


Cisplatin Nephrotoxicity Is Critically Mediated by the Availability of BECLIN1
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Heterozygous Disruption of Becn1 Promotes Cisplatin-Induced Nephropathy In Vivo
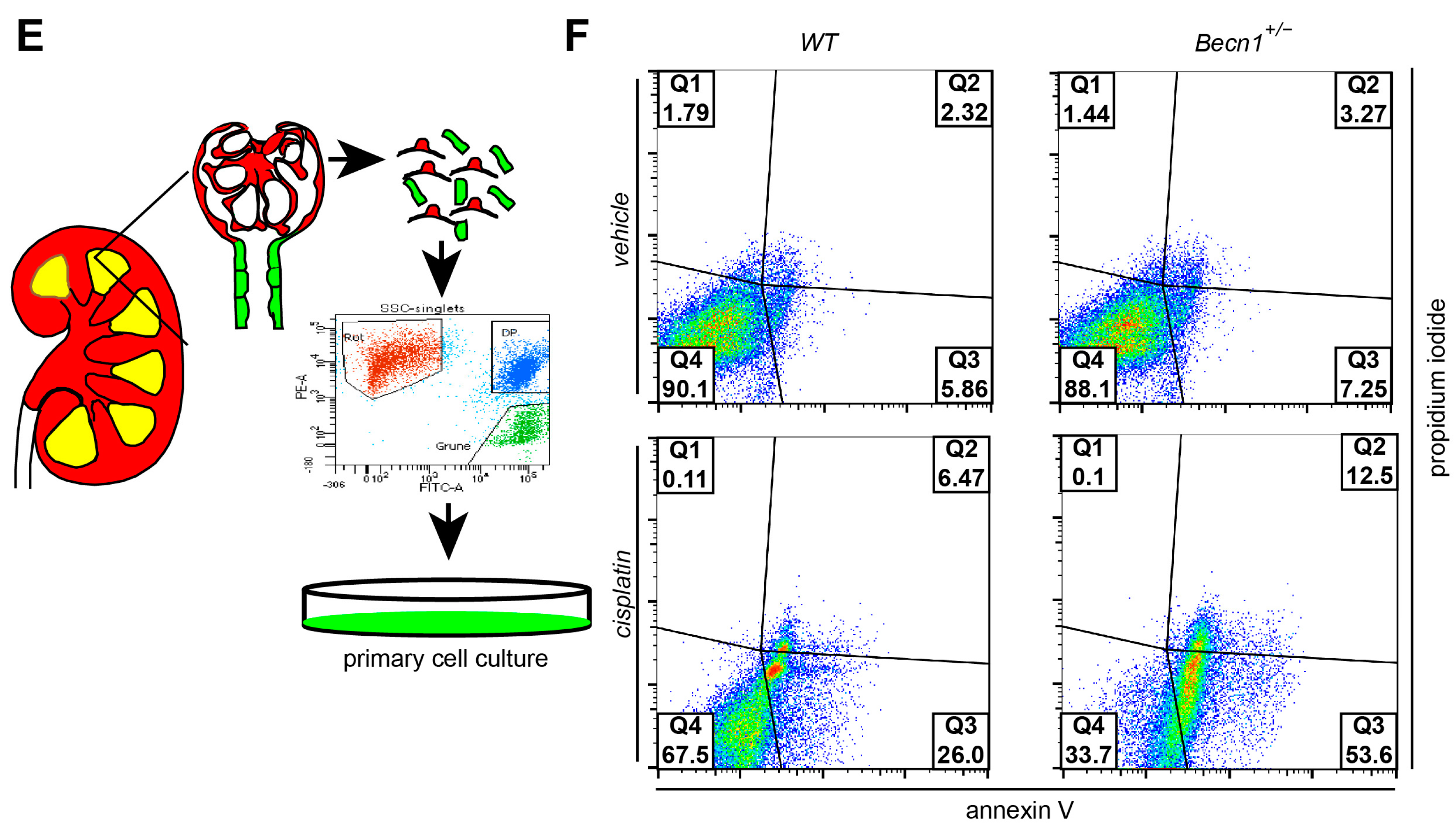
2.2. Heterozygous Disruption of Becn1 Sensitizes to Apoptosis In Vivo and In Vitro
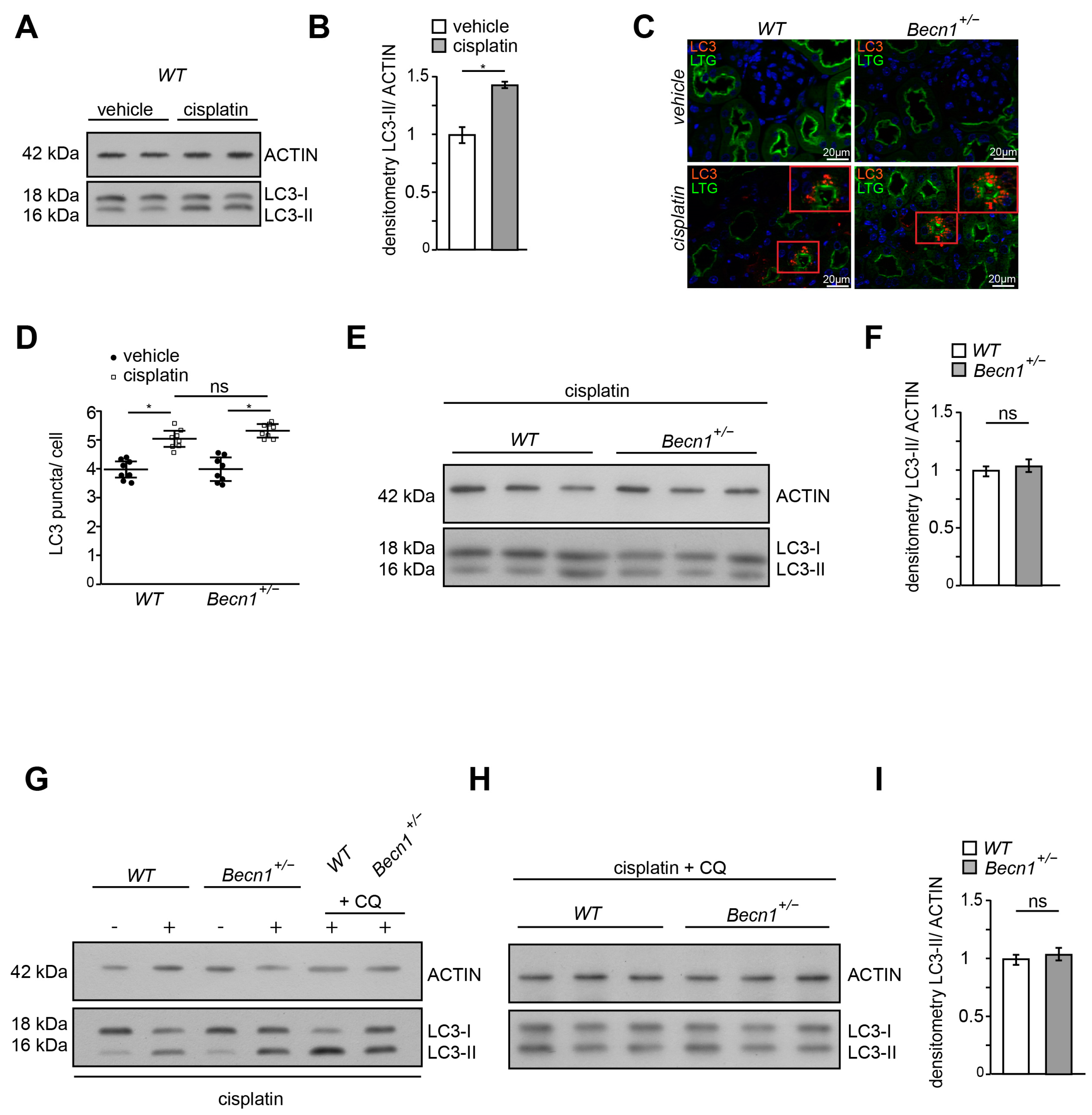
2.3. Cisplatin Induces Autophagy Independent from BECLIN1 Abundance In Vivo
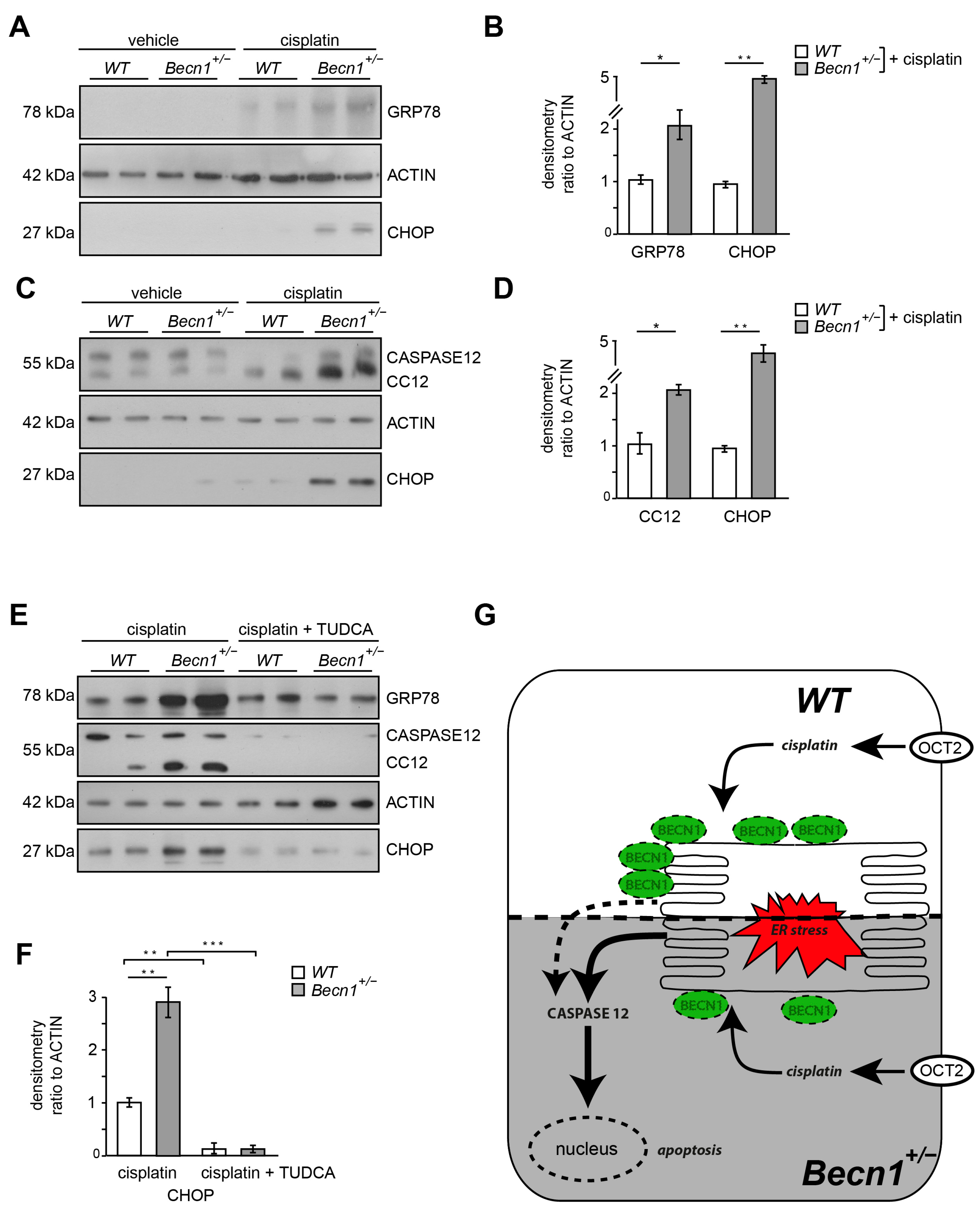
2.4. Cisplatin-Induced ER Stress Depends on BECLIN1 and Activates the Apoptotic Pathway
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Urine and Serum Analysis
4.3. Immunofluorescence Staining
4.4. Histology
4.5. Cell Lysis and Western Blot Procedures
4.6. Primary Cell Isolation and Cell Culture
4.7. Annexin Propidium Iodide Staining
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Kellum, J.A.; Lameire, N. Diagnosis, evaluation, and management of acute kidney injury: A KDIGO summary (Part 1). Crit. Care 2013, 17, 204. [Google Scholar] [CrossRef]
- Wald, R.; Quinn, R.R.; Luo, J.; Li, P.; Scales, D.C.; Mamdani, M.M.; Ray, J.G. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA 2009, 302, 1179–1185. [Google Scholar] [CrossRef]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. JASN 2009, 20, 223–228. [Google Scholar] [CrossRef]
- Einhorn, L.H.; Donohue, J. Cis-diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann. Intern. Med. 1977, 87, 293–298. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, J.; Zhu, Z.; Zhao, W.; Zhou, J.; Wu, C.; Tang, H.; Fan, M.; Li, L.; Lin, Q.; et al. Comparing Paclitaxel Plus Fluorouracil Versus Cisplatin Plus Fluorouracil in Chemoradiotherapy for Locally Advanced Esophageal Squamous Cell Cancer: A Randomized, Multicenter, Phase III Clinical Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 1695–1703. [Google Scholar] [CrossRef]
- Liu, G.F.; Li, G.J.; Zhao, H. Efficacy and Toxicity of Different Chemotherapy Regimens in the Treatment of Advanced or Metastatic Pancreatic Cancer: A Network Meta-Analysis. J. Cell. Biochem. 2018, 119, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, B.; Fu, Z.; Wang, X.; Zhang, Z. Efficacy and safety of oxaliplatin-based regimen versus cisplatin-based regimen in the treatment of gastric cancer: A meta-analysis of randomized controlled trials. Int. J. Clin. Oncol. 