
Chimeric Antigen Receptor T Cell Therapy for Hepatocellular Carcinoma: Where Do We Stand?




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
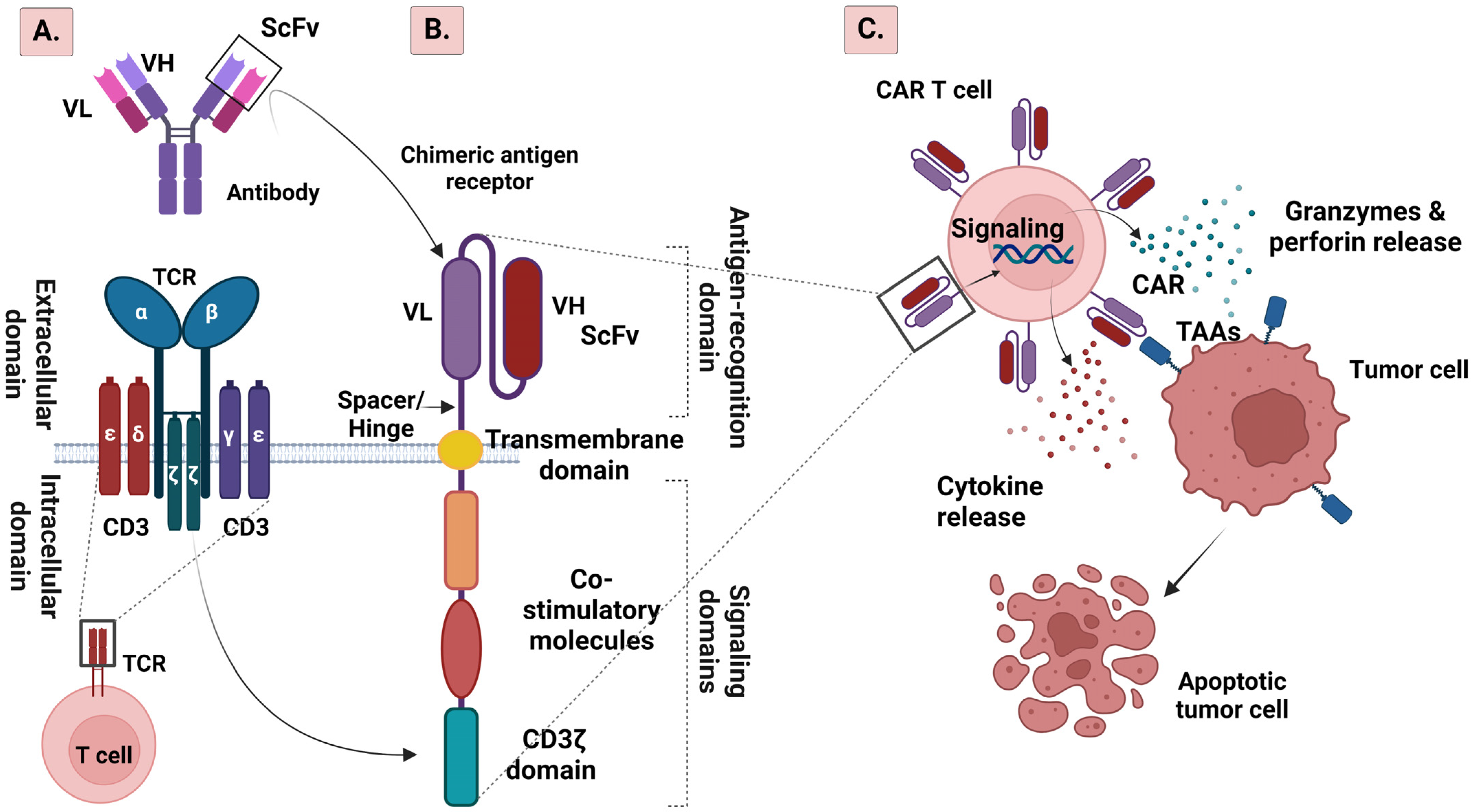
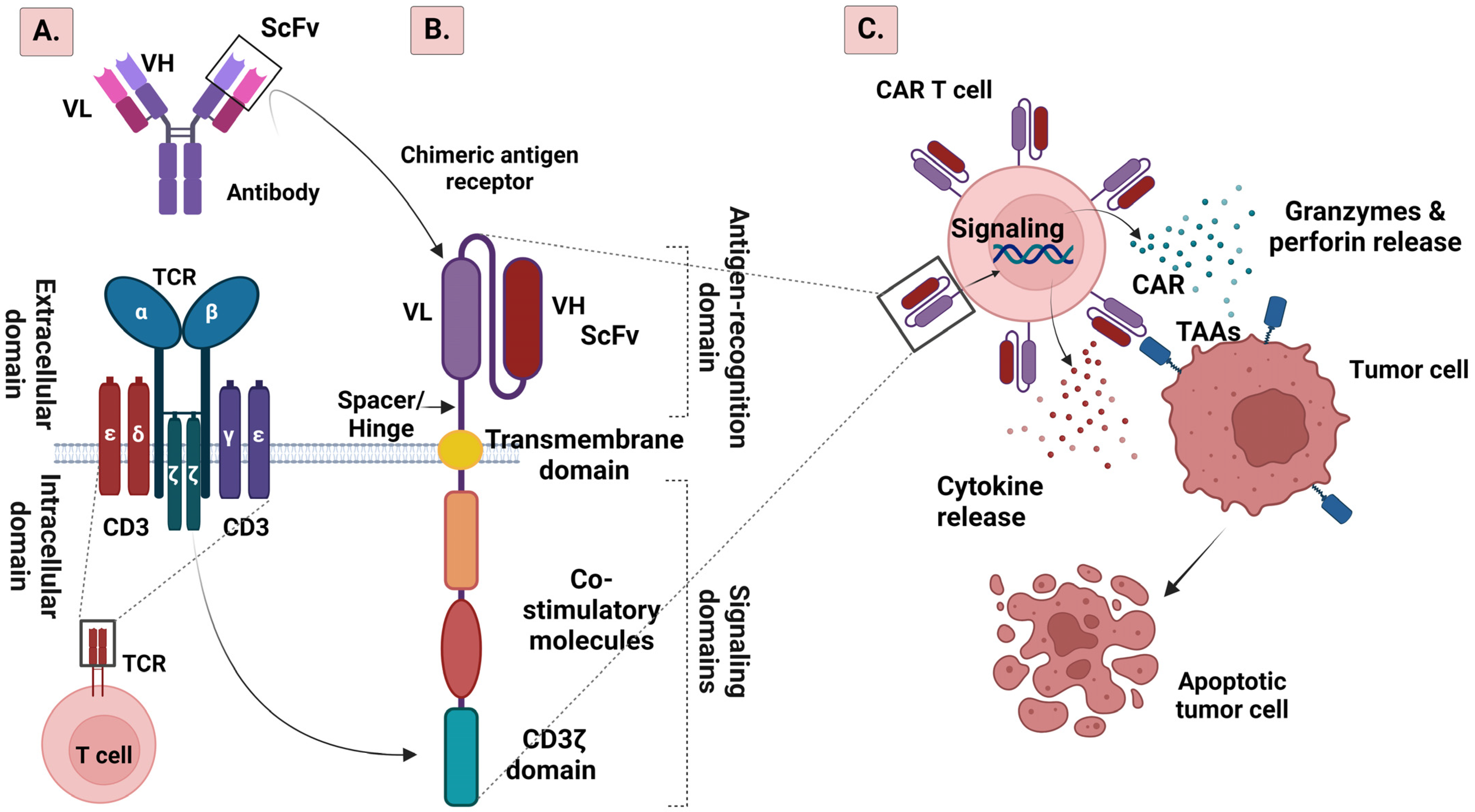
2. CAR T Cell Structure
2.1. From the First to the Fifth Generation of CAR T Cells
2.2. AND Logic Gate CAR
2.3. OR Logic Gate
2.4. NOT Logic Gate
2.5. IF-Better Gate CAR
2.6. SUPRA CAR System
3. CAR T Cell Manufacturing Process and Challenges
4. The Role of Tertiary Lymphoid Structures (TLSs) in CAR T Therapy
5. The Role of T Cell Depletion in CAR T Cell Therapy
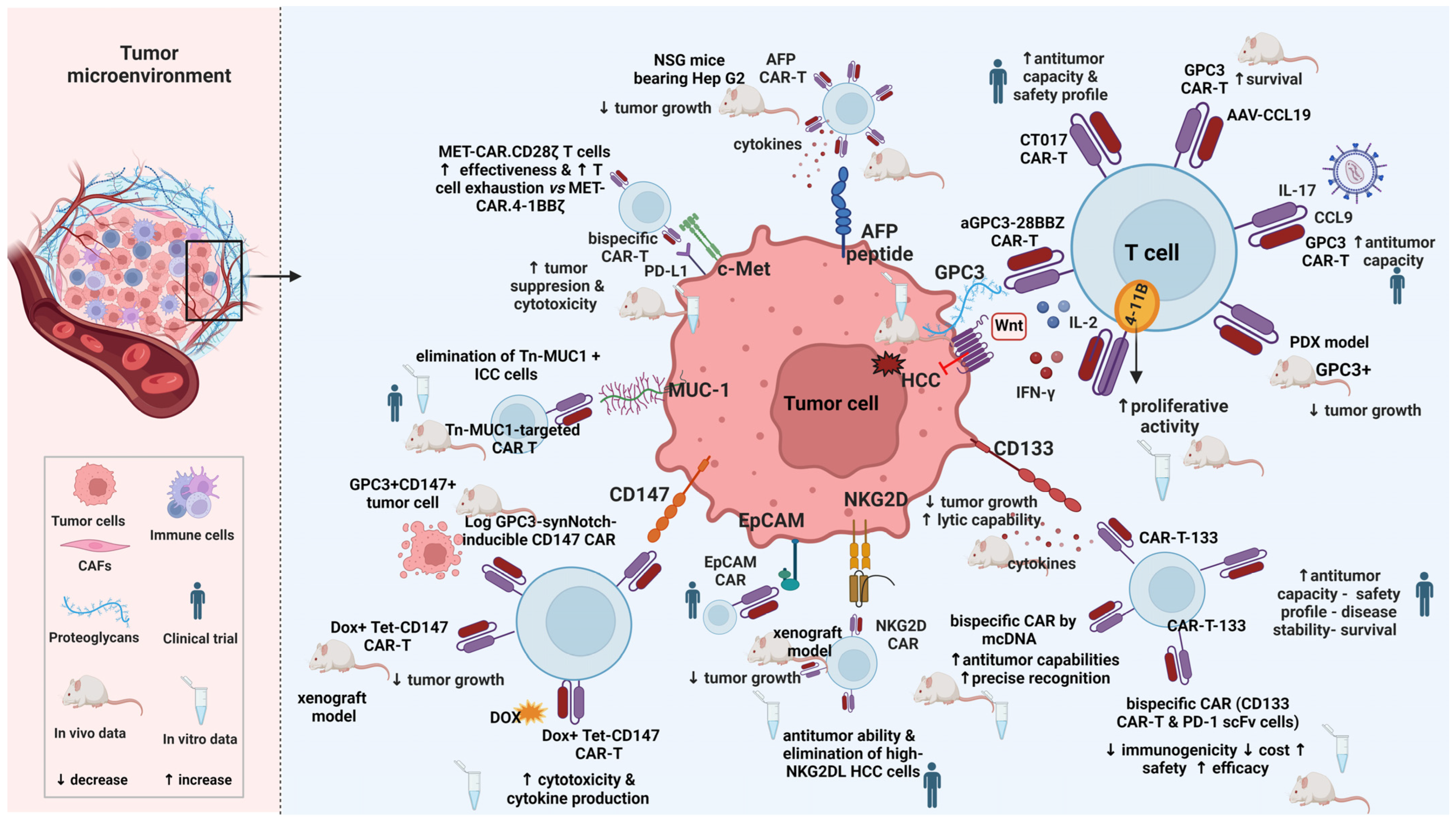
6. CAR T Cell Therapy Targets for HCC
6.1. Glypican-3 (GPC3)
6.2. Alpha-Fetoprotein (AFP)
6.3. CD133
6.4. CD147
6.5. NK Group 2 Member D (NKG2D)
6.6. Epithelial Cell Adhesion Molecule (EpCAM)
6.7. c-Met
6.8. Mucin 1 (MUC1)
6.9. Other Targets
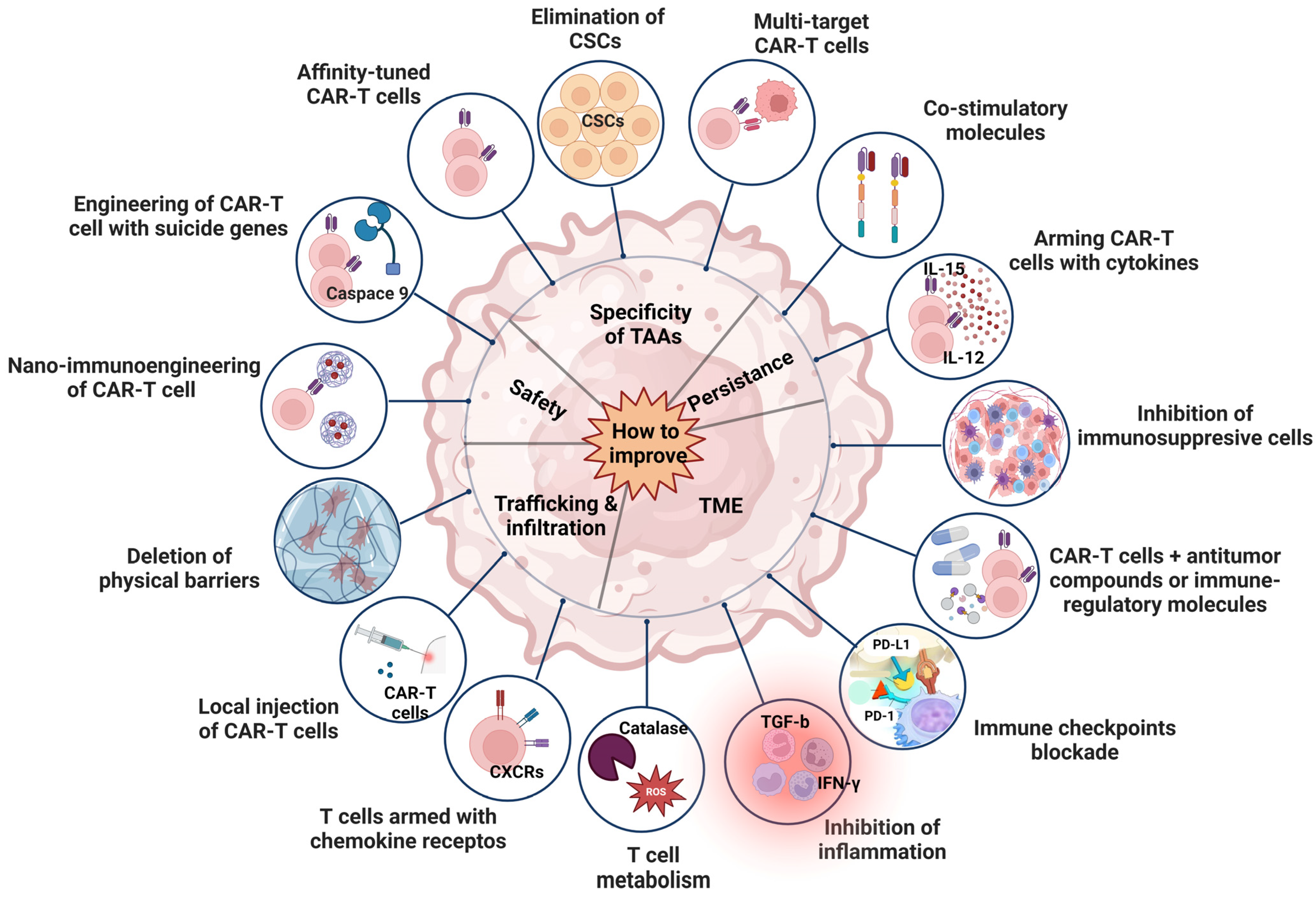
7. Strategies to Improve the Efficacy of CAR T Cell Therapy for HCC
7.1. Improvement of Persistence
7.2. Improvement of TME
7.3. Improvement of Trafficking and Infiltration
7.4. Improvement of Safety
7.5. Improvement of Specificity
7.6. Improvement of Drug Resistance
8. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef]
- Chen, X.; Liu, X.; Du, S. Unveiling the Role of Tumor-Infiltrating T Cells and Immunotherapy in Hepatocellular Carcinoma: A Comprehensive Review. Cancers 2023, 15, 5046. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Splan, M.F.; Weiss, N.S.; McDonald, G.B.; Beretta, L.; Lee, S.P. Incidence and predictors of hepatocellular carcinoma in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 2007, 5, 938–945.e4. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. Clinical states of cirrhosis and competing risks. J. Hepatol. 2018, 68, 563–576. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Correction to: “Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up”. Ann. Oncol. 2019, 30, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-Based Diagnosis, Staging, and Treatment of Patients with Hepatocellular Carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Fessas, P.; Possamai, L.A.; Clark, J.; Daniels, E.; Gudd, C.; Mullish, B.H.; Alexander, J.L.; Pinato, D.J. Immunotoxicity from checkpoint inhibitor therapy: Clinical features and underlying mechanisms. Immunology 2020, 159, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Maher, V.E.; Fernandes, L.L.; Weinstock, C.; Tang, S.; Agarwal, S.; Brave, M.; Ning, Y.M.; Singh, H.; Suzman, D.; Xu, J.; et al. Analysis of the Association between Adverse Events and Outcome in Patients Receiving a Programmed Death Protein 1 or Programmed Death Ligand 1 Antibody. J. Clin. Oncol. 2019, 37, 2730–2737. [Google Scholar] [CrossRef] [PubMed]
- Inno, A.; Metro, G.; Bironzo, P.; Grimaldi, A.M.; Grego, E.; Di Nunno, V.; Picasso, V.; Massari, F.; Gori, S. Pathogenesis, clinical manifestations and management of immune checkpoint inhibitors toxicity. Tumori 2017, 103, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J. Natl. Cancer Inst. 2016, 108, djv439. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, U.; Khan, Z.; Ualiyeva, D.; Amissah, O.B.; Noor, Z.; Khan, A.; Zaman, N.; Khan, M.; Khan, A.; Ali, B. Chimeric antigen receptor T cell structure, its manufacturing, and related toxicities; A comprehensive review. Adv. Cancer Biol.—Metastasis 2022, 4, 100035. [Google Scholar] [CrossRef]
