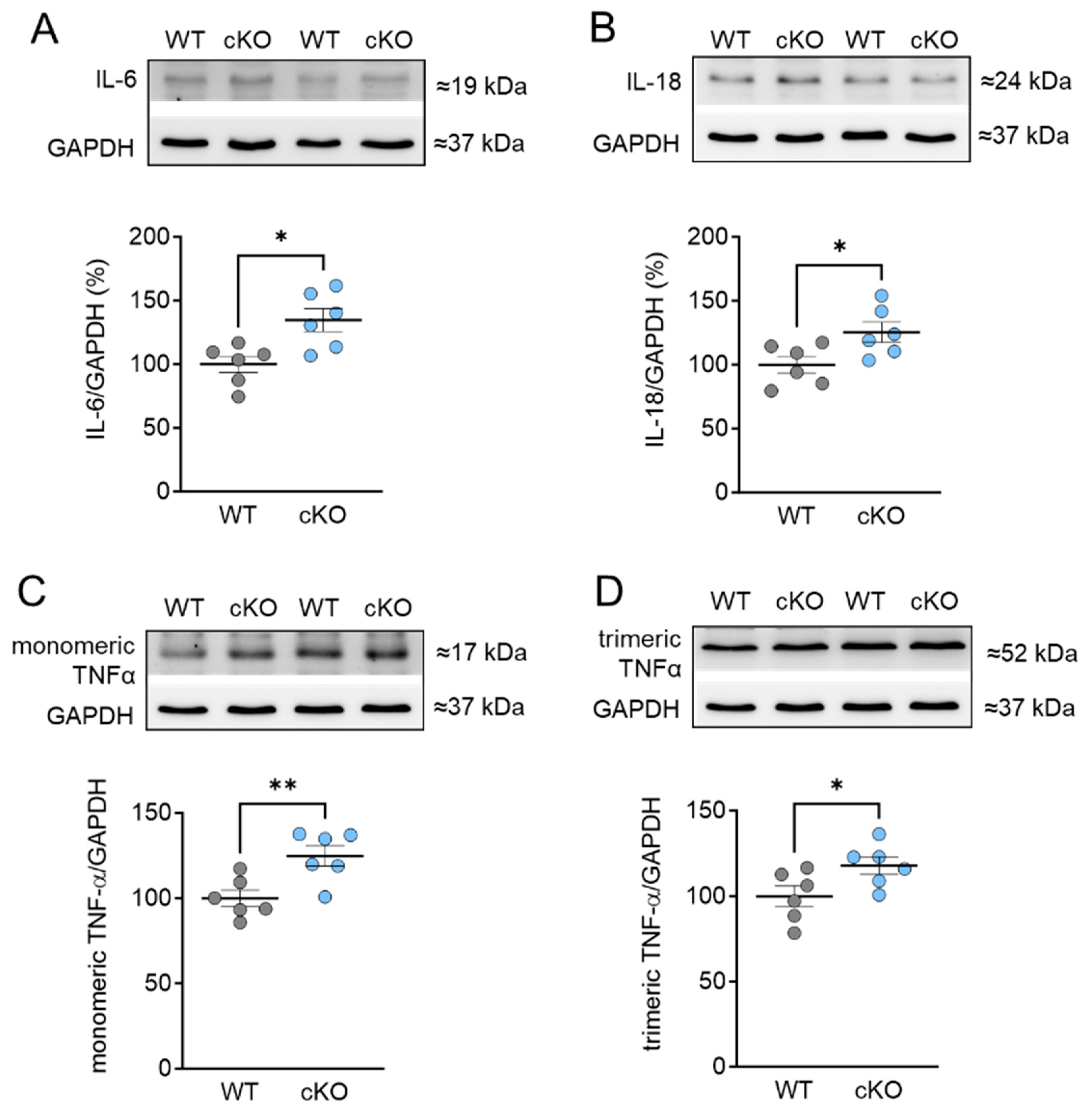
2.4. Increased Inflammation Markers in PKD cKO Mice
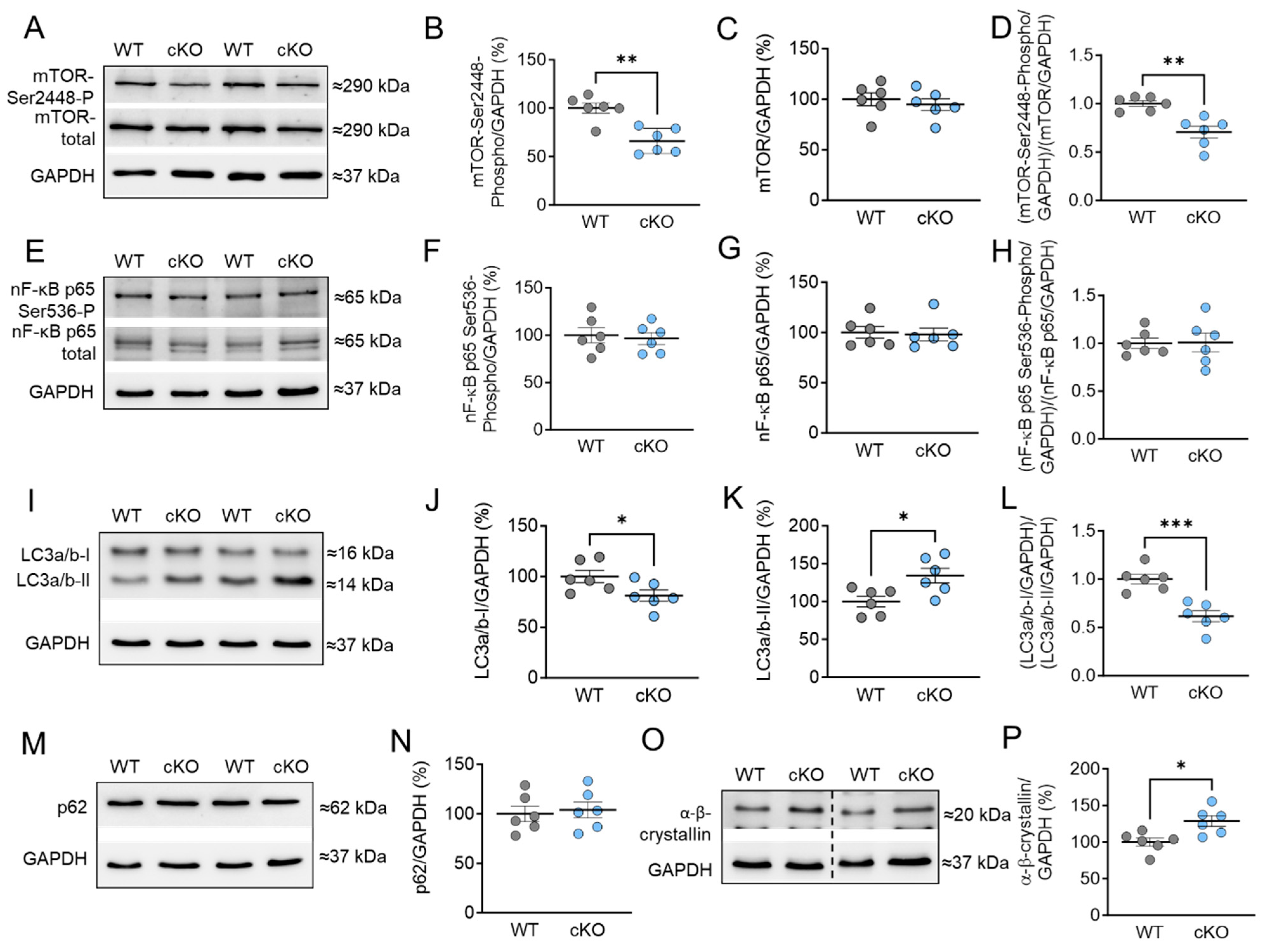
2.5. Stress Signaling and Autophagy Markers in PKD cKO Mice
3. Discussion
3.1. Role of PKD in Regulating Sarcomeric Thin and Thick Filament Function
3.2. PKD and Titin–Role in Cardiomyocyte Function and Mechanics
3.3. Inflammation, Oxidative Stress, Stress Signaling, and PKD1
4. Materials and Methods
4.1. Animal Model and Tissue Sampling
4.2. Force Measurements in Single Skinned Cardiomyocytes
4.3. Protein Isolation and Western Blot Analysis
4.4. Titin Analysis
4.5. ProQ-Diamond and SYPRO-Ruby Staining
4.6. Diagonal Gel Electrophoresis
4.7. Ex Vivo Autophagic Flux Assay Using Western Blot
4.8. Quantification of Glutathione Level in Myocardial Homogenates
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haworth, R.S.; Roberts, N.A.; Cuello, F.; Avkiran, M. Regulation of Protein Kinase D Activity in Adult Myocardium: Novel Counter-Regulatory Roles for Protein Kinase Cepsilon and Protein Kinase A. J. Mol. Cell. Cardiol. 2007, 43, 686–695. [Google Scholar] [CrossRef]
- Avkiran, M.; Rowland, A.J.; Cuello, F.; Haworth, R.S. Protein Kinase D in the Cardiovascular System: Emerging Roles in Health and Disease. Circ. Res. 2008, 102, 157–163. [Google Scholar] [CrossRef]
- Dirkx, E.; Schwenk, R.W.; Coumans, W.A.; Hoebers, N.; Angin, Y.; Viollet, B.; Bonen, A.; Van Eys, G.J.J.M.; Glatz, J.F.C.; Luiken, J.J.F.P. Protein Kinase D1 Is Essential for Contraction-Induced Glucose Uptake but Is Not Involved in Fatty Acid Uptake into Cardiomyocytes. J. Biol. Chem. 2012, 287, 5871–5881. [Google Scholar] [CrossRef]
- Wood, B.M.; Bossuyt, J. Emergency Spatiotemporal Shift: The Response of Protein Kinase D to Stress Signals in the Cardiovascular System. Front. Pharmacol. 2017, 8, 9. [Google Scholar] [CrossRef]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Qi, X.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Requirement of Protein Kinase D1 for Pathological Cardiac Remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 3059–3063. [Google Scholar] [CrossRef]
- Bossuyt, J.; Helmstadter, K.; Wu, X.; Clements-Jewery, H.; Haworth, R.S.; Avkiran, M.; Martin, J.L.; Pogwizd, S.M.; Bers, D.M. Ca2+/Calmodulin-Dependent Protein Kinase IIδ and Protein Kinase D Overexpression Reinforce the Histone Deacetylase 5 Redistribution in Heart Failure. Circ. Res. 2008, 102, 695–702. [Google Scholar] [CrossRef]