2019, 24, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, G.; Boland, A.; Brown, T.; Oyee, J.; Bagust, A.; Dickson, R. A systematic review of the clinical effectiveness of first-line chemotherapy for adult patients with locally advanced or metastatic non-small cell lung cancer. Thorax 2015, 70, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dahal, A.; Bellows, B.K.; Sonpavde, G.; Tantravahi, S.K.; Choueiri, T.K.; Galsky, M.D.; Agarwal, N. Incidence of Severe Nephrotoxicity With Cisplatin Based on Renal Function Eligibility Criteria: Indirect Comparison Meta-analysis. Am. J. Clin. Oncol. 2016, 39, 497–506. [Google Scholar] [CrossRef]
- Latcha, S.; Jaimes, E.A.; Patil, S.; Glezerman, I.G.; Mehta, S.; Flombaum, C.D. Long-Term Renal Outcomes after Cisplatin Treatment. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, S.; Yonezawa, A.; Masuda, S.; Fukatsu, A.; Katsura, T.; Inui, K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 2007, 74, 477–487. [Google Scholar] [CrossRef]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the human organic cation transporter 2. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 3875–3880. [Google Scholar] [CrossRef]
- Safirstein, R.; Miller, P.; Guttenplan, J.B. Uptake and metabolism of cisplatin by rat kidney. Kidney Int. 1984, 25, 753–758. [Google Scholar] [CrossRef]
- Xu, E.Y.; Perlina, A.; Vu, H.; Troth, S.P.; Brennan, R.J.; Aslamkhan, A.G.; Xu, Q. Integrated pathway analysis of rat urine metabolic profiles and kidney transcriptomic profiles to elucidate the systems toxicology of model nephrotoxicants. Chem. Res. Toxicol. 2008, 21, 1548–1561. [Google Scholar] [CrossRef]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiology. Ren. Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar] [CrossRef]
- Liu, H.; Baliga, R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J. Am. Soc. Nephrol. JASN 2005, 16, 1985–1992. [Google Scholar] [CrossRef]
