
Deletion of Gadd45a Expression in Mice Leads to Cognitive and Synaptic Impairment Associated with Alzheimer’s Disease Hallmarks

 ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
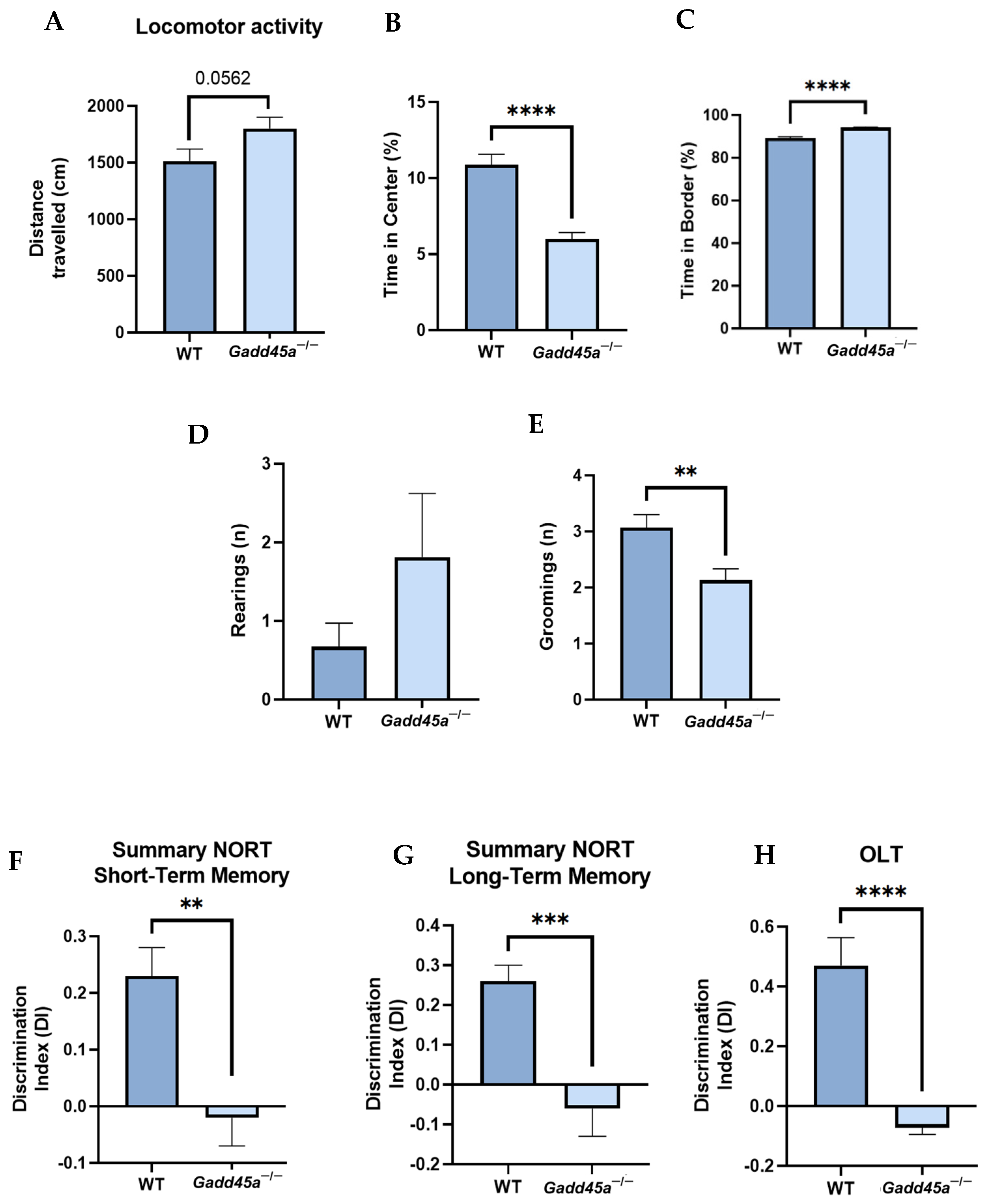
2.1. Gadd45a−/− Mice Show Behavioral and Cognitive Deficiencies
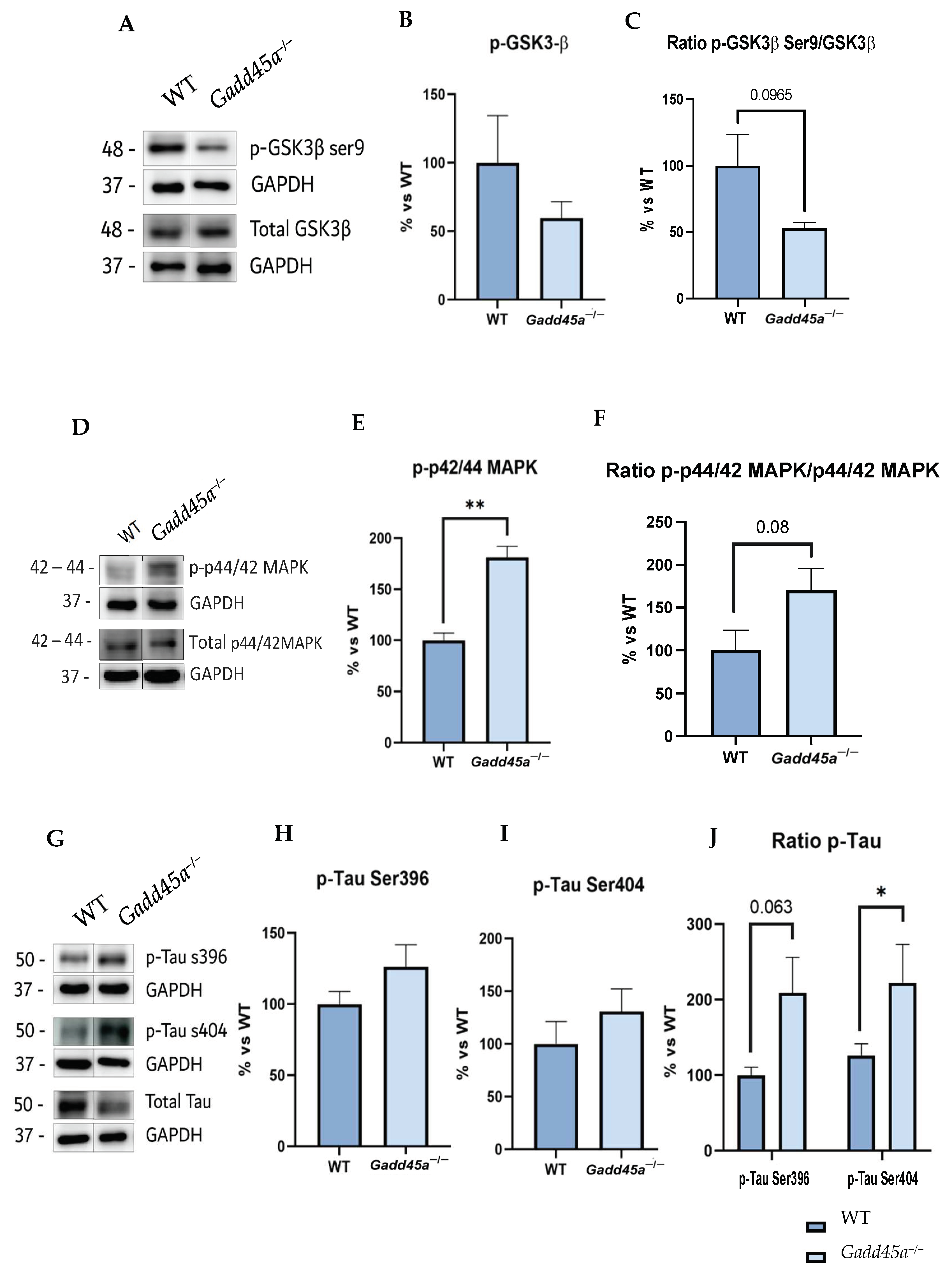
2.2. Alterations in GSK3β and p44/42MAPK (ERK1/2) Promote Tau Hyperphosphorylation in Gadd45a−/− Mice
2.3. Lack of Gadd45a Increases Proinflammatory Target Genes and the NF-kB Pathway
2.4. Lack of Gadd45a Gene Expression Promotes Alterations in Autophagy Markers
2.5. KO-Gadd45a Mice Show Reduced Synaptic Plasticity Genes Accompanied by Dendritic Morphological Abnormalities
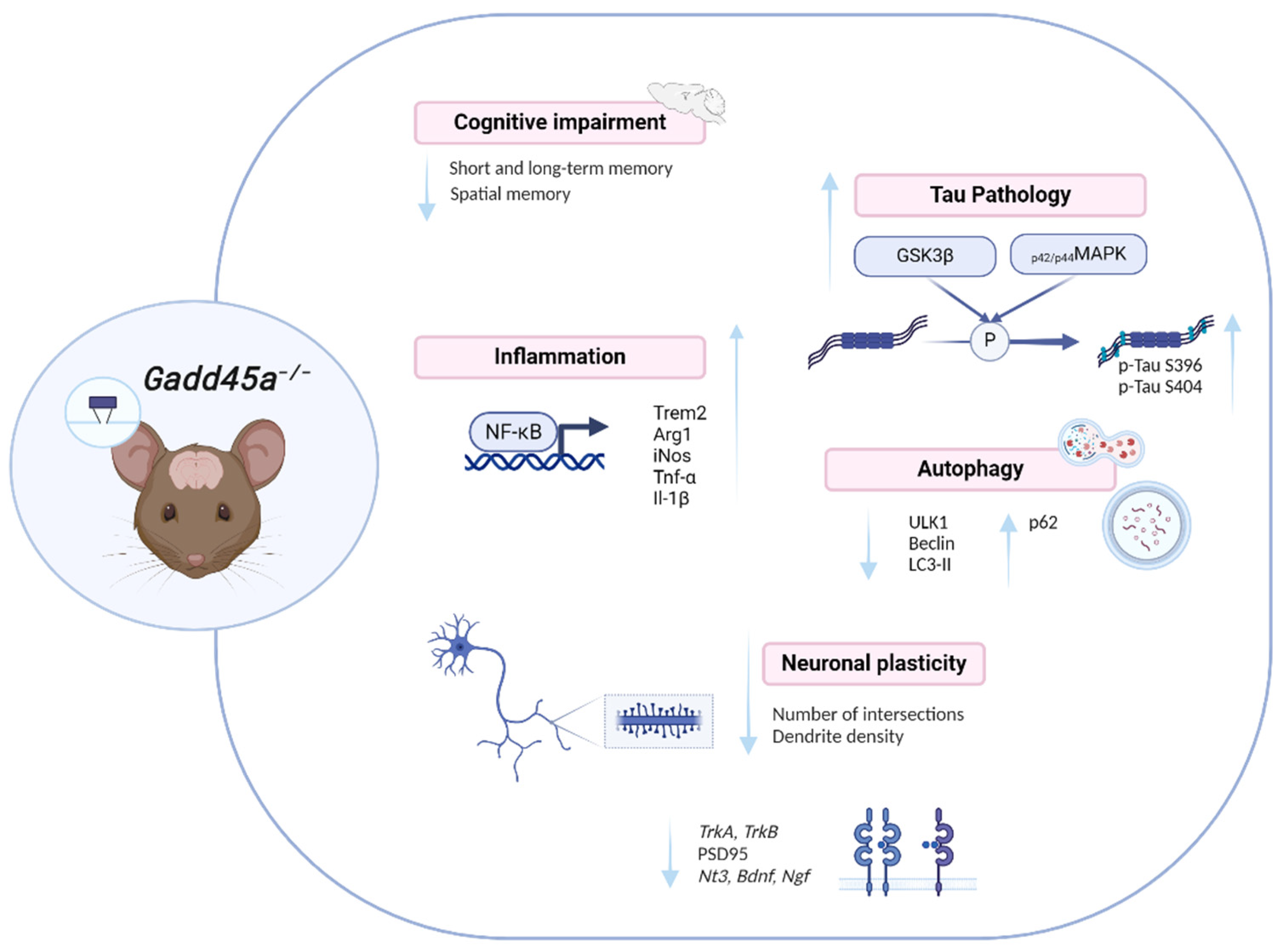
3. Discussion
4. Materials and Methods
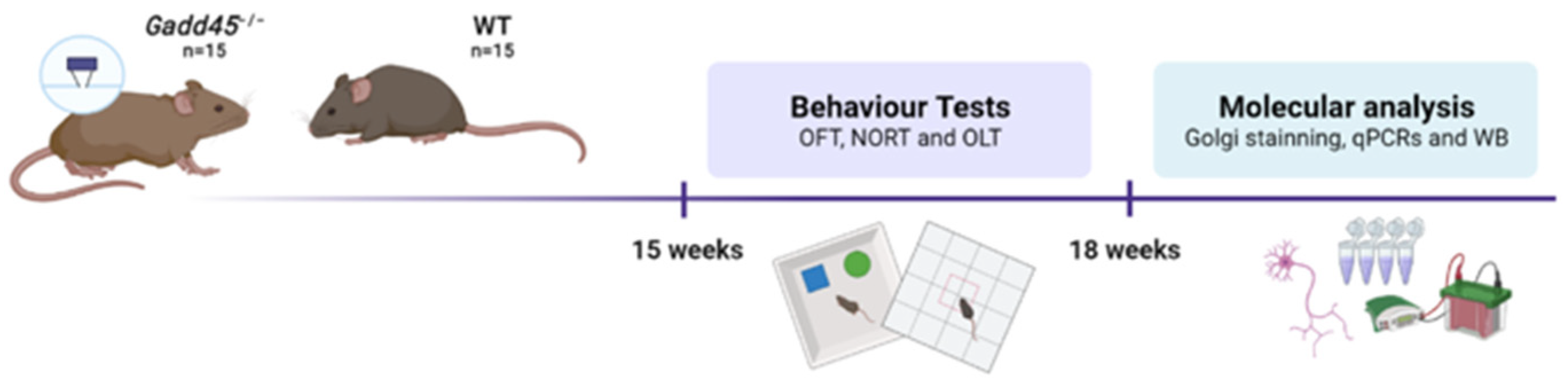
4.1. Animals
4.2. Behavioral and Cognitive Tests
4.2.1. Open Field Test
4.2.2. Novel Object Recognition Test
4.2.3. Object Location Test
4.3. Brain Processing
4.4. Western Blotting (WB)
4.5. RNA Extraction and Gene Expression Determination
4.6. Dendritic Length, Spine Density, and Golgi Staining Protocol
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef]
- da Silva, P.F.L.; Schumacher, B. Principles of the Molecular and Cellular Mechanisms of Aging. J. Investig. Dermatol. 2021, 141, 951–960. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541. [Google Scholar] [CrossRef]
- Rahman, M.M.; Lendel, C. Extracellular protein components of amyloid plaques and their roles in Alzheimer’s disease pathology. Mol. Neurodegener. 