
The Diverse Nature of the Molecular Interactions That Govern the COV-2 Variants’ Cell Receptor Affinity Ranking and Its Experimental Variability

Abstract
1. Introduction
1.1. Hypothesis for Infectivity and Virulence of the VOCs
1.1.1. Spike–ACE2 Receptor Interactions
1.1.2. Spike–Antibody Interactions
2. Results and Discussion
2.1. Ranking Affinity Prediction by a Suite of Protocols of Increasing Complexity
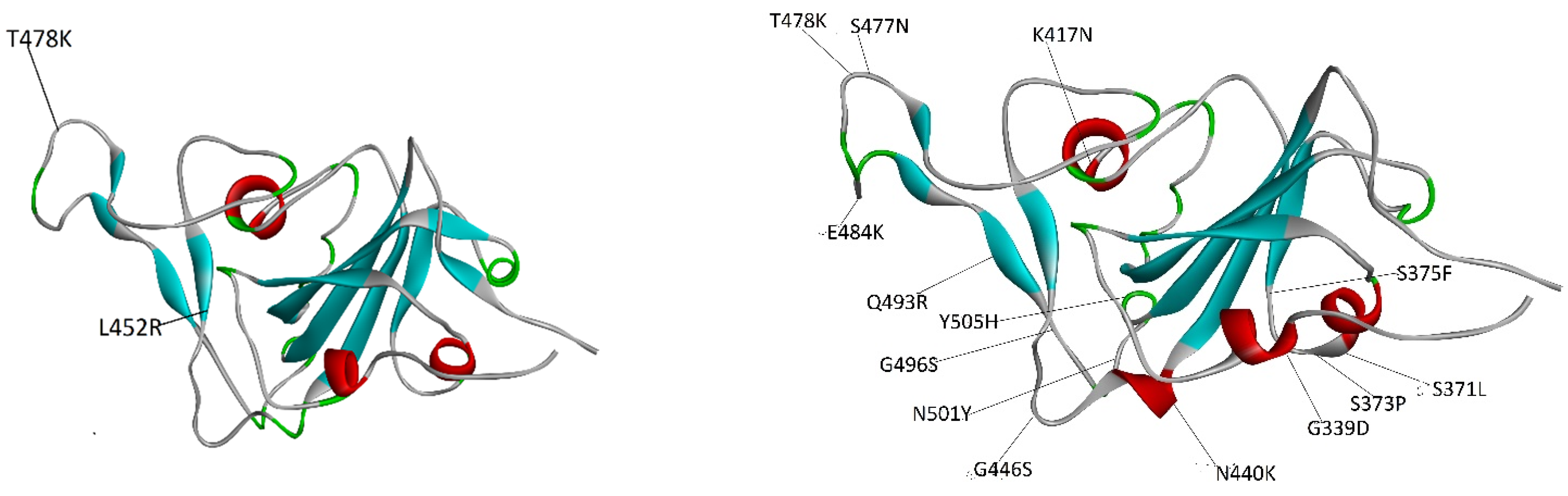
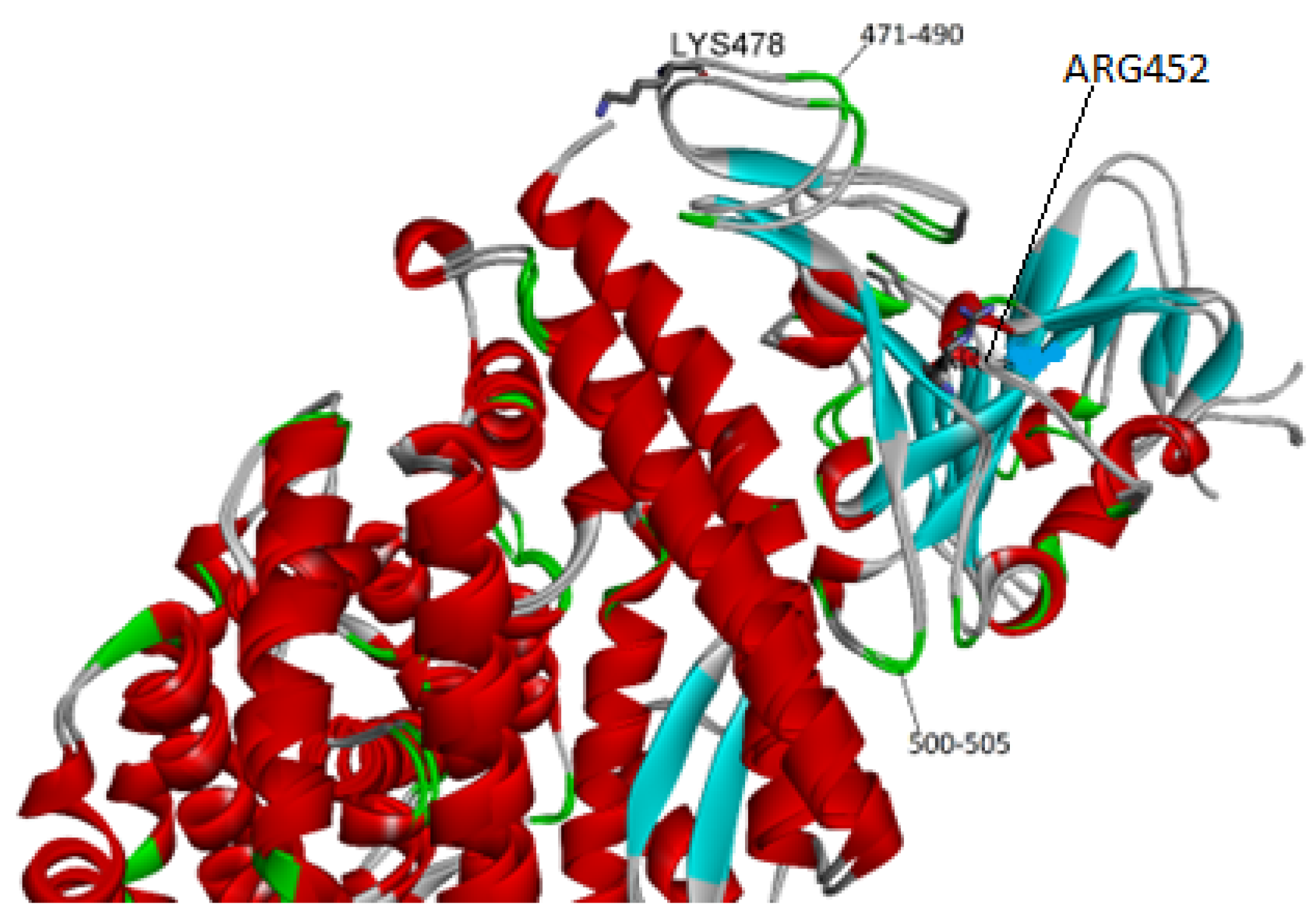
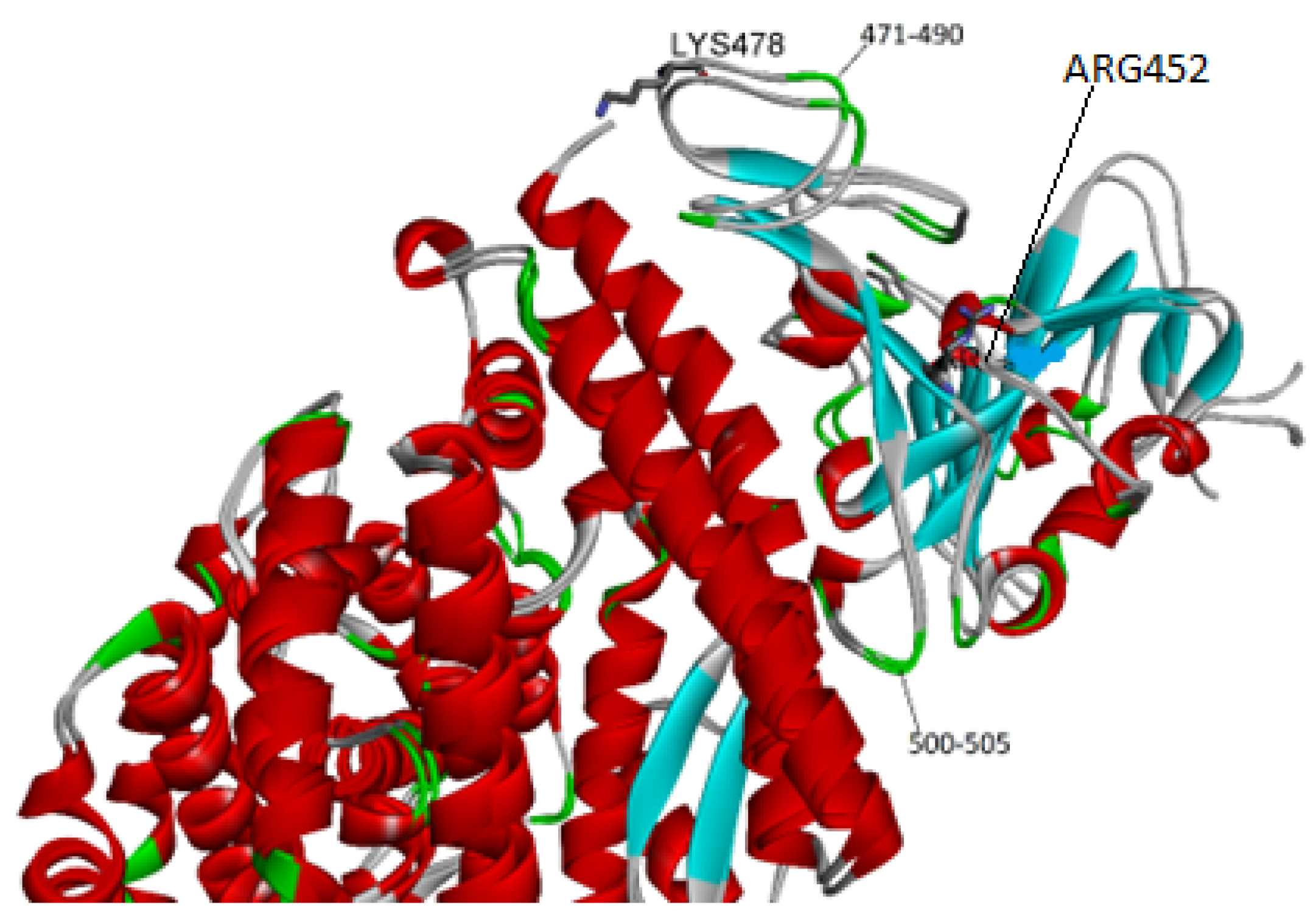
2.2. Affinity Ranking and Structural Variability
3. Materials and Methods
Associated Method Information
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mlcochova, P.; Kemp, S.; Dhar, M.S.; Papa, G.; Meng, B.; Mishra, S.; Whittaker, C.; Mellan, T.; Ferreira, I.; Datir, R.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication, sensitivity to neutralising antibodies and vaccine breakthrough. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, S.; Wu, B.; Yang, Q.; Chen, A.; Li, Y.; Zhang, Y.; Pan, T.; Zhang, H.; He, X. SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Target. Ther. 2021, 6, 430. [Google Scholar] [CrossRef]
- Kim, S.; Liu, Y.; Ziarnik, M.; Cao, Y.; Zhang, X.F.; Im, W. Binding of human ACE2 and RBD of omicron enhanced by unique interaction patterns among SARS-CoV-2 variants of concern. J. Comp. Chem. 2023, 44, 594–601. [Google Scholar] [CrossRef]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Schotsaert, M. The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. MedRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Warshel, A. Critical Differences between the Binding Features of the Spike proteins of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. B 2020, 124, 5907–5912. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Aulic, S.; Fermeglia, M.; Pricl, S. Computational Alanine Scanning and Structural Analysis of the SARS-CoV-2 Spike Protein/Angiotensin-Converting Enzyme 2 Complex. ACS Nano 2020, 14, 11821–11830. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654. [Google Scholar] [CrossRef]
- Verkhivker, G.; Agajanian, S.; Kassab, R.; Krishnan, K. Computer Simulations and Network-Based Profiling of Binding and Allosteric Interactions of SARS-CoV-2 Spike Variant Complexes and the Host Receptor: Dissecting the Mechanistic Effects of the Delta and Omicron Mutations. Int. J. Mol. Sci. 2022, 23, 4376. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.J.; Beh, R.C.; Hung, A.; Karagiannis, T.C. Molecular dynamics simulations highlight the altered binding landscape at the spike-ACE2 interface between the Delta and Omicron variants compared to the SARS-CoV-2 original strain. Comput. Biol. Med. 2022, 149, 106035. [Google Scholar] [CrossRef]
- Khan, A.; Khan, S.A.; Zia, K.; Altowyan, M.S.; Barakat, A.; Ul-Haq, Z. Deciphering the Impact of Mutations on the Binding Efficacy of SARS-CoV-2 Omicron and Delta Variants With Human ACE2 Receptor. Front. Chem. 2022, 10, 892093. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Yang, X.; Luo, M.; Yan, J.; Xu, P.; Zhang, H.; Li, Y.; Wu, S.; Warshel, A.; Bai, C. Mechanistic study of the transmission pattern of the SARS-CoV-2 omicron variant. Proteins 2024, 1–15. [Google Scholar] [CrossRef]