- Sinha, S.; Levine, B. The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene 2008, 27 (Suppl. 1), S137–S148. [Google Scholar] [CrossRef]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Bork, T.; Liang, W.; Kretz, O.; Lagies, S.; Yamahara, K.; Hernando-Erhard, C.; Helmstädter, M.; Schell, C.; Kammerer, B.; Huber, T.B. BECLIN1 Is Essential for Podocyte Secretory Pathways Mediating VEGF Secretion and Podocyte-Endothelial Crosstalk. Int. J. Mol. Sci. 2022, 23, 3825. [Google Scholar] [CrossRef] [PubMed]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef] [PubMed]
- Anwar, T.; Liu, X.; Suntio, T.; Marjamäki, A.; Biazik, J.; Chan, E.Y.W.; Varjosalo, M.; Eskelinen, E.L. ER-Targeted Beclin 1 Supports Autophagosome Biogenesis in the Absence of ULK1 and ULK2 Kinases. Cells 2019, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Lippens, S.; Vanden Berghe, T.; Romagnoli, A.; Fimia, G.M.; Piacentini, M.; Vandenabeele, P. Beclin1: A role in membrane dynamics and beyond. Autophagy 2012, 8, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Klionsky, D.J. Physiological functions of Atg6/Beclin 1: A unique autophagy-related protein. Cell Res. 2007, 17, 839–849. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta 2011, 1813, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, M.; Glab, J.A.; Puthalakath, H. BH3-only proteins: A 20-year stock-take. FEBS J. 2015, 282, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Gawriluk, T.R.; Ko, C.; Hong, X.; Christenson, L.K.; Rucker, E.B., 3rd. Beclin-1 deficiency in the murine ovary results in the reduction of progesterone production to promote preterm labor. Proc. Natl. Acad. Sci. USA 2014, 111, E4194–E4203. [Google Scholar] [CrossRef] [PubMed]
- McKnight, N.C.; Zhong, Y.; Wold, M.S.; Gong, S.; Phillips, G.R.; Dou, Z.; Zhao, Y.; Heintz, N.; Zong, W.X.; Yue, Z. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014, 10, e1004626. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, K.; Luo, J.; Dong, Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am. J. Pathol. 2010, 176, 1181–1192. [Google Scholar] [CrossRef]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef]
- Zhou, Q.; Doherty, J.; Akk, A.; Springer, L.E.; Fan, P.; Spasojevic, I.; Halade, G.V.; Yang, H.; Pham, C.T.N.; Wickline, S.A.; et al. Safety Profile of Rapamycin Perfluorocarbon Nanoparticles for Preventing Cisplatin-Induced Kidney Injury. Nanomaterials 2022, 12, 336. [Google Scholar] [CrossRef]