- Herwig, M.; Kolijn, D.; Lódi, M.; Hölper, S.; Kovács, Á.; Papp, Z.; Jaquet, K.; Haldenwang, P.; Dos Remedios, C.; Reusch, P.H.; et al. Modulation of Titin-Based Stiffness in Hypertrophic Cardiomyopathy via Protein Kinase D. Front. Physiol. 2020, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Haworth, R.S.; Cuello, F.; Herron, T.J.; Franzen, G.; Kentish, J.C.; Gautel, M.; Avkiran, M. Protein Kinase D Is a Novel Mediator of Cardiac Troponin I Phosphorylation and Regulates Myofilament Function. Circ. Res. 2004, 95, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Bardswell, S.C.; Cuello, F.; Rowland, A.J.; Sadayappan, S.; Robbins, J.; Gautel, M.; Walker, J.W.; Kentish, J.C.; Avkiran, M. Distinct Sarcomeric Substrates Are Responsible for Protein Kinase D-Mediated Regulation of Cardiac Myofilament Ca2+ Sensitivity and Cross-Bridge Cycling. J. Biol. Chem. 2010, 285, 5674–5682. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, E.; Cazorla, O.; Schwenk, R.W.; Lorenzen-Schmidt, I.; Sadayappan, S.; van Lint, J.; Carrier, L.; van Eys, G.J.J.M.; Glatz, J.F.C.; Luiken, J.J.F.P. Protein Kinase D Increases Maximal Ca 2+-Activated Tension of Cardiomyocyte Contraction by Phosphorylation of CMyBP-C- Ser 315. Am. J. Physiol.-Heart Circ. Physiol. 2012, 303, 323–331. [Google Scholar] [CrossRef]
- Waldron, R.T.; Rozengurt, E. Protein Kinase C Phosphorylates Protein Kinase D Activation Loop Ser744 and Ser748 and Releases Autoinhibition by the Pleckstrin Homology Domain. J. Biol. Chem. 2003, 278, 154–163. [Google Scholar] [CrossRef]
- Rozengurt, E.; Rey, O.; Waldron, R.T. Protein Kinase D Signaling. J. Biol. Chem. 2005, 280, 13205–13208. [Google Scholar] [CrossRef]
- Jacamo, R.; Sinnett-Smith, J.; Rey, O.; Waldron, R.T.; Rozengurt, E. Sequential Protein Kinase C (PKC)-Dependent and PKC-Independent Protein Kinase D Catalytic Activation via Gq-Coupled Receptors: Differential Regulation of Activation Loop Ser744 and Ser748 Phosphorylation. J. Biol. Chem. 2008, 283, 12877–12887. [Google Scholar] [CrossRef]
- Rybin, V.O.; Guo, J.; Steinberg, S.F. Protein Kinase D1 Autophosphorylation via Distinct Mechanisms at Ser744/Ser748 and Ser916. J. Biol. Chem. 2009, 284, 2332. [Google Scholar] [CrossRef]
- Storz, P.; Toker, A. Protein Kinase D Mediates a Stress-Induced NF-ΚB Activation and Survival Pathway. EMBO J. 2003, 22, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057. [Google Scholar] [CrossRef] [PubMed]