- Kumar, R.; Murugan, N.A.; Srivastava, V. Improved Binding Affinity of Omicron’s Spike Protein for the Human Angiotensin-Converting Enzyme 2 Receptor Is the Key behind Its Increased Virulence. Int. J. Mol. Sci. 2022, 23, 3409. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Di Paola, L. Integrated Biophysical Modeling of the SARS-CoV-2 Spike Protein Binding and Allosteric Interactions with Antibodies. J. Phys. Chem. B 2021, 125, 4596–4619. [Google Scholar] [CrossRef]
- da Costa, C.H.; de Freitas, C.A.; Alves, C.N.; Lameira, J. Assessment of mutations on RBD in the Spike protein of SARS-CoV-2 Alpha, Delta and Omicron variants. Sci. Rep. 2022, 12, 8540. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Key Interacting Residues between RBD of SARS-CoV2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Binding Interactions between Receptor-Binding Domain of Spike Protein and Human Angiotensin Converting Enzyme-2 in Omicron Variant. J. Phys. Chem. Lett. 2022, 13, 3915–3921. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.C.; Roux, B.; Chipot, C. Efficient determination of protein-protein standard binding free energies from first principles. J. Chem. Theory Comput. 2013, 9, 3789–3798. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yan, Y.N.; Yang, M.; Zhang, J.Z. Interaction entropy for protein-protein binding. J. Chem. Phys. 2017, 146, 124124. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Saville, J.W.; Mannar, D.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Demers, J.P.; Zhou, S.; Tuttle, K.S.; Sekirov, I.; Kim, A.; et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat. Commun. 2022, 13, 742. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Shi, K.; Ye, G.; Zhang, W.; Aihara, H.; Li, F. Structural Basis for Human Receptor Recognition by SARS-CoV-2 Omicron. Variant BA.1. J. Virol. 2022, 96, e00249-22. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Kastritis, P.L.; Bonvin, A.M.J.J. On the binding affinity of macromolecular interactions: Daring to ask why proteins interact. J. R. Soc. Interface 2013, 10, 20120835. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from the Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, X.; Hu, B.; Li, D.; Chen, L.; Li, Y.; Tu, Y.; Xiong, S.; Wang, G.; Deng, J.; et al. Mechanisms of SARS-CoV-2 Infection-Induced Kidney Injury: A Literature Review. Front. Cell. Infect. Microbiol. 2022, 12, 838213. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, P.; Dixit, N.M. Modelling how increased Cathepsin B/L and decreased TMPRSS2 usage for cell entry by the SARS-CoV-2 Omicron variant may affect the efficacy and synergy of TMPRSS2 and Cathepsin B/L inhibitors. J. Theo. Biol. 2023, 572, 111568. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Im, W.; Lee, M.S.; Brooks, C.L., III. Generalized Born Model with a Simple Smoothing Function. J. Comput. Chem. 2003, 24, 1691–1702. [Google Scholar] [CrossRef]
- Pastor, R.W. Techniques and Applications of Langevin Dynamics Simulations. In The Molecular Dynamics of Liquid Crystals; Luckhurst, G.R., Veracini, C.A., Eds.; NATO ASI Series; Springer Netherlands: Dordrecht, The Netherlands, 1994; Volume 431, pp. 85–138. [Google Scholar]
- BIOVIA Dassault Systèmes. Discovery Studio Visualizer, Version 3.5; Dassault Systèmes: San Diego, CA, USA, 2018.
