1. Introduction
Obesity has emerged as a public health threat, with drastic increases worldwide. According to the World Health Organization (WHO), about 13% of the global population are obese (body mass index [BMI] ≥ 30 kg/m
2) and the prevalence of obesity is projected to increase to 20% by 2025 [
1]. Obesity and overweight contribute to the rise of various metabolic complications such as inflammation [
2], lipotoxicity [
3], and dyslipidemia [
4]. Also, the adverse effects of obesity and overweight contribute to the rise of non-communicable diseases such as diabetes [
5], cardiovascular diseases (CVDs) [
6], and different types of cancers [
7]. Therefore, there is an urgent need for innovative therapeutic strategies to curb the prevalence of obesity and its underlying metabolic complications.
In recent years, natural products have gained considerable attention due to their potential health benefits [
8,
9]. Curcumin, a bioactive compound derived from the rhizome of the turmeric plant, otherwise called
Curcuma longa, which belongs to the Zingiberaceae family, has shown promising anti-obesity properties through its ability to modulate various cellular pathways involved in lipid metabolism [
10,
11], adipogenesis [
12,
13,
14], and inflammation [
15,
16]. While curcumin holds significant potential as an anti-obesity agent, its poor bioavailability and limited systemic exposure have hindered its clinical applications [
17]. To address these challenges, studies have focused on the development of synthetic curcumin derivatives that are designed to improve the pharmacokinetic properties and enhance efficacy of curcumin. It is important to note that the cost of developing synthetic derivatives of curcumin depends on the complexity of the structure. In addition, the extraction of natural compounds is associated with a low yield and purity. As such, we previously reported that curcumin derivatives have the potential to ameliorate obesity and obesity-related metabolic complications even more effectively than curcumin [
18].
The use of computational techniques in drug discovery and development has shown to be a useful tool for predicting the pharmacological properties of synthetic compounds, thus providing a cost-effective and time-efficient method of assessing their therapeutic potential [
19]. Hence, in this study, we used computational techniques to explore the anti-obesity potential of curcumin and three previously synthesized curcumin derivatives. In a study by Ooko at al., (2016) [
20], two chloro curcumin derivatives, specifically (1E,6E)-4-chloro-1,7-bis(3,4-dimethoxyphenyl)hepta-1,6-diene-3,5-dione (1A6) and (1E,6E)-4-chloro-1,7-bis(3-hydroxyphenyl)hepta-1,6-diene-3,5-dione (1A8), along with an asymmetric curcumin derivative, (1E,6E,8E)-1-(3,4-dimethoxyphenyl)-9-phenyldeca-1,6,8-triene-3,5-dione (1B8), were synthesized using a chemical reaction involving acetyl acetone or 3-chloroacetylacetone with boric oxide. Subsequently, the synthetic compounds underwent characterization through various spectroscopic techniques, including mass spectrometry (MS), infrared spectroscopy (FTIR), and proton (1H)-nuclear magnetic resonance (NMR) spectroscopy [
20]. Moreover, these compounds demonstrated potential anti-cancer properties [
20].
These findings lay the foundation to further evaluate the bioactivity of these synthetic curcumin derivatives, particularly for their anti-obesity potential. In this context, the present study aims to assess the pharmacokinetics and pharmacodynamics profiles of curcumin and synthetic curcumin derivatives (1A6, 1A8, and 1B8) using in silico approaches to identify and evaluate targeted obesity mechanisms. By leveraging computational models, we will evaluate their physicochemical properties, druglikeness, toxicity, and ADME properties, including absorption through the gastrointestinal tract, distribution to target tissues, and metabolism by hepatic enzymes. Furthermore, we explored the interactions between these compounds and their molecular targets involved in adipogenesis, lipid metabolism, and inflammation to predict their potential efficacy in combating obesity.
3. Discussion
Natural compounds continue to play a crucial role in drug discovery due to their vast chemical diversity and potential therapeutic value. Additionally, natural products such as plant extracts have been a significant source of bioactive compounds throughout history [
24,
25]. Many drugs currently on the market have their roots in natural products, including antibiotics, anticancer agents, and immunosuppressants. Their biological activity makes them valuable starting points for the development of new drugs [
26,
27,
28,
29]. Curcumin, a bioactive compound derived from the rhizome of
Curcuma longa, has reportedly shown great potential as an anti-obesity therapeutic [
30,
31,
32] due to its ability to modulate various cellular pathways involved in obesity including adipogenesis [
12,
13,
14], lipid metabolism [
10,
11], and inflammation [
15,
16]. In this study, the pharmacological profiles of curcumin and its synthetic derivatives (1A6, 1A8, and 1B8) were explored to unveil insights into their potential anti-obesity activity using molecular recognition techniques.
Firstly, the characterization of curcumin and the synthetic curcumin derivatives was carried out to evaluate the pharmacokinetic, physicochemical, and toxicity properties using online predictive databases. We centered our pharmacokinetic analysis on the LRo5, which serves as a widely recognized standard for evaluating the druglikeness of chemical compounds [
33]. This framework, established by Owens and Lipinski in 2003 [
34] and further discussed by Pollastri et al. (2010) [
35], is fundamental in characterizing potential drug candidates and has become a cornerstone in the field of drug discovery [
34,
35]. Our findings revealed that curcumin and its synthetic derivatives align with the LRo5 criteria, indicating their potential as drug-like substances without any violations. Lipophilicity, as an important physicochemical property of drugs and bioactive compounds, measures the affinity of a compound for lipid environments, which can affect its ability to permeate biological membranes and reach target sites. A higher lipophilicity value suggests a greater affinity for lipid-rich environments [
36]. Curcumin had a lipophilicity value of 3.2, which clearly indicates that curcumin falls within the moderately lipophilic range [
37,
38]. Interestingly, this reporting is in line with the experimental data previously reported by Clariano et al. (2023) [
39] and Shereen et al. (2019) [
40], who evaluated the mechanistic delivery of curcumin in humans and estimated the lipophilicity of curcumin to be in a similar range (~LogP 3.6). The synthetic derivatives 1A6 and 1B8 demonstrated a higher lipophilicity than curcumin, potentially indicating improved membrane permeability which is advantageous for drug development to enhance bioavailability. In contrast, 1A8, notably with the lowest lipophilicity, might face challenges crossing membrane barriers effectively.
Moreover, these compounds are synthetic in nature and exhibit a favorable toxicity profile. It is worth noting that while curcumin along with 1A6 and 1B8 displayed activity towards toxicity endpoints such as hepatotoxicity and immunotoxicity at lethal dosages, their overall toxicity properties were promising. This suggests that further refinement of these compounds may mitigate these specific endpoint effects. In fact, the approach of compound refinement, aimed at reducing toxicity while enhancing effectiveness, is common in several studies [
24,
41,
42]. The synthetic derivative 1A8 was predicted to be the safest since it did not exhibit any toxicity towards the endpoints evaluated. The observed variation in the toxicity and pharmacokinetics outcomes among the compounds could be attributed to structural modifications such as the methoxy, chloro, and phenolic functional groups on the curcumin scaffold, which explains the importance of understanding how a compound’s structure influences its biological effects [
43].