- Ozer, M.; Goksu, S.Y. Adoptive Cell Therapy in Hepatocellular Carcinoma: A Review of Clinical Trials. Cancers 2023, 15, 1808. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Loskog, A.; Giandomenico, V.; Rossig, C.; Pule, M.; Dotti, G.; Brenner, M. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia 2006, 20, 1819–1828. [Google Scholar] [CrossRef]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Oertle, J.; Warren, D.; Prato, D. Chimeric antigen receptor (CAR) T cell therapy for malignant cancers: Summary and perspective. J. Cell. Immunother. 2016, 2, 59–68. [Google Scholar] [CrossRef]
- Acuto, O.; Michel, F. CD28-mediated co-stimulation: A quantitative support for TCR signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert. Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Asmamaw Dejenie, T.; Tiruneh, G.M.M.; Dessie Terefe, G.; Tadele Admasu, F.; Wale Tesega, W.; Chekol Abebe, E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccin. Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef]
- Moreno, C.; Haynie, C. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines 2022, 10, 1493. [Google Scholar] [CrossRef]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J., Jr. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef]
- Savanur, M.A.; Weinstein-Marom, H.; Gross, G. Implementing Logic Gates for Safer Immunotherapy of Cancer. Front. Immunol. 2021, 12, 780399. [Google Scholar] [CrossRef]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef]
- Haubner, S.; Mansilla-Soto, J.; Nataraj, S.; He, X.; Park, J.H.; Wang, X.; Rivière, I.; Sadelain, M. IF-Better Gating: Combinatorial Targeting and Synergistic Signaling for Enhanced CAR T Cell Efficacy. Blood 2021, 138, 2774. [Google Scholar] [CrossRef]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [PubMed]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- Stroncek, D.F.; Ren, J.; Lee, D.W.; Tran, M.; Frodigh, S.E.; Sabatino, M.; Khuu, H.; Merchant, M.S.; Mackall, C.L. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy 2016, 18, 893–901. [Google Scholar] [CrossRef]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.-T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, ra132–ra153. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Scott, C.D.; Leonardi, A.J.; Yamamoto, T.N.; Cruz, A.C.; Ouyang, C.; Ramaswamy, M.; Roychoudhuri, R.; Ji, Y.; Eil, R.L.; et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig. 2016, 126, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi, S.; Nunez-Cruz, S.; O’Connor, R.S.; Fraietta, J.A.; Patel, P.R.; Scholler, J.; Barrett, D.M.; Lundh, S.M.; Davis, M.M.; Bedoya, F.; et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol. Res. 2018, 6, 1100–1109. [Google Scholar] [CrossRef]
- Bai, Y.; Kan, S.; Zhou, S.; Wang, Y.; Xu, J.; Cooke, J.P.; Wen, J.; Deng, H. Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Discov. 2015, 1, 15040. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Hückelhoven, A.; Sellner, L.; Stock, S.; Schmitt, A.; Kleist, C.; Gern, U.; Loskog, A.; et al. Differences in Expansion Potential of Naive Chimeric Antigen Receptor T Cells from Healthy Donors and Untreated Chronic Lymphocytic Leukemia Patients. Front. Immunol. 2017, 8, 1956. [Google Scholar] [CrossRef]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.C.; Goc, J.; Giraldo, N.A.; Sautès-Fridman, C.; Fridman, W.H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014, 35, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kratz, A.; Campos-Neto, A.; Hanson, M.S.; Ruddle, N.H. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J. Exp. Med. 1996, 183, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Pipi, E.; Nayar, S.; Gardner, D.H.; Colafrancesco, S.; Smith, C.; Barone, F. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front. Immunol. 2018, 9, 1952. [Google Scholar] [CrossRef] [PubMed]