2021, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Duff, K. Linking Abeta and tau in late-onset Alzheimer’s disease: A dual pathway hypothesis. Neuron 2008, 60, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Samudra, N.; Lane-Donovan, C.; VandeVrede, L.; Boxer, A.L. Tau pathology in neurodegenerative disease: Disease mechanisms and therapeutic avenues. J. Clin. Investig. 2023, 133, e168553. [Google Scholar] [CrossRef]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Recuero, M.; Serrano, E.; Bullido, M.J.; Valdivieso, F. Aβ production as consequence of cellular death of a human neuroblastoma overexpressing APP. FEBS Lett. 2004, 570, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Litwiniuk, A.; Juszczak, G.R.; Stankiewicz, A.M.; Urbańska, K. The role of glial autophagy in Alzheimer’s disease. Mol. Psychiatry 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, M.; Rashidy-Pour, A. Association between chronic stress and Alzheimer’s disease: Therapeutic effects of Saffron. Biomed. Pharmacother. 2021, 133, 110995. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Hoffman, B. Gadd45 in stress signaling. J. Mol. Signal 2008, 3, 15. [Google Scholar] [CrossRef]
- Sultan, F.A.; Sweatt, J.D. The role of the Gadd45 family in the nervous system: A focus on neurodevelopment, neuronal injury, and cognitive neuroepigenetics. Adv. Exp. Med. Biol. 2013, 793, 81–119. [Google Scholar] [PubMed]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar]
- Sytnikova, Y.A.; Kubarenko, A.V.; Schäfer, A.; Weber, A.N.R.; Niehrs, C. Gadd45a Is an RNA Binding Protein and Is Localized in Nuclear Speckles. PLoS ONE 2011, 6, e14500. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Smit-McBride, Z.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Tacutu, R.; Fraifeld, V.E. Gadd45 proteins: Relevance to aging, longevity and age-related pathologies. Ageing Res. Rev. 2012, 11, 51. [Google Scholar] [CrossRef]