Examining the metabolism and elimination patterns in the liver is a customary practice in the field of drug discovery and development. This method is crucial for anticipating the amount of a drug that effectively reaches its biological targets following oral administration, commonly referred to as the liver first-pass effect [
44]. In our study, we evaluated the potential of curcumin and its synthetic derivatives to be substrates of the enzymes involved in the liver first-pass effect. The findings revealed the activity of these compounds to be common against the CYP3A4 and CYP2C9 enzymes. Of note, these enzymes are part of the cytochrome P450 family and play a crucial role in the biotransformation of various substances, including drugs in the liver [
45]. Consequently, our findings suggest that curcumin and its synthetic derivatives may undergo metabolism in the liver, potentially influencing their pharmacokinetics and interaction with other drugs. These findings, especially for curcumin, greatly correlate with those previously articulated in the literature [
46] and therefore open avenues for further structural modifications to steer these compounds away from becoming substrates for these biotransformation enzymes. For instance, the modification of curcumin structure with hydroxylation, methoxylation, cyclization, fluorination, or alkylation can be implemented to enhance its properties for improved stability, solubility, and biological activity.
In general, the enhanced permeability of orally administered drugs in the intestines tends to correlate with improved pharmacokinetic properties [
47]. Our study illustrated that curcumin and its synthetic derivatives exhibit high absorption in the intestines, demonstrating their potential to pass into the circulatory system. The literature also indicates that curcumin metabolites such as dihydrocurcumin and tetrahydrocurcumin are traced in the intestines [
48]. However, our in silico prediction for the intestinal absorption of curcumin does not correlate with previously reported in vitro and in vivo findings [
49]. This may be attributed to the intestinal efflux of the P-gp enzyme on curcumin [
49]. The P-gp is a vital membrane protein in the liver, intestines, and other tissues that expels substances from cells [
50]. Our study predicted that curcumin and its synthetic derivatives are not substrates for the P-gp enzyme, indicating that this enzyme does not play a role in expelling these compounds from cells. Unfortunately, these predictive findings, mainly on curcumin, are opposed by experimental data in the literature [