- Grützner, A.; Garcia-Manyes, S.; Kötter, S.; Badilla, C.L.; Fernandez, J.M.; Linke, W.A. Modulation of Titin-Based Stiffness by Disulfide Bonding in the Cardiac Titin N2-B Unique Sequence. Biophys. J. 2009, 97, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernández, J.M. S-Glutathionylation of Cryptic Cysteines Enhances Titin Elasticity by Blocking Protein Folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Lovelock, J.D.; Monasky, M.M.; Jeong, E.M.; Lardin, H.A.; Liu, H.; Patel, B.G.; Taglieri, D.M.; Gu, L.; Kumar, P.; Pokhrel, N.; et al. Ranolazine Improves Cardiac Diastolic Dysfunction through Modulation of Myofilament Calcium Sensitivity. Circ. Res. 2012, 110, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Budde, H.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Herwig, M.; Varatnitskaya, M.; Sieme, M.; Delalat, S.; Sultana, I.; Kolijn, D.; et al. The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin i and Myosin Binding Protein c in End-Stage Human Failing Hearts. Antioxidants 2021, 10, 1134. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Kimchi, A. PKD Is a Kinase of Vps34 That Mediates ROS-Induced Autophagy Downstream of DAPk. Cell Death Differ. 2011, 19, 788–797. [Google Scholar] [CrossRef]
- Jeong, E.M.; Monasky, M.M.; Gu, L.; Taglieri, D.M.; Patel, B.G.; Liu, H.; Wang, Q.; Greener, I.; Dudley, S.C.; Solaro, R.J. Tetrahydrobiopterin Improves Diastolic Dysfunction by Reversing Changes in Myofilament Properties. J. Mol. Cell. Cardiol. 2013, 56, 44–54. [Google Scholar] [CrossRef]
- Utter, M.S.; Warren, C.M.; John Solaro, R. Impact of Anesthesia and Storage on Posttranslational Modifications of Cardiac Myofilament Proteins. Physiol. Rep. 2015, 3, e12393. [Google Scholar] [CrossRef]
- McDonagh, B. Diagonal Electrophoresis for the Detection of Protein Disulfides. Methods Mol. Biol. 2012, 869, 309–315. [Google Scholar] [CrossRef]
- Saraswat, R.; McDonagh, B. Diagonal Electrophoresis for the Detection of Proteins Involved in Disulfide Bonds. Methods Mol. Biol. 2019, 1855, 279–286. [Google Scholar] [CrossRef]
- Sommer, A.; Traut, R.R. Diagonal Polyacrylamide-Dodecyl Sulfate Gel Electrophoresis for the Identification of Ribosomal Proteins Crosslinked with Methyl-4-Mercaptobutyrimidate. Proc. Natl. Acad. Sci. USA 1974, 71, 3946–3950. [Google Scholar] [CrossRef]