- Nasr, I.W.; Reel, M.; Oberbarnscheidt, M.H.; Mounzer, R.H.; Baddoura, F.K.; Ruddle, N.H.; Lakkis, F.G. Tertiary lymphoid tissues generate effector and memory T cells that lead to allograft rejection. Am. J. Transplant. 2007, 7, 1071–1079. [Google Scholar] [CrossRef]
- Cai, D.; Yu, H.; Wang, X.; Mao, Y.; Liang, M.; Lu, X.; Shen, X. Turning Tertiary Lymphoid Structures (TLS) into Hot Spots: Values of TLS in Gastrointestinal Tumors. Cancers 2023, 15, 367. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Lawand, M.; Giraldo, N.A.; Kaplon, H.; Germain, C.; Fridman, W.H.; Dieu-Nosjean, M.C. Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front. Immunol. 2016, 7, 407. [Google Scholar] [CrossRef]
- Khanal, S.; Wieland, A.; Gunderson, A.J. Mechanisms of tertiary lymphoid structure formation: Cooperation between inflammation and antigenicity. Front. Immunol. 2023, 14, 1267654. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Petitprez, F. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Calderaro, J.; Petitprez, F.; Becht, E.; Laurent, A.; Hirsch, T.Z.; Rousseau, B.; Luciani, A.; Amaddeo, G.; Derman, J.; Charpy, C.; et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J. Hepatol. 2019, 70, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Vanhersecke, L.; Brunet, M.; Guégan, J.P.; Rey, C.; Bougouin, A.; Cousin, S.; Moulec, S.L.; Besse, B.; Loriot, Y.; Larroquette, M.; et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2021, 2, 794–802. [Google Scholar] [CrossRef]
- Zhu, G.; Falahat, R.; Wang, K.; Mailloux, A.; Artzi, N.; Mulé, J.J. Tumor-Associated Tertiary Lymphoid Structures: Gene-Expression Profiling and Their Bioengineering. Front. Immunol. 2017, 8, 767. [Google Scholar] [CrossRef]
- Johansson-Percival, A.; Ganss, R. Therapeutic Induction of Tertiary Lymphoid Structures in Cancer Through Stromal Remodeling. Front. Immunol. 2021, 12, 674375. [Google Scholar] [CrossRef] [PubMed]
- Gago da Graça, C.; van Baarsen, L.G.M. Tertiary Lymphoid Structures: Diversity in Their Development, Composition, and Role. J. Immunol. 2021, 206, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Q.; Han, Z.; Zhu, Y.; Shen, H.; Liu, Z.; Zhou, Z.; Ding, W.; Han, S.; He, J.; et al. IL-7 and CCR2b Co-Expression-Mediated Enhanced CAR-T Survival and Infiltration in Solid Tumors. Front. Oncol. 2021, 11, 734593. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Su, J.; Sun, R.; Sun, Y.; Wang, Y.; Dong, Y.; Shi, B.; Jiang, H.; Li, Z. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion. Clin. Cancer Res. 2020, 26, 5494–5505. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Loiacono, F.; Di Carlo, E.; Scaramuccia, A.; Mora, M.; Conte, R.; Benelli, R.; Spaggiari, G.M.; Cantoni, C.; Campana, S.; et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015, 6, 8280. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Hammerschmidt, S.I.; Moschovakis, G.L.; Förster, R. Chemokines and Chemokine Receptors in Lymphoid Tissue Dynamics. Annu. Rev. Immunol. 