49]. In fact, a recent in silico study suggested that synthetic curcumin derivatives interact with the P-gp enzyme as antagonist [
51].
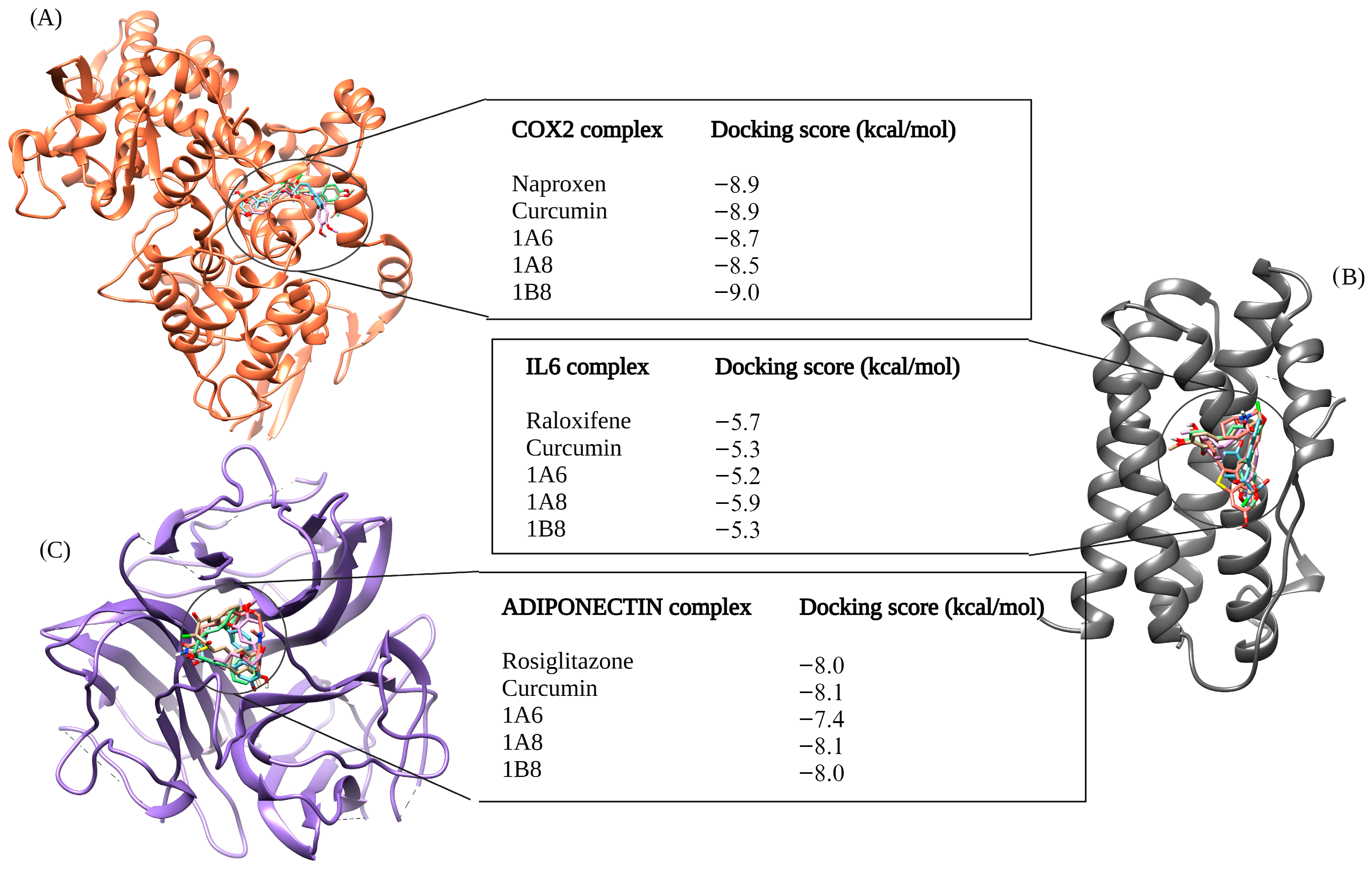
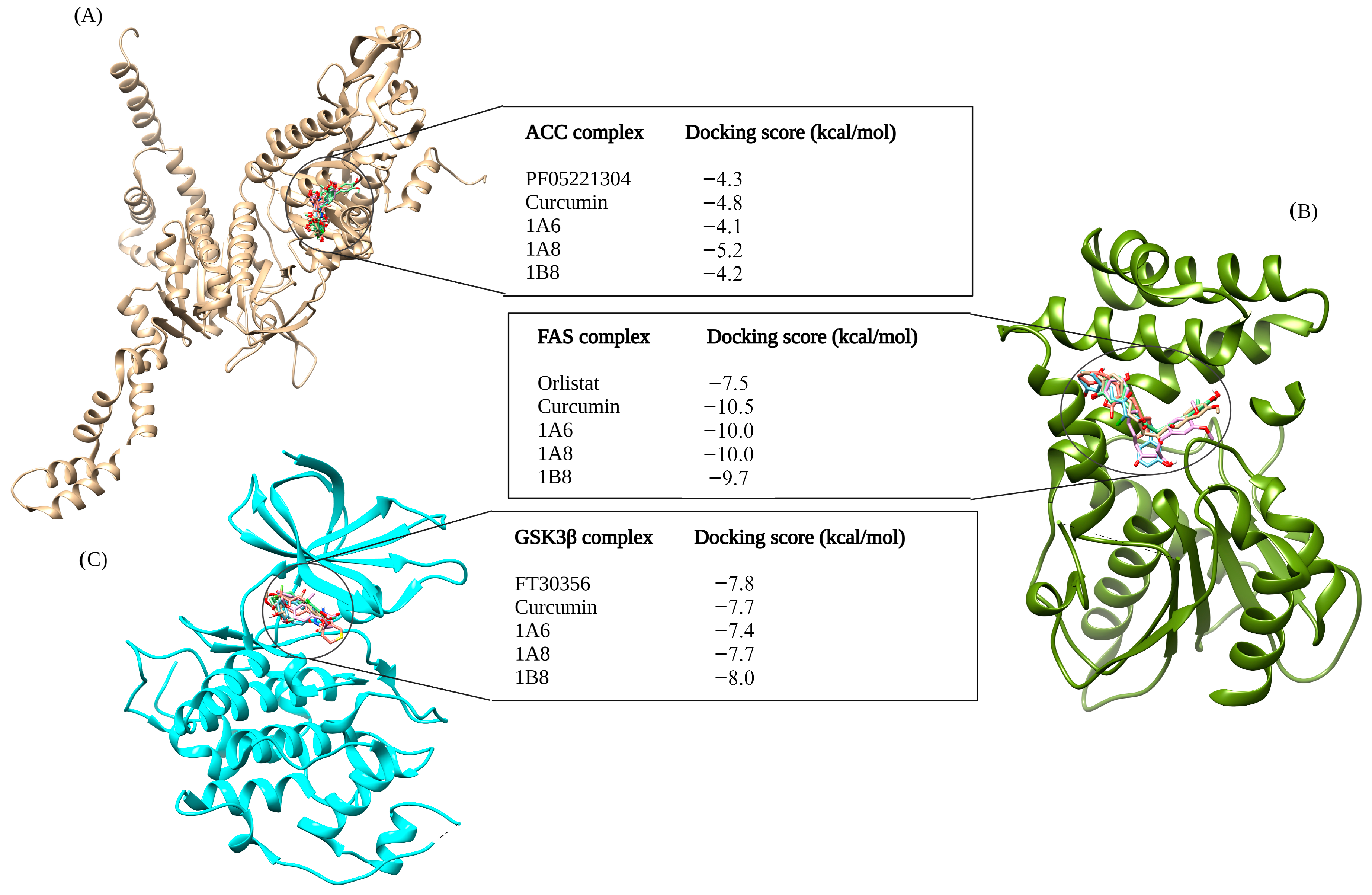
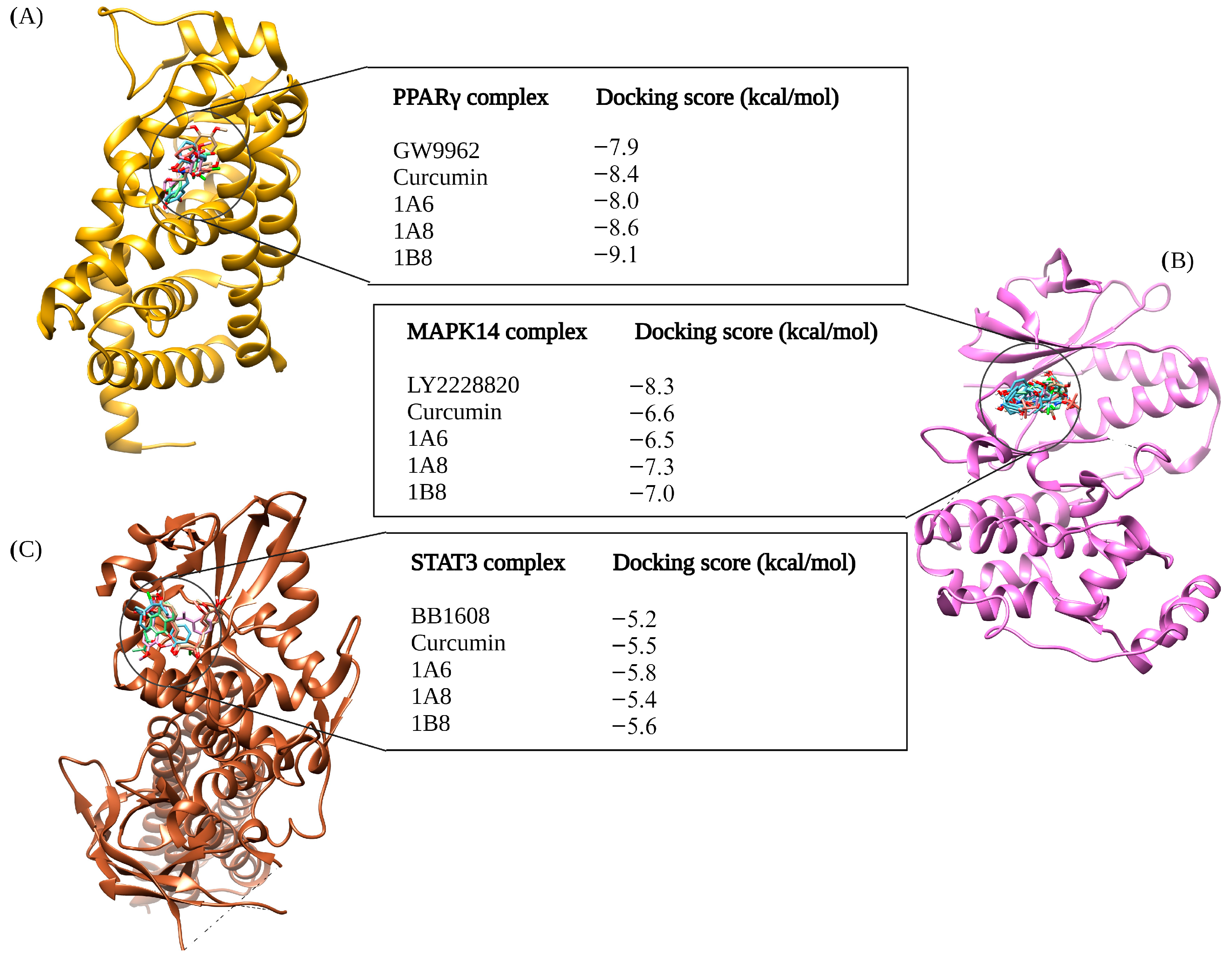
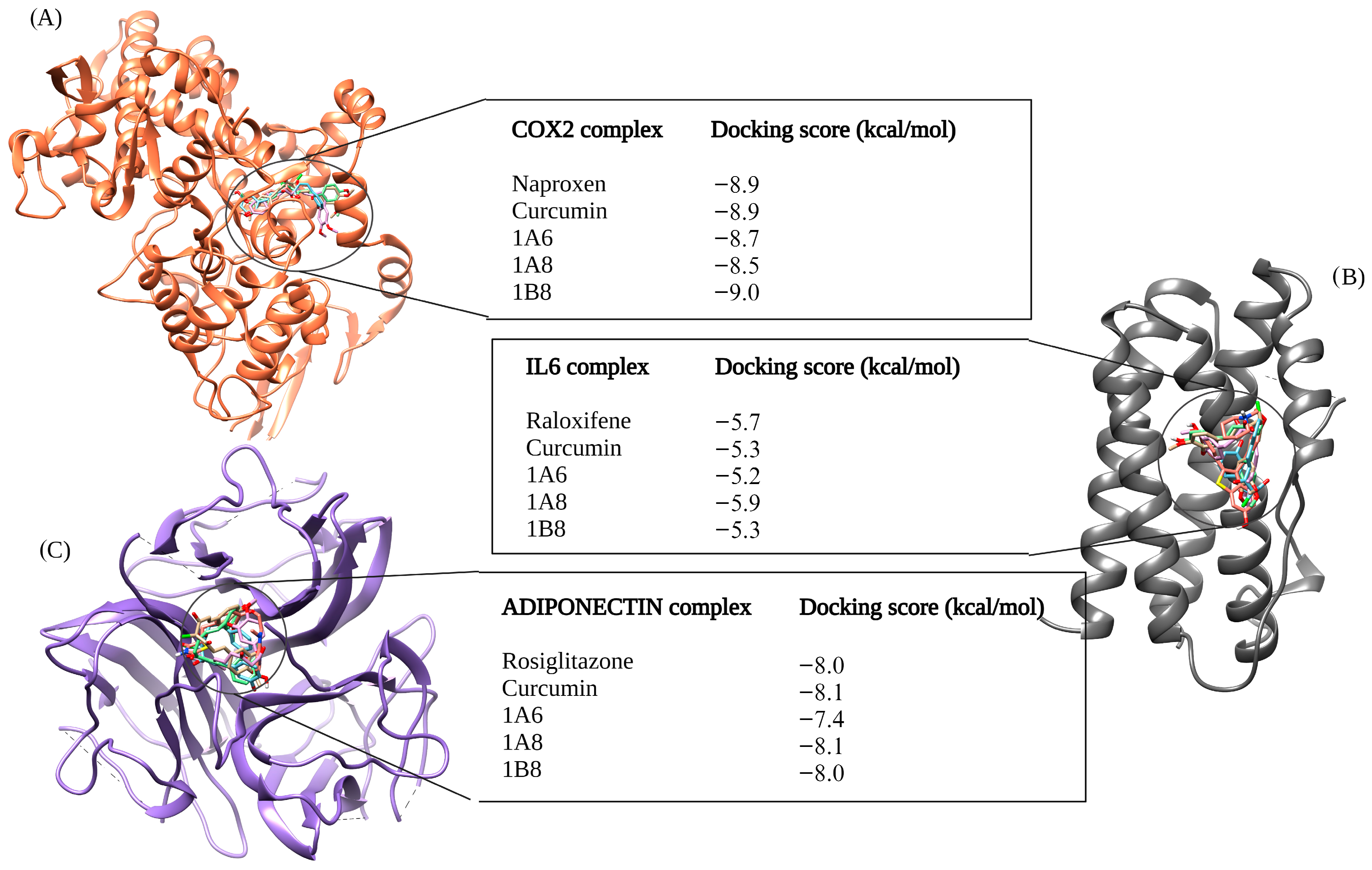
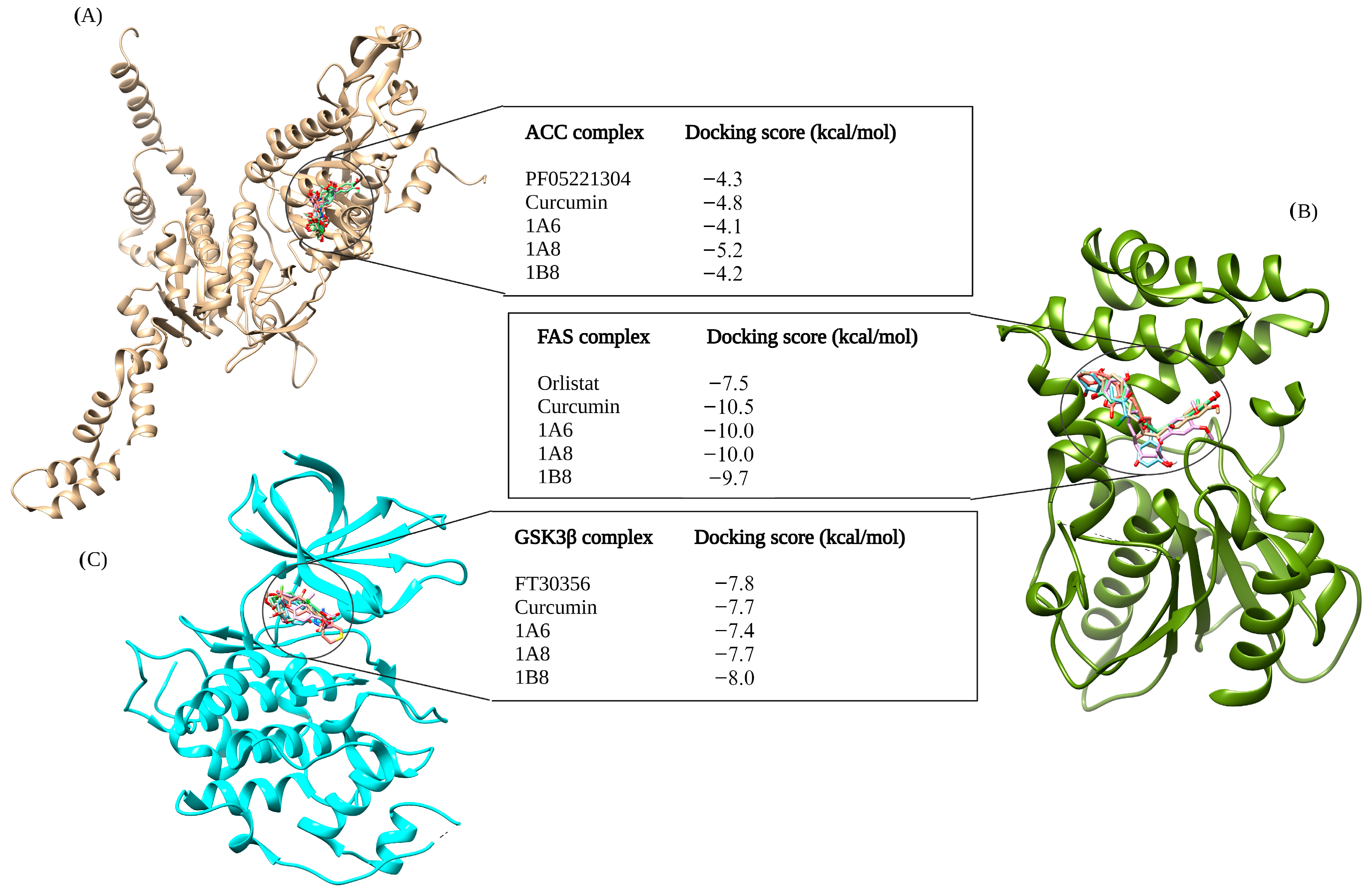
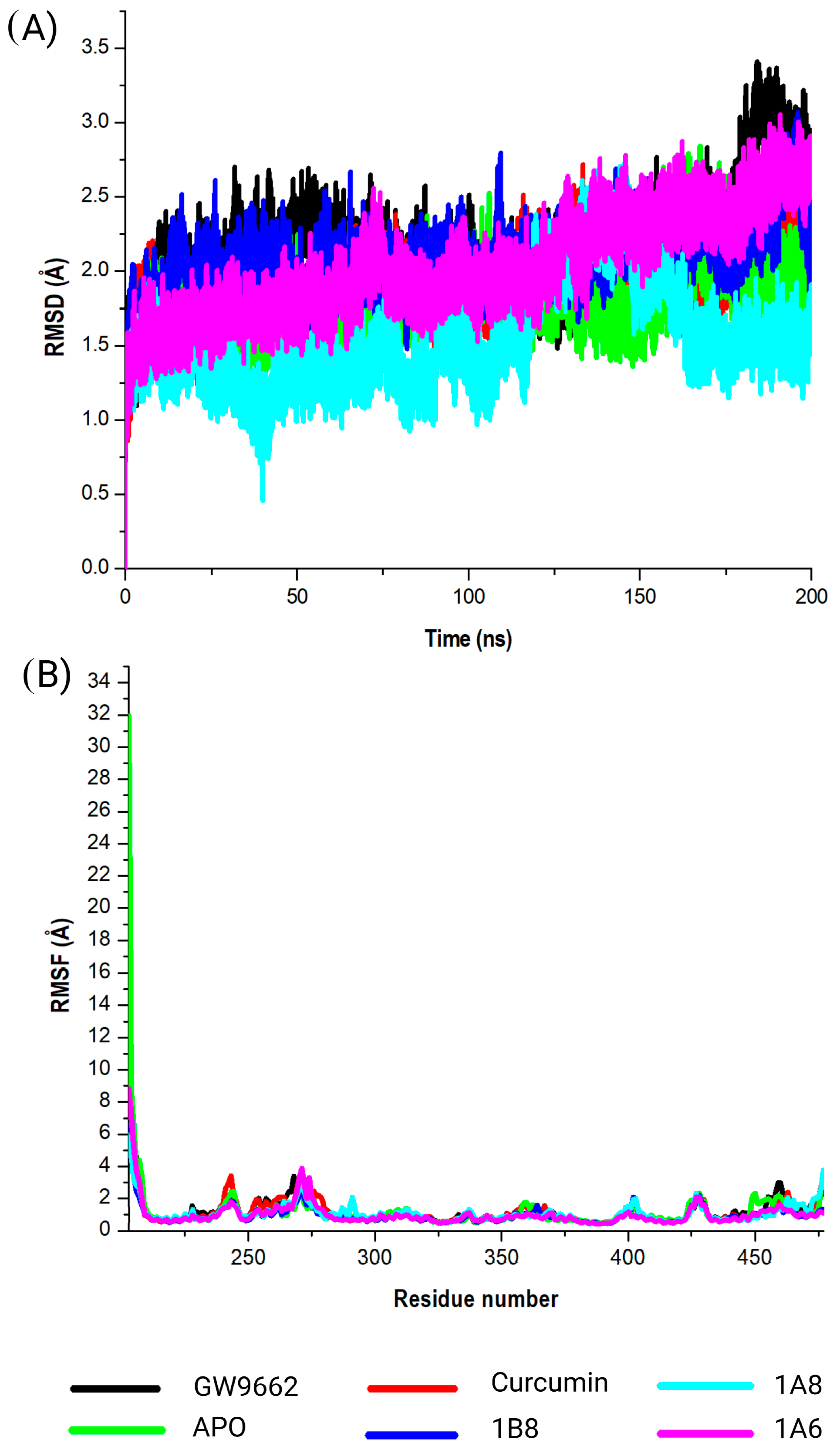
The selection of protein targets in this study was vital for investigating the mechanism of action of curcumin and its synthetic derivatives and predicting their potential as anti-obesity therapeutic agents. The systematic approach used predicted targets that are shared by all compounds and are relevant to the metabolic pathways involved in obesity including adipogenesis, lipid metabolism, and inflammation. Based on molecular docking results (binding interaction and docking score), three target proteins, PPARγ, COX2, and FAS, were best docked to curcumin and the synthetic derivatives among the nine target proteins that were selected and evaluated for molecular docking. Moreover, the results showed that the binding interactions and docking affinity between curcumin and the synthetic derivatives were similar for PPARγ, COX2, and FAS. This similarity in binding modes indicates that these derivatives share a common mechanism of action with curcumin, possibly due to structural similarity. Additionally, these compounds exhibited comparable binding affinity to PPARγ, COX2, and FAS when compared to the respective reference ligands (GW9962, Orlistat, and Naproxen).
In the PPARγ complex, curcumin and its synthetic derivatives maintained favorable binding to the target protein. In contrast, GW9662, which served as a reference control, exhibited the lowest binding affinity to PPARγ when compared to curcumin and its synthetic derivatives. This suggests that curcumin and its synthetic derivatives may be potential ligands for PPARγ. PPARγ is a nuclear receptor transcription factor that plays a key role in adipogenesis [
52] and is a major therapeutic target for various diseases including obesity, dyslipidemia, type 2 diabetes mellitus, neurodegenerative disorders, and cancers [
53]. In experimental studies, curcumin and synthetic analogs of curcumin were demonstrated to induce PPARγ expression in glial and carcinoma cells, thereby providing neuroprotective and anti-cancer effects [
54,
55]. This further suggests that other than being potential ligands, curcumin and its derivatives may regulate the expression of PPARγ.