- Rinalducci, S.; Murgiano, L.; Zolla, L. Redox Proteomics: Basic Principles and Future Perspectives for the Detection of Protein Oxidation in Plants. J. Exp. Bot. 2008, 59, 3781–3801. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Baglioni, C. The Active Form of Tumor Necrosis Factor Is a Trimer. J. Biol. Chem. 1987, 262, 6951–6954. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 Prevents Maturation of Autophagic Vacuoles by Inhibiting Fusion between Autophagosomes and Lysosomes in Rat Hepatoma Cell Line, H-4-II-E Cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 Disrupts Autophagic Flux by Inhibiting Both V-ATPase-Dependent Acidification and Ca-P60A/SERCA-Dependent Autophagosome-Lysosome Fusion. Autophagy 2015, 11, 1437. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Andres, A.M.; Sin, J.; Taylor, D.P.J. Untangling Autophagy Measurements: All Fluxed Up. Circ. Res. 2015, 116, 504. [Google Scholar] [CrossRef]
- Wu, Y.C.; Wu, W.K.K.; Li, Y.; Yu, L.; Li, Z.J.; Wong, C.C.M.; Li, H.T.; Sung, J.J.Y.; Cho, C.H. Inhibition of Macroautophagy by Bafilomycin A1 Lowers Proliferation and Induces Apoptosis in Colon Cancer Cells. Biochem. Biophys. Res. Commun. 2009, 382, 451–456. [Google Scholar] [CrossRef]
- Cohen, P. The Origins of Protein Phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Solaro, R.J. Multiplex Kinase Signaling Modifies Cardiac Function at the Level of Sarcomeric Proteins. J. Biol. Chem. 2008, 283, 26829. [Google Scholar] [CrossRef] [PubMed]
- Cuello, F.; Bardswell, S.C.; Haworth, R.S.; Yin, X.; Lutz, S.; Wieland, T.; Mayr, M.; Kentish, J.C.; Avkiran, M. Protein Kinase D Selectively Targets Cardiac Troponin I and Regulates Myofilament Ca2+ Sensitivity in Ventricular Myocytes. Circ. Res. 2007, 100, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Noland, T.A.; Guo, X.; Raynor, R.L.; Jideama, N.M.; Averyhart-Fullard, V.; Solaro, R.J.; Kuo, J.F. Cardiac Troponin I Mutants. J. Biol. Chem. 1995, 270, 25445–25454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhao, J.J.; Potter, J.D. Phosphorylation of Both Serine Residues in Cardiac Troponin I Is Required to Decrease the Ca2+ Affinity of Cardiac Troponin C. J. Biol. Chem. 1995, 270, 30773–30780. [Google Scholar] [CrossRef] [PubMed]
- Pfitzer, G.; Rüegg, J.C.; Flockerzi, V.; Hofmann, F. CGMP-Dependent Protein Kinase Decreases Calcium Sensitivity of Skinned Cardiac Fibers. FEBS Lett. 1982, 149, 171–175. [Google Scholar] [CrossRef]