2016, 34, 203–242. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.M.; Chen, L.; Brzana, E.A.; Patil, P.R.; Taylor, J.L.; Fabian, K.L.; Wallace, C.T.; Jones, S.D.; Watkins, S.C.; Lu, B.; et al. Tbet and IL-36γ cooperate in therapeutic DC-mediated promotion of ectopic lymphoid organogenesis in the tumor microenvironment. Oncoimmunology 2017, 6, e1322238. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chin, R.K.; Christiansen, P.; Sun, Y.; Tumanov, A.V.; Wang, J.; Chervonsky, A.V.; Fu, Y.X. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity 2006, 25, 499–509. [Google Scholar] [CrossRef]
- Aoyama, S.; Nakagawa, R.; Mulé, J.J.; Mailloux, A.W. Inducible Tertiary Lymphoid Structures: Promise and Challenges for Translating a New Class of Immunotherapy. Front. Immunol. 2021, 12, 675538. [Google Scholar] [CrossRef]
- Or-Geva, N.; Reisner, Y. The evolution of T-cell depletion in haploidentical stem-cell transplantation. Br. J. Haematol. 2016, 172, 667–684. [Google Scholar] [CrossRef]
- Booth, C.; Veys, P. T cell depletion in paediatric stem cell transplantation. Clin. Exp. Immunol. 2013, 172, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Snook, A.E.; Magee, M.S.; Schulz, S.; Waldman, S.A. Selective antigen-specific CD4(+) T-cell, but not CD8(+) T- or B-cell, tolerance corrupts cancer immunotherapy. Eur. J. Immunol. 2014, 44, 1956–1966. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.J.; Gottschalk, S.; Talleur, A.C. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front. Immunol. 2020, 11, 618427. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh Kozani, P.; Safarzadeh Kozani, P.; Rahbarizadeh, F.; Khoshtinat Nikkhoi, S. Strategies for Dodging the Obstacles in CAR T Cell Therapy. Front. Oncol. 2021, 11, 627549. [Google Scholar] [CrossRef] [PubMed]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Thomas, X.; Paubelle, E. Tisagenlecleucel-T for the treatment of acute lymphocytic leukemia. Expert Opin. Biol. Ther. 2018, 18, 1095–1106. [Google Scholar] [CrossRef]
- Bouchkouj, N.; Kasamon, Y.L.; de Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef]
- Zhou, F.; Shang, W.; Yu, X. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef]
- Capurro, M.; Wanless, I.R.; Sherman, M.; Deboer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Nakatsura, T.; Yoshitake, Y.; Senju, S.; Monji, M.; Komori, H.; Motomura, Y.; Hosaka, S.; Beppu, T.; Ishiko, T.; Kamohara, H.; et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem. Biophys. Res. Commun. 2003, 306, 16–25. [Google Scholar] [CrossRef]
- Gao, W.; Ho, M. The role of glypican-3 in regulating Wnt in hepatocellular carcinomas. Cancer Rep. 2011, 1, 14–19. [Google Scholar]
- Montalbano, M.; Rastellini, C.; McGuire, J.T.; Prajapati, J.; Shirafkan, A.; Vento, R.; Cicalese, L. Role of Glypican-3 in the growth, migration and invasion of primary hepatocytes isolated from patients with hepatocellular carcinoma. Cell Oncol. 2018, 41, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Haruyama, Y.; Yorita, K.; Yamaguchi, T.; Kitajima, S.; Amano, J.; Ohtomo, T.; Ohno, A.; Kondo, K.; Kataoka, H. High preoperative levels of serum glypican-3 containing N-terminal subunit are associated with poor prognosis in patients with hepatocellular carcinoma after partial hepatectomy. Int. J. Cancer 2015, 137, 1643–1651. [Google Scholar] [CrossRef]