- Yang, Z.; Song, L.; Huang, C. Gadd45 Proteins as Critical Signal Transducers Linking NF-κB to MAPK Cascades. Curr. Cancer Drug Targets 2009, 9, 915. [Google Scholar] [CrossRef]
- Torp, R.; Su, J.H.; Deng, G.; Cotman, C.W. GADD45 is induced in Alzheimer’s disease, and protects against apoptosis in vitro. Neurobiol. Dis. 1998, 5, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bgatova, N.; Dubatolova, T.; Omelyanchuk, L.; Plyusnina, E.; Shaposhnikov, M.; Moskalev, A. Gadd45 expression correlates with age dependent neurodegeneration in Drosophila melanogaster. Biogerontology 2015, 16, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.M.; Bullard, S.A.; Basisty, N.; Marcotte, G.R.; Skopec, Z.P.; Dierdorff, J.M.; Al-Zougbi, A.; Tomcheck, K.C.; DeLau, A.D.; Rathmacher, J.A.; et al. Activating transcription factor 4 (ATF4) promotes skeletal muscle atrophy by forming a heterodimer with the transcriptional regulator C/EBPβ. J. Biol. Chem. 2020, 295, 2787–2803. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Sheikh, M.S.; Bulavin, D.V.; Lundgren, K.; Augeri-Henmueller, L.; Shehee, R.; Molinaro, T.A.; Kim, K.E.; Tolosa, E.; Ashwell, J.D.; et al. Genomic instability in Gadd45a-deficient mice. Nat. Genet. 1999, 23, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Jiménez, P.; Fernández-Messina, L.; Ovejero-Benito, M.C.; Chicharro, P.; Vera-Tomé, P.; Vara, A.; Cibrian, D.; Martínez-Fleta, P.; Jiménez-Fernández, M.; Sánchez-García, I.; et al. Growth arrest and DNA damage-inducible proteins (GADD45) in psoriasis. Sci. Rep. 2021, 11, 14579. [Google Scholar] [CrossRef]
- Schäfer, A. Gadd45 proteins: Key players of repair-mediated DNA demethylation. Adv. Exp. Med. Biol. 2013, 793, 35–50. [Google Scholar]