- Copeland, O.; Sadayappan, S.; Messer, A.E.; Steinen, G.J.M.; Van der Velden, J.; Marston, S.B. Analysis of Cardiac Myosin Binding Protein-C Phosphorylation in Human Heart Muscle. J. Mol. Cell. Cardiol. 2010, 49, 1003–1011. [Google Scholar] [CrossRef]
- El-Armouche, A.; Pohlmann, L.; Schlossarek, S.; Starbatty, J.; Yeh, Y.H.; Nattel, S.; Dobrev, D.; Eschenhagen, T.; Carrier, L. Decreased Phosphorylation Levels of Cardiac Myosin-Binding Protein-C in Human and Experimental Heart Failure. J. Mol. Cell. Cardiol. 2007, 43, 223–229. [Google Scholar] [CrossRef] [PubMed]
- El-Armouche, A.; Boknik, P.; Eschenhagen, T.; Carrier, L.; Knaut, M.; Ravens, U.; Dobrev, D. Molecular Determinants of Altered Ca2+ Handling in Human Chronic Atrial Fibrillation. Circulation 2006, 114, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulou, K.; Wittig, I.; Heidler, J.; Piasecki, A.; Richter, F.; Diering, S.; Van Der Velden, J.; Buck, F.; Donzelli, S.; Schröder, E.; et al. S-Glutathiolation Impairs Phosphoregulation and Function of Cardiac Myosin-Binding Protein C in Human Heart Failure. FASEB J. 2016, 30, 1849–1864. [Google Scholar] [CrossRef] [PubMed]
- Chakouri, N.; Reboul, C.; Boulghobra, D.; Kleindienst, A.; Nottin, S.; Gayrard, S.; Roubille, F.; Matecki, S.; Lacampagne, A.; Cazorla, O. Stress-Induced Protein S-Glutathionylation and Phosphorylation Crosstalk in Cardiac Sarcomeric Proteins-Impact on Heart Function. Int. J. Cardiol. 2018, 258, 207–216. [Google Scholar] [CrossRef]
- Ahmed, S.H.; Lindsey, M.L. Titin Phosphorylation: Myocardial Passive Stiffness Regulated by the Intracellular Giant. Circ. Res. 2009, 105, 611. [Google Scholar] [CrossRef] [PubMed]
- Koser, F.; Loescher, C.; Linke, W.A. Posttranslational Modifications of Titin from Cardiac Muscle: How, Where, and What For? FEBS J. 2019, 286, 2240–2260. [Google Scholar] [CrossRef]
- Giganti, D.; Yan, K.; Badilla, C.L.; Fernandez, J.M.; Alegre-Cebollada, J. Disulfide Isomerization Reactions in Titin Immunoglobulin Domains Enable a Mode of Protein Elasticity. Nat. Commun. 2018, 9, 185. [Google Scholar] [CrossRef]
- Cowell, C.F.; Döppler, H.; Yan, I.K.; Hausser, A.; Umazawa, Y.; Storz, P. Mitochondrial Diacylglycerol Initiates Protein-Kinase D1-Mediated ROS Signaling. J. Cell Sci. 2009, 122, 919–928. [Google Scholar] [CrossRef]