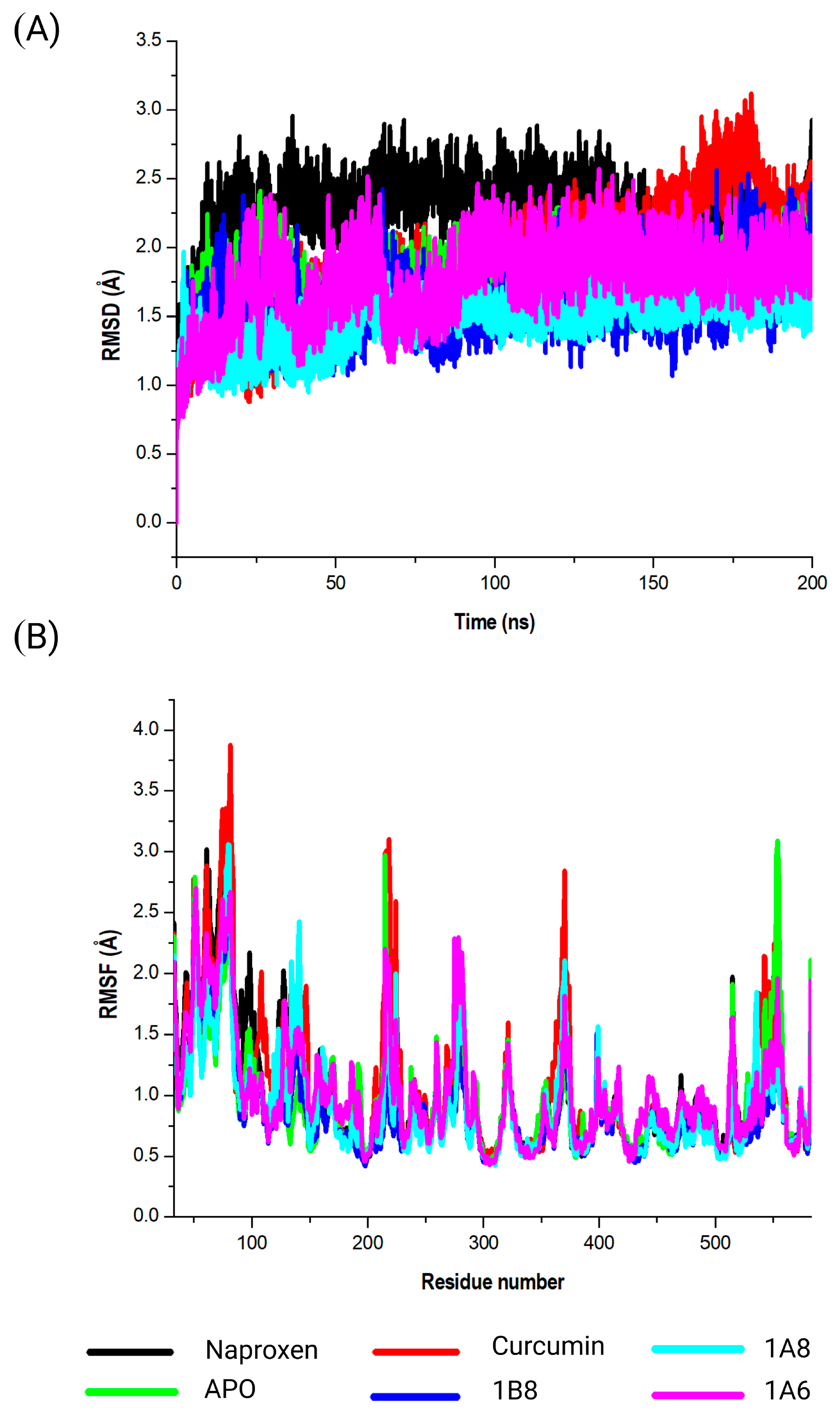
In the COX2 complex, 1B8 demonstrated a high docking score compared to Naproxen, while curcumin indicated a similar binding affinity to Naproxen. However, 1A6 and 1A8 demonstrated the lowest docking scores. This result suggests that curcumin and its derivative 1B8 may have a higher binding affinity for COX2 than Naproxen. Compared to our study, Sohilait et al. (2017) [
56] showed that analogs of curcumin strongly bind with COX2. Although 1B8 had the most favorable affinity towards COX2 compared to curcumin and other synthetic derivatives, these findings suggest that 1B8 can be a potential ligand for COX2 regulation.
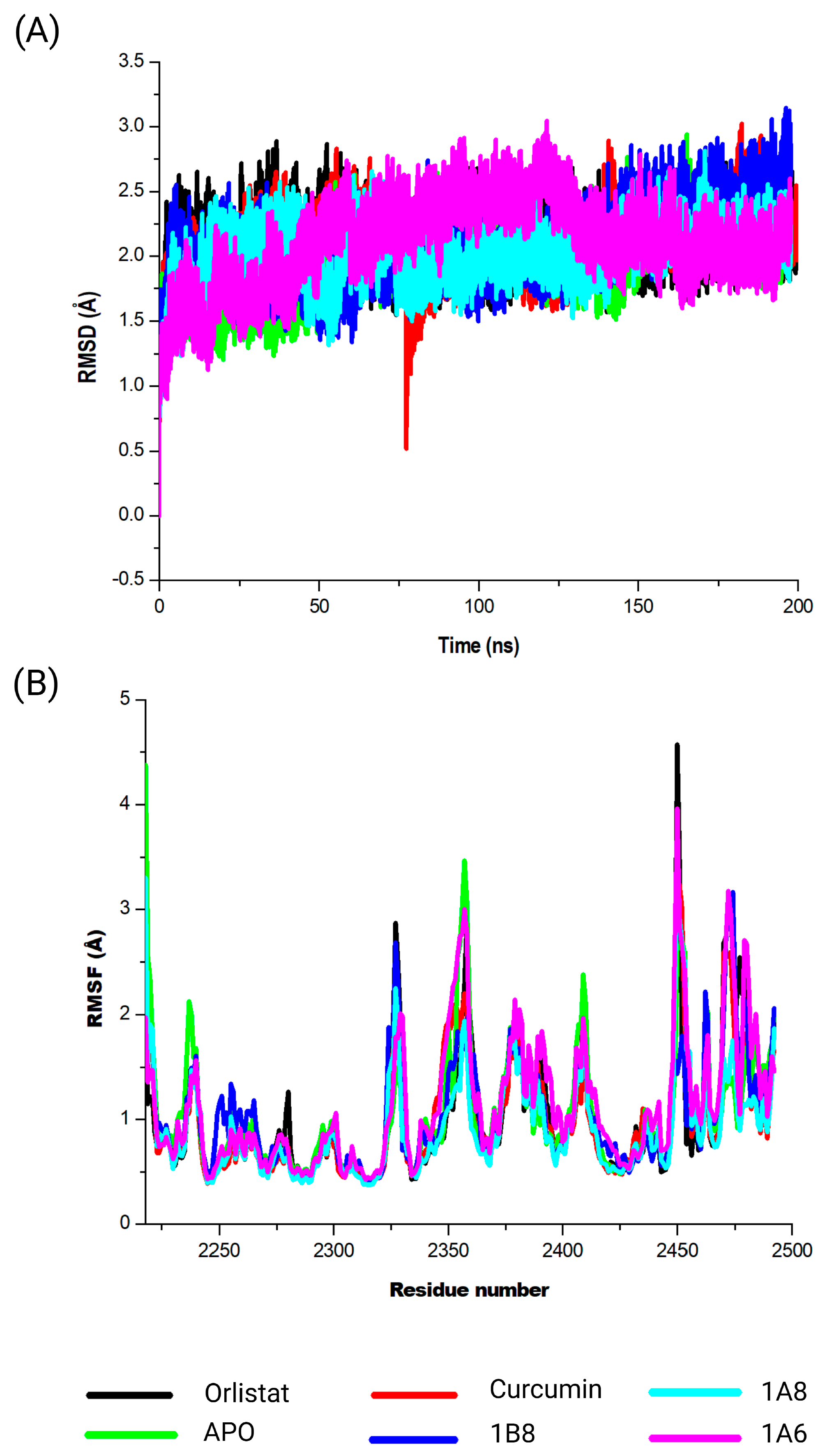
Orlistat, used as the reference control, exhibited the weakest binding affinity for the FAS protein when compared to curcumin and its derivatives. FAS is an enzyme involved in the synthesis of fatty acids by catalyzing the synthesis of long-chain fatty acids from acetyl-CoA and malonyl-CoA [
57]. HoweverMoreover, Orlistat, an anti-obesity agent that inhibits gastric and pancreatic lipases, has been experimentally shown to inhibit FAS expression [
58]. This implies that Orlistat is particularly effective in binding to FAS, making it a potent inhibitor of this enzyme as recently demonstrated [
59]. An in vivo study by Maithilikarpagaselvi et al. (2016) [
60] demonstrated that curcumin facilitates its anti-obesity effects by suppressing fat accumulation through the inhibition of FAS enzymes.