- Corey-Bloom, J. The ABC of Alzheimer’s disease: Cognitive changes and their management in Alzheimer’s disease and related dementias. Int. Psychogeriatr. 2002, 14 (Suppl. S1), 51–75. [Google Scholar] [CrossRef]
- Aparisi Rey, A.; Karaulanov, E.; Sharopov, S.; Arab, K.; Schäfer, A.; Gierl, M.; Guggenhuber, S.; Brandes, C.; Pennella, L.; Gruhn, W.H.; et al. Gadd45α modulates aversive learning through post-transcriptional regulation of memory-related mRNAs. EMBO Rep. 2019, 20, e46022. [Google Scholar] [CrossRef]
- Li, X.; Marshall, P.R.; Leighton, L.J.; Zajaczkowski, E.L.; Wang, Z.; Madugalle, S.U.; Yin, J.; Bredy, T.W.; Wei, W. The DNA Repair-Associated Protein Gadd45γ Regulates the Temporal Coding of Immediate Early Gene Expression within the Prelimbic Prefrontal Cortex and Is Required for the Consolidation of Associative Fear Memory. J. Neurosci. 2019, 39, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell Neurosci. 2020, 14, 485621. [Google Scholar] [CrossRef]
- Leugers, C.J.; Koh, J.Y.; Hong, W.; Lee, G. Tau in MAPK Activation. Front. Neurol. 2013, 4, 161. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S141–S144. [Google Scholar] [CrossRef]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. p38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.; Alessandrini, A.; Cole, A.J.; Yee, A.G.; Furshpan, E.J. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc. Natl. Acad. Sci. USA 1998, 95, 11975. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2020, 17, 157–172. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388. [Google Scholar] [CrossRef]