- Zhou, Q.; Quirk, J.D.; Hu, Y.; Yan, H.; Gaut, J.P.; Pham, C.T.N.; Wickline, S.A.; Pan, H. Rapamycin Perfluorocarbon Nanoparticle Mitigates Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2023, 24, 6086. [Google Scholar] [CrossRef]
- Hu, X.; Ma, Z.; Wen, L.; Li, S.; Dong, Z. Autophagy in Cisplatin Nephrotoxicity during Cancer Therapy. Cancers 2021, 13, 5618. [Google Scholar] [CrossRef]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.M.; Dong, Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008, 74, 631–640. [Google Scholar] [CrossRef]
- Inoue, K.; Kuwana, H.; Shimamura, Y.; Ogata, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T.; Morita, T.; Sasaki, S.; et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin. Exp. Nephrol. 2010, 14, 112–122. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Goemans, G.C.; Skepper, J.N.; Tolkovsky, A.M. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009, 28, 2128–2141. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Mau, T.; O’Brien, M.; Garg, S.; Yung, R. Impaired autophagy activity is linked to elevated ER-stress and inflammation in aging adipose tissue. Aging 2016, 8, 2525–2537. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Zhang, C.W.; Singh, B.K.; Sinha, R.A.; Moe, K.T.; DeSilva, D.A.; Yen, P.M. Hyperhomocysteinemia causes ER stress and impaired autophagy that is reversed by Vitamin B supplementation. Cell Death Dis. 2016, 7, e2513. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Zhang, Z.; Duan, K.; Shi, W.; Huang, R.; Wang, B.; Luo, L.; Zhang, Y.; Ruan, H.; Huang, H. Beclin 1 deficiency causes hepatic cell apoptosis via endoplasmic reticulum stress in zebrafish larvae. FEBS Lett. 2020, 594, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Shu, S.; Guo, C.; Tang, C.; Dong, Z. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann. Med. 2018, 50, 381–390. [Google Scholar] [CrossRef]
- Inagi, R. Endoplasmic reticulum stress as a progression factor for kidney injury. Curr. Opin. Pharmacol. 2010, 10, 156–165. [Google Scholar] [CrossRef]
- Fan, Y.; Xiao, W.; Lee, K.; Salem, F.; Wen, J.; He, L.; Zhang, J.; Fei, Y.; Cheng, D.; Bao, H.; et al. Inhibition of Reticulon-1A-Mediated Endoplasmic Reticulum Stress in Early AKI Attenuates Renal Fibrosis Development. J. Am. Soc. Nephrol. JASN 2017, 28, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Peyrou, M.; Hanna, P.E.; Cribb, A.E. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 99, 346–353. [Google Scholar] [CrossRef]