- Storz, P.; Döppler, H.; Toker, A. Activation Loop Phosphorylation Controls Protein Kinase D-Dependent Activation of Nuclear Factor ΚB. Mol. Pharmacol. 2004, 66, 870–879. [Google Scholar] [CrossRef]
- Döppler, H.; Storz, P. A Novel Tyrosine Phosphorylation Site in Protein Kinase D Contributes to Oxidative Stress-Mediated Activation. J. Biol. Chem. 2007, 282, 31873–31881. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Döppler, H.; Toker, A. Protein Kinase D Mediates Mitochondrion-to-Nucleus Signaling and Detoxification from Mitochondrial Reactive Oxygen Species. Mol. Cell. Biol. 2005, 25, 8520. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Döppler, H.; Storz, P.; Li, J.; Comb, M.J.; Toker, A. A Phosphorylation State-Specific Antibody Recognizes Hsp27, a Novel Substrate of Protein Kinase D. J. Biol. Chem. 2005, 280, 15013–15019. [Google Scholar] [CrossRef] [PubMed]
- Linke, W.A.; Hamdani, N. Gigantic Business: Titin Properties and Function through Thick and Thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef]
- Kötter, S.; Unger, A.; Hamdani, N.; Lang, P.; Vorgerd, M.; Nagel-Steger, L.; Linke, W.A. Human Myocytes Are Protected from Titin Aggregation-Induced Stiffening by Small Heat Shock Proteins. J. Cell Biol. 2014, 204, 187–202. [Google Scholar] [CrossRef]
- Hassoun, R.; Budde, H.; Zhazykbayeva, S.; Herwig, M.; Sieme, M.; Delalat, S.; Mostafi, N.; Gömöri, K.; Tangos, M.; Jarkas, M.; et al. Stress Activated Signalling Impaired Protein Quality Control Pathways in Human Hypertrophic Cardiomyopathy. Int. J. Cardiol. 2021, 344, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377. [Google Scholar] [CrossRef]
- Orogo, A.M.; Gustafsson, Å.B. Therapeutic Targeting of Autophagy: Potential and Concerns in Treating Cardiovascular Disease. Circ. Res. 2015, 116, 489. [Google Scholar] [CrossRef]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why Should Autophagic Flux Be Assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef]
- Fass, E.; Shvets, E.; Degani, I.; Hirschberg, K.; Elazar, Z. Microtubules Support Production of Starvation-Induced Autophagosomes but Not Their Targeting and Fusion with Lysosomes. J. Biol. Chem. 2006, 281, 36303–36316. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Gertner, M.; Pontarelli, F.; Court-Vazquez, B.; Bennett, M.V.L.; Ofengeim, D.; Zukin, R.S. Global Ischemia Induces Lysosomal-Mediated Degradation of MTOR and Activation of Autophagy in Hippocampal Neurons Destined to Die. Cell Death Differ. 