- Sun, L.; Gao, F.; Gao, Z.; Ao, L.; Li, N.; Ma, S.; Jia, M.; Li, N.; Lu, P.; Sun, B.; et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J. Immunother. Cancer 2021, 9, e001875. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef]
- Li, W.; Guo, L.; Rathi, P.; Marinova, E.; Gao, X.; Wu, M.F.; Liu, H.; Dotti, G.; Gottschalk, S.; Metelitsa, L.S.; et al. Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum. Gene Ther. 2017, 28, 437–448. [Google Scholar] [CrossRef]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef]
- Meng, M.; Wu, Y.C. Combination of AAV-CCL19 and GPC3 CAR-T Cells in the Treatment of Hepatocellular Carcinoma. J. Immunol. Res. 2021, 2021, 1782728. [Google Scholar] [CrossRef]
- Fu, Q.; Zheng, Y.; Fang, W.; Zhao, Q.; Zhao, P.; Liu, L.; Zhai, Y.; Tong, Z.; Zhang, H.; Lin, M.; et al. RUNX-3-expressing CAR T cells targeting glypican-3 in patients with heavily pretreated advanced hepatocellular carcinoma: A phase I trial. EClinicalMedicine 2023, 63, 102175. [Google Scholar] [CrossRef]
- Li, D.; Qin, J.; Zhou, T.; Li, Y.; Cheng, X.; Chen, Z.; Chen, J.; Zheng, W.V. Bispecific GPC3/PD-1 CAR-T cells for the treatment of HCC. Int. J. Oncol. 2023, 62, 53. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef]
- Gillespie, J.R.; Uversky, V.N. Structure and function of alpha-fetoprotein: A biophysical overview. Biochim. Biophys. Acta 2000, 1480, 41–56. [Google Scholar] [CrossRef]
- Xue, J.; Cao, Z.; Cheng, Y.; Wang, J.; Liu, Y.; Yang, R.; Li, H.; Jiang, W.; Li, G.; Zhao, W.; et al. Acetylation of alpha-fetoprotein promotes hepatocellular carcinoma progression. Cancer Lett. 2020, 471, 12–26. [Google Scholar] [CrossRef]
- Li, W.; Liu, K.; Chen, Y.; Zhu, M.; Li, M. Role of Alpha-Fetoprotein in Hepatocellular Carcinoma Drug Resistance. Curr. Med. Chem. 2021, 28, 1126–1142. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, Y.; Xiang, J.; Long, L.; Green, S.; Yang, Z.; Zimdahl, B.; Lu, J.; Cheng, N.; Horan, L.H.; et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin. Cancer Res. 2017, 23, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Vallera, D.A. CD133, Selectively Targeting the Root of Cancer. Toxins 2016, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- Kohga, K.; Tatsumi, T.; Takehara, T.; Tsunematsu, H.; Shimizu, S.; Yamamoto, M.; Sasakawa, A.; Miyagi, T.; Hayashi, N. Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. J. Hepatol. 2010, 52, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, H.; Tao, K.; Li, R.; Song, Z.; Zhao, Q.; Zhang, F.; Dou, K. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int. J. Clin. Pract. 2008, 62, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- Yang, C.; You, J.; Pan, Q.; Tang, Y.; Cai, L.; Huang, Y.; Gu, J.; Wang, Y.; Yang, X.; Du, Y.; et al. Targeted delivery of a PD-1-blocking scFv by CD133-specific CAR-T cells using nonviral Sleeping Beauty transposition shows enhanced antitumour efficacy for advanced hepatocellular carcinoma. BMC Med. 2023, 21, 327. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Ye, X.; Ju, Y.; Cao, N.; Wang, S.; Cai, J. Nonviral mcDNA-mediated bispecific CAR T cells kill tumor cells in an experimental mouse model of hepatocellular carcinoma. BMC Cancer 2022, 22, 814. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Rao, D.; Jin, Q.; Lai, M.; Zhang, J.; Lai, Z.; Shen, H.; Zhong, T. Role of CD147 in the development and diagnosis of hepatocellular carcinoma. Front. Immunol. 2023, 14, 1149931. [Google Scholar] [CrossRef]