- Long, Y.; Zhen, X.; Zhu, F.; Hu, Z.; Lei, W.; Li, S.; Zha, Y.; Nie, J. Hyperhomocysteinemia Exacerbates Cisplatin-induced Acute Kidney Injury. Int. J. Biol. Sci. 2017, 13, 219–231. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef] [PubMed]
- Ruck, A.; Attonito, J.; Garces, K.T.; Núnez, L.; Palmisano, N.J.; Rubel, Z.; Bai, Z.; Nguyen, K.C.; Sun, L.; Grant, B.D.; et al. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy 2011, 7, 386–400. [Google Scholar] [CrossRef]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Safaei, R.; Katano, K.; Larson, B.J.; Samimi, G.; Holzer, A.K.; Naerdemann, W.; Tomioka, M.; Goodman, M.; Howell, S.B. Intracellular localization and trafficking of fluorescein-labeled cisplatin in human ovarian carcinoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 756–767. [Google Scholar] [CrossRef]
- Katano, K.; Safaei, R.; Samimi, G.; Holzer, A.; Tomioka, M.; Goodman, M.; Howell, S.B. Confocal microscopic analysis of the interaction between cisplatin and the copper transporter ATP7B in human ovarian carcinoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 4578–4588. [Google Scholar] [CrossRef]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, H.B.; Fang, Y.L.; Zhang, Z.R.; Shao, J.; Hong, M.; Huang, C.J.; Liu, J.; Chen, R.Q. Cisplatin-induced necroptosis in TNFα dependent and independent pathways. Cell. Signal. 2017, 31, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Onyshchenko, K.; Wang, L.; Gaedicke, S.; Grosu, A.L.; Firat, E.; Niedermann, G. Necroptosis-dependent Immunogenicity of Cisplatin: Implications for Enhancing the Radiation-induced Abscopal Effect. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023, 29, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Alassaf, N.; Attia, H. Autophagy and necroptosis in cisplatin-induced acute kidney injury: Recent advances regarding their role and therapeutic potential. Front. Pharmacol. 2023, 14, 1103062. [Google Scholar] [CrossRef]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef]
- Ramesh, G.; Reeves, W.B. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842. [Google Scholar] [CrossRef]