2016, 24, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wang, W.; Wang, H.; Peng, H.; Liu, X.; Guo, W.; Su, G.; Zhao, Z. PKD Knockdown Inhibits Pressure Overload-Induced Cardiac Hypertrophy by Promoting Autophagy via AKT/MTOR Pathway. Int. J. Biol. Sci. 2017, 13, 276–285. [Google Scholar] [CrossRef]
- Kim, M.-S.; Fielitz, J.; McAnally, J.; Shelton, J.M.; Lemon, D.D.; McKinsey, T.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Protein Kinase D1 Stimulates MEF2 Activity in Skeletal Muscle and Enhances Muscle Performance. Mol. Cell. Biol. 2008, 28, 3600–3609. [Google Scholar] [CrossRef] [PubMed]
- Agah, R.; Frenkel, P.A.; French, B.A.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. Gene Recombination in Postmitotic Cells: Targeted Expression of Cre Recombinase Provokes Cardiac-Restricted, Site-Specific Rearrangement in Adult Ventricular Muscle in Vivo. J. Clin. Investig. 1997, 100, 169–179. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; Von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged Myofilament Phosphorylation and Function in Experimental Heart Failure with Preserved Ejection Fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef]
- Hamdani, N.; Franssen, C.; Lourenço, A.; Falca o-Pires, I.; Fontoura, D.; Leite, S.; Plettig, L.; Lopez, B.; Ottenheijm, C.A.; Becher, P.M.; et al. Myocardial Titin Hypophosphorylation Importantly Contributes to Heart Failure with Preserved Ejection Fraction in a Rat Metabolic Risk Model. Circ. Heart Fail. 2013, 6, 1239–1249. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Company | Cat. No. | Dilution |
|---|---|---|---|
| SQSTM1/p62 | Cell Signaling Technology (Leiden, The Netherlands) | 39749S | 1:1000 |
| LC3a/b | Cell Signaling Technology | 12741S | 1:1000 |
| mTor-total | Cell Signaling Technology | 2972S | 1:1000 |
| Phospho-mTor (Ser2448) | Cell Signaling Technology | 2971S | 1:1000 |
| Ubiquitin | Cell Signaling Technology | 43124S | 1:1000 |
| Phospho-Serine/Threonine antibody | ECM Bioscience LLC (Versailles, KY, USA) | PP2551 | 1:500 |
| α-GSH | Abcam (Cambridge, UK) | ab19534 | 1:1000 |
| IL-6 | Invitrogen (Waltham, MA, USA) | P620 | 1:1000 |
| IL-18 | Invitrogen | PA5-80719 | 1:1000 |
| TNF-α | Invitrogen | AMC3012 | 1:1000 |
| NF-κB p65 | Cell Signaling Technology | 8242S | 1:1000 |