- Sun, J.; Hemler, M.E. Regulation of MMP-1 and MMP-2 production through CD147/extracellular matrix metalloproteinase inducer interactions. Cancer Res. 2001, 61, 2276–2281. [Google Scholar] [PubMed]
- Zhu, S.; Li, Y.; Zhang, Y.; Wang, X.; Gong, L.; Han, X.; Yao, L.; Lan, M.; Zhang, W. Expression and clinical implications of HAb18G/CD147 in hepatocellular carcinoma. Hepatol. Res. 2015, 45, 97–106. [Google Scholar] [CrossRef]
- Fan, W.; Wu, Y.; Lu, M.; Yao, W.; Cui, W.; Zhao, Y.; Wang, Y.; Li, J. A meta-analysis of the efficacy and safety of iodine [(131)I] metuximab infusion combined with TACE for treatment of hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.; Wei, D.; Liu, Z.K.; Yong, Y.L.; Wei, W.; Zhang, Z.Y.; Lv, J.J.; Zhang, Z.; Chen, Z.N.; Bian, H. Doxycycline Inducible Chimeric Antigen Receptor T Cells Targeting CD147 for Hepatocellular Carcinoma Therapy. Front. Cell Dev. Biol. 2019, 7, 233. [Google Scholar] [CrossRef]
- Tseng, H.-c.; Xiong, W.; Badeti, S.; Yang, Y.; Ma, M.; Liu, T.; Ramos, C.A.; Dotti, G.; Fritzky, L.; Jiang, J.-g.; et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat. Commun. 2020, 11, 4810. [Google Scholar] [CrossRef]
- Zhang, J.; Basher, F.; Wu, J.D. NKG2D Ligands in Tumor Immunity: Two Sides of a Coin. Front. Immunol. 2015, 6, 97. [Google Scholar] [CrossRef]
- Sun, B.; Yang, D.; Dai, H.-j.; Liu, X.; Jia, R.; Cui, X.; Li, W.; Cai, C.; Xu, J.; Zhao, X. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol. Res. 2019, 7, 1813–1823. [Google Scholar] [CrossRef]
- Tay, J.C.K.; Wang, J.; Du, Z.; Ng, Y.Y.; Li, Z.; Ren, Y.; Zhang, C.; Zhu, J.; Xu, X.H.; Wang, S. Manufacturing NKG2D CAR-T cells with piggyBac transposon vectors and K562 artificial antigen-presenting cells. Mol. Ther. Methods Clin. Dev. 2021, 21, 107–120. [Google Scholar] [CrossRef]
- Curio, S.; Jonsson, G.; Marinović, S.; Young European Federation of Immunological Societies. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 2021, 1, ltab018. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, Y. The EpCAM overexpression is associated with clinicopathological significance and prognosis in hepatocellular carcinoma patients: A systematic review and meta-analysis. Int. J. Surg. 2018, 56, 274–280. [Google Scholar] [CrossRef]
- Terris, B.; Cavard, C.; Perret, C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J. Hepatol. 2010, 52, 280–281. [Google Scholar] [CrossRef]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.X.; et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.L.; Li, D.; Gong, Y.L.; Huang, Y.; Qin, D.Y.; Jiang, L.; Liang, X.; Yang, X.; Gou, H.F.; Wang, Y.S.; et al. Preclinical Evaluation of Chimeric Antigen Receptor-Modified T Cells Specific to Epithelial Cell Adhesion Molecule for Treating Colorectal Cancer. Hum. Gene Ther. 2019, 30, 402–412. [Google Scholar] [CrossRef]
- Migliore, C.; Giordano, S. Molecular cancer therapy: Can our expectation be MET? Eur. J. Cancer 2008, 44, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.G.; Factor, V.M.; Sánchez, A.; Uchida, K.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. USA 2004, 101, 4477–4482. [Google Scholar] [CrossRef]
- Qi, X.S.; Guo, X.Z.; Han, G.H.; Li, H.Y.; Chen, J. MET inhibitors for treatment of advanced hepatocellular carcinoma: A review. World J. Gastroenterol. 2015, 21, 5445–5453. [Google Scholar] [CrossRef]