Overall, the difference in binding affinity between curcumin and its derivatives was significantly minimal, which could be due to the similarity in the structural scaffold. This structural similarity likely results in comparable binding interactions with the target protein. The observation of similar binding affinities among curcumin and its derivatives can have important implications for drug development. It suggests that while structural modifications might lead to improvements in other aspects (e.g., pharmacokinetics, toxicity, or selectivity), they may not have a pronounced effect on the binding affinity.
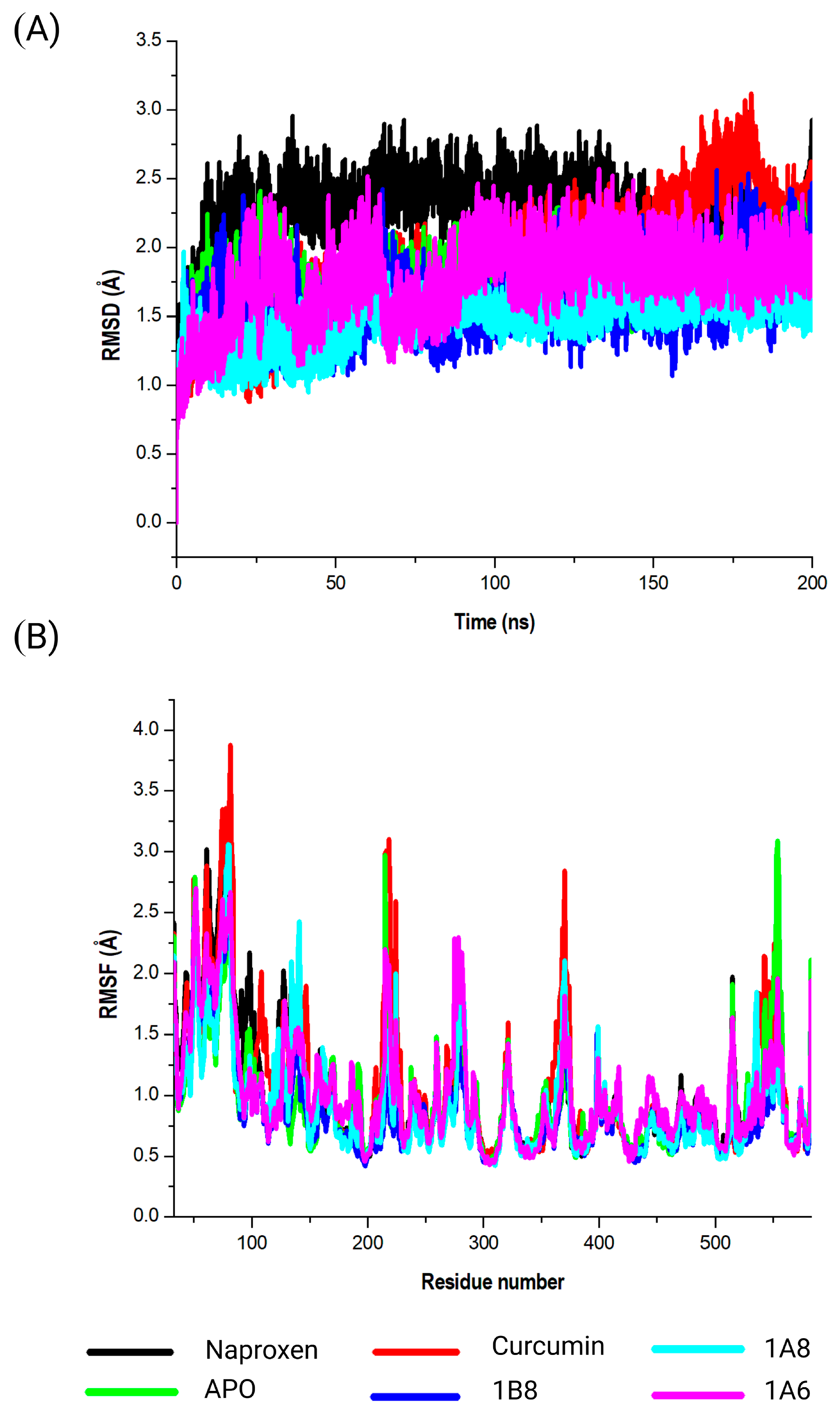
To elucidate on the stability of these interactions, molecular dynamics simulations were conducted for both unbound and bound forms of PPARγ, COX2, and FAS. The results revealed that all simulated systems remained stable throughout the 200 nanoseconds of the simulation. Stability was confirmed by minimal atomic deviations, which were within the generally accepted threshold range of 1 to 3 angstroms for good stability [
61]. This stability implies that the interactions between the compounds and their respective protein targets were robust and maintained throughout the simulation, providing confidence in the reliability of the binding interactions. In the specific case of PPARγ, the analysis indicated that curcumin, synthetic curcumin derivatives, and GW9662 induced structural flexibility in similar regions of the target protein. This flexibility was characterized by increased fluctuations in these regions when compared to the unbound state of the target. Such fluctuations suggest that the binding of these compounds caused PPARγ to adopt a more flexible conformation, which may have functional implications related to its mode of action. Generally, conformational dynamics of PPARγ inhibitors can be influenced by factors such as ligand structure, binding site interactions, and solvent environment [
62,
63]. Different conformations of inhibitors can have varying degrees of affinity and specificity towards the PPARγ protein [
64].