- Souder, D.C.; Anderson, R.M. An expanding GSK3 network: Implications for aging research. Geroscience 2019, 41, 369. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Vázquez, S.; Pallàs, M.; Griñán-Ferré, C. Glial cell reactivity and oxidative stress prevention in Alzheimer’s disease mice model by an optimized NMDA receptor antagonist. Sci. Rep. 2022, 12, 17908. [Google Scholar] [CrossRef]
- Ott, L.W.; Resing, K.A.; Sizemore, A.W.; Heyen, J.W.; Cocklin, R.R.; Pedrick, N.M.; Woods, H.C.; Chen, J.Y.; Goebl, M.G.; Witzmann, F.A.; et al. Tumor Necrosis Factor-α -and Interleukin-1-Induced Cellular Responses: Coupling Proteomic and Genomic Information. J. Proteome Res. 2007, 6, 2176. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 424429. [Google Scholar] [CrossRef]
- Vahsen, B.F.; Ribas, V.T.; Sundermeyer, J.; Boecker, A.; Dambeck, V.; Lenz, C.; Shomroni, O.; Caldi Gomes, L.; Tatenhorst, L.; Barski, E.; et al. Inhibition of the autophagic protein ULK1 attenuates axonal degeneration in vitro and in vivo, enhances translation, and modulates splicing. Cell Death Differ. 2020, 27, 2810–2827. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.Y.; Tian, C.; Wang, H.; Xu, Y.; Ren, K.; Zhang, B.Y.; Gao, C.; Shi, Q.; Meng, G.; Zhang, L.B.; et al. Activation of the AMPK-ULK1 pathway plays an important role in autophagy during prion infection. Sci. Rep. 2015, 5, 14728. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell Biol. 2013, 15, 727. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA Damage Response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, P.; Saito, T.; Komatsu, M. p62/SQSTM1: “Jack of all trades” in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Ha, H.; Lee, B.S.; Kim, B.H.; Song, H.K.; Kim, Y.K. LC3B is an RNA-binding protein to trigger rapid mRNA degradation during autophagy. Nat. Commun. 