- Woo, H.Y.; Heo, J. The role of c-MET inhibitors in advanced hepatocellular carcinoma: Now and future. Ann. Transl. Med. 2020, 8, 1617. [Google Scholar] [CrossRef]
- Jiang, W.; Li, T.; Guo, J.; Wang, J.; Jia, L.; Shi, X.; Yang, T.; Jiao, R.; Wei, X.; Feng, Z.; et al. Bispecific c-Met/PD-L1 CAR-T Cells Have Enhanced Therapeutic Effects on Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 546586. [Google Scholar] [CrossRef]
- Huang, X.; Guo, J.; Li, T.; Jia, L.; Tang, X.; Zhu, J.; Tang, Q.; Feng, Z. c-Met-targeted chimeric antigen receptor T cells inhibit hepatocellular carcinoma cells in vitro and in vivo. J. Biomed. Res. 2021, 36, 10–21. [Google Scholar] [CrossRef]
- Qin, A.; Qin, Y.; Lee, J.; Musket, A.; Ying, M.; Krenciute, G.; Marincola, F.M.; Yao, Z.Q.; Musich, P.R.; Xie, Q. Tyrosine kinase signaling-independent MET-targeting with CAR-T cells. J. Transl. Med. 2023, 21, 682. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, G.; Li, Q.; Wang, F.; Xie, F.; Zhai, R.; Guo, Y.; Chen, T.; Zhang, N.; Ni, W.; et al. Mucin1 promotes the migration and invasion of hepatocellular carcinoma cells via JNK-mediated phosphorylation of Smad2 at the C-terminal and linker regions. Oncotarget 2015, 6, 19264–19278. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, Z.; Gong, R.; Li, L.; Wu, H.; Jin, H.; Qian, Q. Specific cytotoxicity of MUC1 chimeric antigen receptor-engineered Jurkat T cells against hepatocellular carcinoma. Acad. J. Second. Mil. Med. Univ. 2014, 35, 1177–1182. [Google Scholar] [CrossRef]
- Mao, L.; Su, S.; Li, J.; Yu, S.; Gong, Y.; Chen, C.; Hu, Z.; Huang, X. Development of Engineered CAR T Cells Targeting Tumor-Associated Glycoforms of MUC1 for the Treatment of Intrahepatic Cholangiocarcinoma. J. Immunother. 2023, 46, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Nakamura, K.; Hijioka, S.; Kamei, A.; Ikari, T.; Ishikawa, Y.; Shinozaki, E.; Mizunuma, N.; Hatake, K.; Miyajima, A. Dlk-1, a cell surface antigen on foetal hepatic stem/progenitor cells, is expressed in hepatocellular, colon, pancreas and breast carcinomas at a high frequency. J. Biochem. 2010, 148, 85–92. [Google Scholar] [CrossRef]
- Zhai, Y.; He, K. DLK1-directed chimeric antigen receptor T-cell therapy for hepatocellular carcinoma. Liver Int. 2022, 42, 2524–2537. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Morine, Y.; Ikemoto, T.; Imura, S.; Higashijima, J.; Iwahashi, S.; Saito, Y.U.; Takasu, C.; Yamada, S.; Ishikawa, D.; et al. Elevated Preoperative Serum CEA Level Is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma Through the Epithelial-Mesenchymal Transition. Anticancer Res. 2017, 37, 1169–1175. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]
- Katz, S.C.; Hardaway, J.; Prince, E.; Guha, P.; Cunetta, M.; Moody, A.; Wang, L.J.; Armenio, V.; Espat, N.J.; Junghans, R.P. HITM-SIR: Phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA(+) liver metastases. Cancer Gene Ther. 2020, 27, 341–355. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Huang, X.; Huang, X.; Chen, W.; Hao, L.; Chen, Z. Chimeric antigen receptor modified T cell (CAR-T) co-expressed with ICOSL-41BB promote CAR-T proliferation and tumor rejection. Biomed. Pharmacother. 2019, 118, 109333. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Di, S.; Shi, B.; Jiang, H.; Shi, Z.; Liu, Y.; Wang, Y.; Luo, H.; Yu, M.; Wu, X.; et al. Increased antitumor activities of glypican-3-specific chimeric antigen receptor-modified T cells by coexpression of a soluble PD1-CH3 fusion protein. Cancer Immunol. Immunother. 2018, 67, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.-q. Reconsidering CAR-T cell therapy for solid tumors. J. Med. Postgrad. 2019, 12, 337–340. [Google Scholar]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Joseph Espat, N.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef]