- Chen, Y.; Zhao, X.; Zhou, H.; Zhu, H.; Jiang, S.; Wang, P. Broadly neutralizing antibodies to SARS-CoV-2 and other human coronaviruses. Nat. Rev. Immunol. 2023, 23, 189–199. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Source | Wild Type | Delta | Omicron |
|---|---|---|---|
| Mannar et al. (kd, nM) [4] | 3.0 ± 0.3 | 2.0 ± 0.2 | 2.1 ± 0.3 |
| Kim et al. (Kd, nM) [9] | 27.5 ± 4.8 | 21.5 ± 2.9 | 5.5 ± 1.4 |
| Han et al. (Kd, nM) [7] | 24.63 ± 5.00 | 25.07 ± 6.70 | 31.40 ± 11.62 |
| Zhang et al. (Kd, nM) [8] | ------ | 2.9 | 8.9 |
| VOC (PDB Entry) | BSA (a) | Binding Energy (b) |
|---|---|---|
| Wild Type (6m0j) | −1719.2 | −43.0 |
| Omicron (7u0n) | −1720.6 | −43.0 |
| Omicron (7wbp) | −1733.1 | −43.3 |
| Omicron (7wpb) | −1710.7 | −42.8 |
| Omicron (7t9l) | −1678.2 | −42.0 |
| Delta (7tew) | −1807.6 | −45.2 |
| VOC/PDB | BSA (c) | Binding Energy (a) |
|---|---|---|
| Wild Type (6M0J) | −1750.9 (32.0) (b) | −43.7 (0.8) (b) |
| Delta (7TEW) | −1849.4 (19.6) | −46.2 (0.5) |
| Omicron (7T9L) | −1708.0 (48.0) | −42.7 (1.2) |
| Omicron (7U0N) | −1682.5 (50.9) | −42.1(1.3) |
| VOC | PDB Entry (b) | Binding Energy (Pisa) (a) |
|---|---|---|
| Wild Type | 6m0j | −5.1 |
| Delta | 7tew | −6.3 |
| Omicron | 7t9l | −6.1 |
| Omicron | 7wpb | −3.4 |
| Omicron | 7u0n | −4.4 |
| Omicron | 7wbp | −6.2 |
| VOC | PDB Entry | Binding Energy (a) |
|---|---|---|
| Native | 6m0j | −76.7(1.4) (b) |
| Delta | 7tew | −87.3 (4.3) |
| Omicron | 7u0n | −67.0 (2.0) |
| Omicron | 7t9l | −83.9 (1.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sussman, F.; Villaverde, D.S. The Diverse Nature of the Molecular Interactions That Govern the COV-2 Variants’ Cell Receptor Affinity Ranking and Its Experimental Variability. Int. J. Mol. Sci. 2024, 25, 2585. https://doi.org/10.3390/ijms25052585
Sussman F, Villaverde DS. The Diverse Nature of the Molecular Interactions That Govern the COV-2 Variants’ Cell Receptor Affinity Ranking and Its Experimental Variability. International Journal of Molecular Sciences. 2024; 25(5):2585. https://doi.org/10.3390/ijms25052585
Chicago/Turabian StyleSussman, Fredy, and Daniel S. Villaverde. 2024. "The Diverse Nature of the Molecular Interactions That Govern the COV-2 Variants’ Cell Receptor Affinity Ranking and Its Experimental Variability" International Journal of Molecular Sciences 25, no. 5: 2585. https://doi.org/10.3390/ijms25052585
APA StyleSussman, F., & Villaverde, D. S. (2024). The Diverse Nature of the Molecular Interactions That Govern the COV-2 Variants’ Cell Receptor Affinity Ranking and Its Experimental Variability. International Journal of Molecular Sciences, 25(5), 2585. https://doi.org/10.3390/ijms25052585