2022, 13, 1436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, W.; Li, D.; Fu, M.; Chen, R.; Zhan, Q. GADD45A inhibits autophagy by regulating the interaction between BECN1 and PIK3C3. Autophagy 2015, 11, 2247. [Google Scholar] [CrossRef]
- Conroy, J.N.; Coulson, E.J. High-affinity TrkA and p75 neurotrophin receptor complexes: A twisted affair. J. Biol. Chem. 2022, 298, 101568. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Z.; Wei, W.; Zhang, T.; Li, Z.; Chen, J.; Liu, K. Gadd45α is involved in regulating activity-dependent and exon-specific BDNF expression in postmitotic cortical neurons. Neuroreport 2021, 32, 1147–1152. [Google Scholar] [CrossRef]
- Brito, D.V.C.; Kupke, J.; Gulmez Karaca, K.; Oliveira, A.M.M. Regulation of neuronal plasticity by the DNA repair associated Gadd45 proteins. Curr. Res. Neurobiol. 2022, 3, 100031. [Google Scholar] [CrossRef]
- Boros, B.D.; Greathouse, K.M.; Gearing, M.; Herskowitz, J.H. Dendritic spine remodeling accompanies Alzheimer’s disease pathology and genetic susceptibility in cognitively normal aging. Neurobiol. Aging 2019, 73, 92–103. [Google Scholar] [CrossRef]
- Reza-Zaldivar, E.E.; Hernández-Sápiens, M.A.; Minjarez, B.; Gómez-Pinedo, U.; Sánchez-González, V.J.; Márquez-Aguirre, A.L.; Canales-Aguirre, A.A. Dendritic Spine and Synaptic Plasticity in Alzheimer’s Disease: A Focus on MicroRNA. Front. Cell Dev. Biol. 2020, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, J.; Miyamoto, Y.; Murabe, M.; Fujiwara, Y.; Sanbe, A.; Fujita, Y.; Murase, S.; Tanoue, A. Gadd45a, the gene induced by the mood stabilizer valproic acid, regulates neurite outgrowth through JNK and the substrate paxillin in N1E-115 neuroblastoma cells. Exp. Cell Res. 2007, 313, 1886–1896. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griñán-Ferré, C.; Jarne-Ferrer, J.; Bellver-Sanchis, A.; Ribalta-Vilella, M.; Barroso, E.; Salvador, J.M.; Jurado-Aguilar, J.; Palomer, X.; Vázquez-Carrera, M.; Pallàs, M. Deletion of Gadd45a Expression in Mice Leads to Cognitive and Synaptic Impairment Associated with Alzheimer’s Disease Hallmarks. Int. J. Mol. Sci. 2024, 25, 2595. https://doi.org/10.3390/ijms25052595
Griñán-Ferré C, Jarne-Ferrer J, Bellver-Sanchis A, Ribalta-Vilella M, Barroso E, Salvador JM, Jurado-Aguilar J, Palomer X, Vázquez-Carrera M, Pallàs M. Deletion of Gadd45a Expression in Mice Leads to Cognitive and Synaptic Impairment Associated with Alzheimer’s Disease Hallmarks. International Journal of Molecular Sciences. 2024; 25(5):2595. https://doi.org/10.3390/ijms25052595
Chicago/Turabian StyleGriñán-Ferré, Christian, Júlia Jarne-Ferrer, Aina Bellver-Sanchis, Marta Ribalta-Vilella, Emma Barroso, Jesús M. Salvador, Javier Jurado-Aguilar, Xavier Palomer, Manuel Vázquez-Carrera, and Mercè Pallàs. 2024. "Deletion of Gadd45a Expression in Mice Leads to Cognitive and Synaptic Impairment Associated with Alzheimer’s Disease Hallmarks" International Journal of Molecular Sciences 25, no. 5: 2595. https://doi.org/10.3390/ijms25052595
APA StyleGriñán-Ferré, C., Jarne-Ferrer, J., Bellver-Sanchis, A., Ribalta-Vilella, M., Barroso, E., Salvador, J. M., Jurado-Aguilar, J., Palomer, X., Vázquez-Carrera, M., & Pallàs, M. (2024). Deletion of Gadd45a Expression in Mice Leads to Cognitive and Synaptic Impairment Associated with Alzheimer’s Disease Hallmarks. International Journal of Molecular Sciences, 25(5), 2595. https://doi.org/10.3390/ijms25052595