- Tang, X.J.; Sun, X.Y.; Huang, K.M.; Zhang, L.; Yang, Z.S.; Zou, D.D.; Wang, B.; Warnock, G.L.; Dai, L.J.; Luo, J. Therapeutic potential of CAR-T cell-derived exosomes: A cell-free modality for targeted cancer therapy. Oncotarget 2015, 6, 44179–44190. [Google Scholar] [CrossRef]
- Ma, W.; Zhu, D.; Li, J.; Chen, X.; Xie, W.; Jiang, X.; Wu, L.; Wang, G.; Xiao, Y.; Liu, Z.; et al. Coating biomimetic nanoparticles with chimeric antigen receptor T cell-membrane provides high specificity for hepatocellular carcinoma photothermal therapy treatment. Theranostics 2020, 10, 1281–1295. [Google Scholar] [CrossRef]
- Siriwon, N.; Kim, Y.J.; Siegler, E.; Chen, X.; Rohrs, J.A.; Liu, Y.; Wang, P. CAR-T Cells Surface-Engineered with Drug-Encapsulated Nanoparticles Can Ameliorate Intratumoral T-cell Hypofunction. Cancer Immunol. Res. 2018, 6, 812–824. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, Q.; Dukhovlinova, E.; Chen, G.; Ahn, S.; Wang, C.; Ogunnaike, E.A.; Ligler, F.S.; Dotti, G.; Gu, Z. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv. Mater. 2019, 31, e1900192. [Google Scholar] [CrossRef]
- Elazar, A.; Chandler, N.J.; Davey, A.S.; Weinstein, J.Y.; Nguyen, J.V.; Trenker, R. De novo-designed transmembrane domains tune engineered receptor functions. eLife 2022, 11, e75660. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiang, W.; Feng, Z.-Q. The research advances of CAR-T cell therapy in solid tumor. J. Med. Postgrad. 2019, 12, 886–890. [Google Scholar]
- Zhao, W.; Jia, L.; Zhang, M.; Huang, X.; Qian, P.; Tang, Q.; Zhu, J.; Feng, Z. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am. J. Cancer Res. 2019, 9, 1846–1856. [Google Scholar] [PubMed]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Mishra, A.; Maiti, R. Antigen loss following CAR-T cell therapy: Mechanisms, implications, and potential solutions. Eur. J. Haematol. 2024, 112, 211–222. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggeletopoulou, I.; Kalafateli, M.; Triantos, C. Chimeric Antigen Receptor T Cell Therapy for Hepatocellular Carcinoma: Where Do We Stand? Int. J. Mol. Sci. 2024, 25, 2631. https://doi.org/10.3390/ijms25052631
Aggeletopoulou I, Kalafateli M, Triantos C. Chimeric Antigen Receptor T Cell Therapy for Hepatocellular Carcinoma: Where Do We Stand? International Journal of Molecular Sciences. 2024; 25(5):2631. https://doi.org/10.3390/ijms25052631
Chicago/Turabian StyleAggeletopoulou, Ioanna, Maria Kalafateli, and Christos Triantos. 2024. "Chimeric Antigen Receptor T Cell Therapy for Hepatocellular Carcinoma: Where Do We Stand?" International Journal of Molecular Sciences 25, no. 5: 2631. https://doi.org/10.3390/ijms25052631
APA StyleAggeletopoulou, I., Kalafateli, M., & Triantos, C. (2024). Chimeric Antigen Receptor T Cell Therapy for Hepatocellular Carcinoma: Where Do We Stand? International Journal of Molecular Sciences, 25(5), 2631. https://doi.org/10.3390/ijms25052631