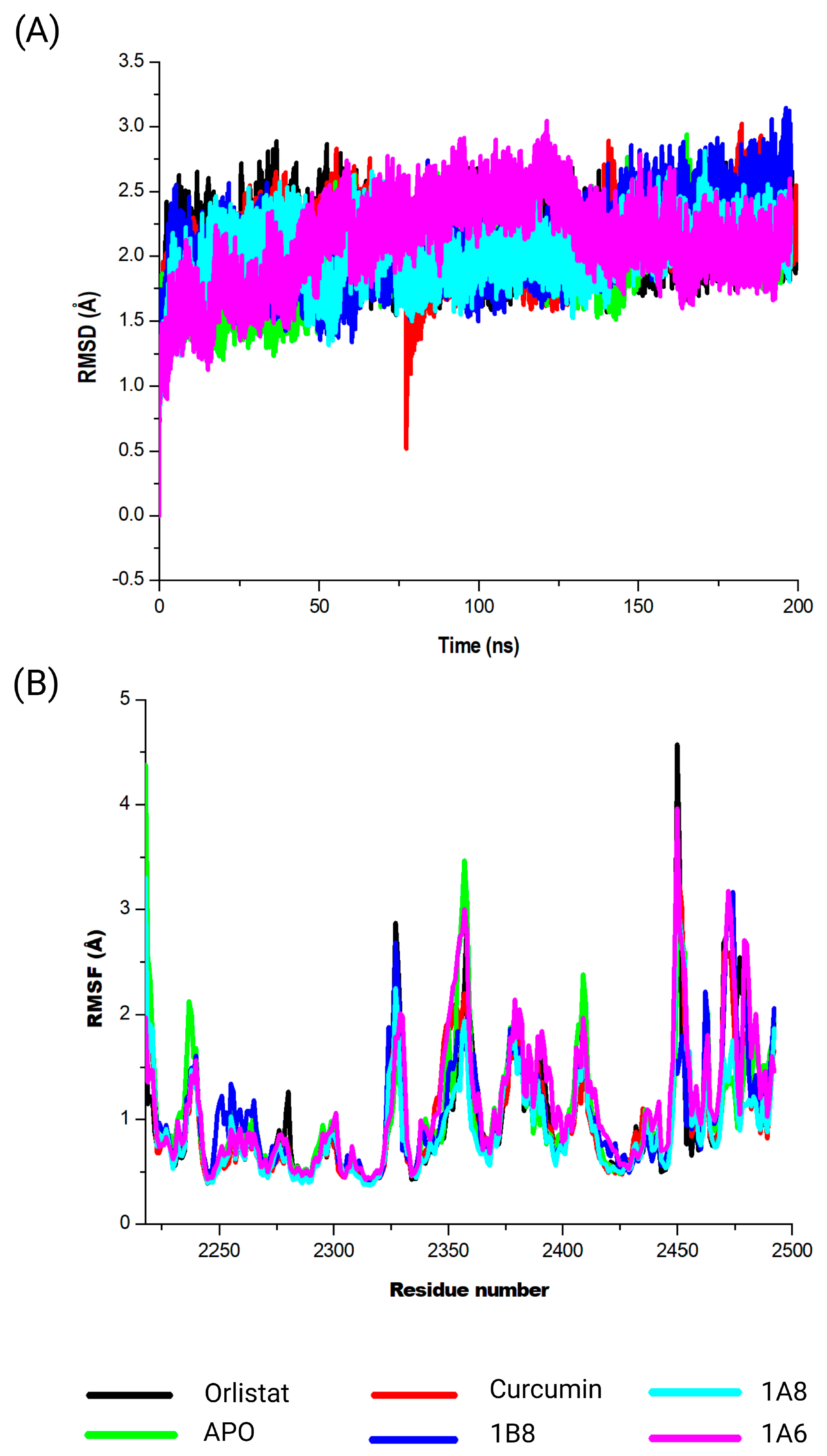
In the COX2 and FAS systems, curcumin and the synthetic curcumin derivatives showed similar conformational dynamics on each of the targets, which was characterized by increased structural flexibility and a less compact structure in contrast to the unbound targets. The findings could therefore suggest the protein is undergoing more dynamic conformational changes, allowing it to explore a wider range of structural configurations [
65,
66]. Similarly, FAS and COX2 inhibitors exhibited varying conformational changes to their respective targets depending on the structures of their respective inhibitors [
67,
68]. Overall, curcumin and the synthetic curcumin derivatives had a similar impact on the conformational dynamics of the target proteins, which is attributed to the similarity in their structures.
In addition, we assessed the amino acids within the binding site responsible for stabilizing molecular interactions following molecular dynamics simulations. This analysis is crucial in drug discovery to tailor molecules that selectively interact with the target protein [
69]. Our results revealed that compounds interacting with the binding site of PPARγ consistently engage in specific interactions with residues like Ser88, affirming their association with the localized region of the PPARγ binding site. Notably, the binding of curcumin was sustained through hydrophilic interactions with seventeen amino acid residues and one hydrogen interaction in the binding site, potentially contributing to the observed high binding free energies. The interaction between curcumin and PPARγ has been well-documented in various studies [
70,
71,
72]. Furthermore, synthetic derivatives of curcumin exhibited stronger binding free energies compared to the reference compound (GW9662), supported by their hydrophilic and hydrogen interactions and amino acid residues within the binding site. This is speculated to result from the structural similarity shared by curcumin and its derivatives. One study suggested that curcumin analogs interact with PPARγ to suppress adipogenesis more effectively than curcumin [
73]. This implies that derivatives of curcumin may outperform curcumin in targeting the PPARγ isoform regulating adipogenesis, while activating the PPARγ isoform associated with inflammation and lipid metabolism suppression. The observed variations underscore the complexity of the molecular interactions and highlight the potential therapeutic implications of curcumin derivatives in modulating PPARγ isoforms.
In this study, we demonstrated consistent interaction of the ligand compounds with amino acid residues like Ser321, Ser498, Arg89, and Tyr323, forming conserved interactions within the binding site of COX2. Remarkably, the synthetic derivative 1A8 exhibited the highest binding free energy, greater than the reference compound (Naproxen). This strong interaction was substantiated by nine hydrophilic and three hydrogen interactions with amino acid residues in the COX2 binding site. The tendency of curcumin analogs to strongly interact with the COX2 binding site has been previously reported [
56], while curcumin derivatives have also been reported to selectively interact with COX2 [
74]. This specificity may account for the superior interaction demonstrated by 1A8 compared to 1A6 and 1B8. Despite 1A8 outperforming other compounds, it is essential to recognize that curcumin exhibited strong interactions with COX2, surpassing the control inhibitor (Naproxen). Compared to this study, an in vivo study demonstrated that curcumin inhibits the expression of COX2 [
75,
76].
While common interactions were observed for PPARγ and COX2, the ligand compounds did not engage with shared amino acid residues in the binding site of FAS. However, curcumin demonstrated two hydrogen interactions with Tyr130 (2.71 Å) and Glu214 (2.59 Å) residues within the binding site residue of FAS. Additionally, the interaction of curcumin within the FAS binding site was stabilized by ten hydrophilic interactions. This interaction was validated by high binding free energies, comparable to the reference compound (Orlistat). Interestingly, an in vitro study on the breast cancer cell line SKBR3 also demonstrated that curcumin decreases the activity and expression of the FAS enzyme [
77]. It is crucial to highlight that curcumin displayed substantial interactions with both PPARγ and FAS, possibly due to the involvement of these proteins in lipid metabolism. Curcumin derivatives have been shown to decrease FAS expression, thus suppressing lipid accumulation in HepG2 cells [
78]. This indicates that curcumin and its synthetic derivatives potentially target FAS and regulate its expression.
While our study presents promising findings in the comprehensive analysis of curcumin and its synthetic derivatives, several limitations should be acknowledged. The reliance on computational analyses, without direct in vitro and in vivo validation, leaves a gap in understanding the clinical implications of our results. Furthermore, the scope of toxicity assessment is primarily focused on acute effects, necessitating extended studies to capture potential long-term impacts and interactions with other physiological systems. In addition, caution is warranted in generalizing the findings, as the study centers on specific derivatives (1A6, 1A8, and 1B8), while variability among curcumin derivatives may exist. Despite overall promising results, specific endpoint effects observed at lethal dosages highlight the need for refinement strategies to enhance the safety profile of the compounds. Additionally, the focus of the study on a select few anti-obesity target proteins may limit the generalizability of the results, and further exploration of interactions with a broader range of relevant proteins is recommended. Lastly, while the study identifies potential interactions with key liver enzymes, a more detailed exploration of the metabolic pathways undergone by these compounds is necessary.
This in-depth investigation combines computational methodologies with molecular recognition techniques to unravel the potential anti-obesity activity of curcumin and its synthetic derivatives. The findings provide a foundation for further experimental validation, emphasizing the need for a holistic understanding of the pharmacological profiles of these compounds for effective drug development. The structural insights and mechanistic understanding garnered from this study pave the way for future investigations bringing research close to harnessing the therapeutic potential of curcumin and its derivatives in the fight against obesity.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}