| Phospho-NF-κB p65 (S536) | Cell Signaling Technology | 3033S | 1:1000 |
| Myosin-binding protein C | Invitrogen | PA571701 | 1:2000 |
| Phospho-cardiac troponin I (Ser23/24) antibody | Cell Signaling Technology | 4004S | 1:1000 |
| Phospho-cardiac troponin I (phospho Ser43) antibody | Abcam | ab196005 | 1:1000 |
| Phospho-cardiac troponin I (phospho Thr143) antibody | Abcam | ab58546 | 1:1000 |
| Troponin I | Cell Signaling Technology | 4002S | 1:1000 |
| α-β-crystallin | Abcam | ab13497 | 1:1000 |
| GAPDH | Cell Signaling Technology | 97166 | 1:2000 |
| GAPDH | Cell Signaling Technology | 2118 | 1:2000 |
| Secondary Antibody | Company | Cat. No. | Dilution |
|---|---|---|---|
| Anti-Rabbit | Cell Signaling Technology | 7074 | 1:10,000 |
| Anti-mouse | Cell Signaling Technology | 7076 | 1:10,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herwig, M.; Begovic, M.; Budde, H.; Delalat, S.; Zhazykbayeva, S.; Sieme, M.; Schneider, L.; Jaquet, K.; Mügge, A.; Akin, I.; et al. Protein Kinase D Plays a Crucial Role in Maintaining Cardiac Homeostasis by Regulating Post-Translational Modifications of Myofilament Proteins. Int. J. Mol. Sci. 2024, 25, 2790. https://doi.org/10.3390/ijms25052790
Herwig M, Begovic M, Budde H, Delalat S, Zhazykbayeva S, Sieme M, Schneider L, Jaquet K, Mügge A, Akin I, et al. Protein Kinase D Plays a Crucial Role in Maintaining Cardiac Homeostasis by Regulating Post-Translational Modifications of Myofilament Proteins. International Journal of Molecular Sciences. 2024; 25(5):2790. https://doi.org/10.3390/ijms25052790
Chicago/Turabian StyleHerwig, Melissa, Merima Begovic, Heidi Budde, Simin Delalat, Saltanat Zhazykbayeva, Marcel Sieme, Luca Schneider, Kornelia Jaquet, Andreas Mügge, Ibrahim Akin, and et al. 2024. "Protein Kinase D Plays a Crucial Role in Maintaining Cardiac Homeostasis by Regulating Post-Translational Modifications of Myofilament Proteins" International Journal of Molecular Sciences 25, no. 5: 2790. https://doi.org/10.3390/ijms25052790
APA StyleHerwig, M., Begovic, M., Budde, H., Delalat, S., Zhazykbayeva, S., Sieme, M., Schneider, L., Jaquet, K., Mügge, A., Akin, I., El-Battrawy, I., Fielitz, J., & Hamdani, N. (2024). Protein Kinase D Plays a Crucial Role in Maintaining Cardiac Homeostasis by Regulating Post-Translational Modifications of Myofilament Proteins. International Journal of Molecular Sciences, 25(5), 2790. https://doi.org/10.3390/ijms25052790