- Zhang, B.; Ramesh, G.; Norbury, C.C.; Reeves, W.B. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44. [Google Scholar] [CrossRef]
- Zhang, B.; Ramesh, G.; Uematsu, S.; Akira, S.; Reeves, W.B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol. JASN 2008, 19, 923–932. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, Z.; He, L.; Wang, Y. CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget 2016, 7, 31652–31662. [Google Scholar] [CrossRef]
- Liu, P.; Li, X.; Lv, W.; Xu, Z. Inhibition of CXCL1-CXCR2 axis ameliorates cisplatin-induced acute kidney injury by mediating inflammatory response. Biomed. Pharmacother. = Biomed. Pharmacother. 2020, 122, 109693. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef]
- Faruqi, M. Peptides: Activating autophagy. Nat. Rev. Drug Discov. 2013, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Rubera, I.; Poujeol, C.; Bertin, G.; Hasseine, L.; Counillon, L.; Poujeol, P.; Tauc, M. Specific Cre/Lox recombination in the mouse proximal tubule. J. Am. Soc. Nephrol. JASN 2004, 15, 2050–2056. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Conway, E.M.; Harris, R.C. Survivin mediates renal proximal tubule recovery from AKI. J. Am. Soc. Nephrol. JASN 2013, 24, 2023–2033. [Google Scholar] [CrossRef]
- Yakulov, T.A.; Todkar, A.P.; Slanchev, K.; Wiegel, J.; Bona, A.; Groß, M.; Scholz, A.; Hess, I.; Wurditsch, A.; Grahammer, F.; et al. CXCL12 and MYC control energy metabolism to support adaptive responses after kidney injury. Nat. Commun. 2018, 9, 3660. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bork, T.; Hernando-Erhard, C.; Liang, W.; Tian, Z.; Yamahara, K.; Huber, T.B. Cisplatin Nephrotoxicity Is Critically Mediated by the Availability of BECLIN1. Int. J. Mol. Sci. 2024, 25, 2560. https://doi.org/10.3390/ijms25052560
Bork T, Hernando-Erhard C, Liang W, Tian Z, Yamahara K, Huber TB. Cisplatin Nephrotoxicity Is Critically Mediated by the Availability of BECLIN1. International Journal of Molecular Sciences. 2024; 25(5):2560. https://doi.org/10.3390/ijms25052560
Chicago/Turabian StyleBork, Tillmann, Camila Hernando-Erhard, Wei Liang, Zhejia Tian, Kosuke Yamahara, and Tobias B. Huber. 2024. "Cisplatin Nephrotoxicity Is Critically Mediated by the Availability of BECLIN1" International Journal of Molecular Sciences 25, no. 5: 2560. https://doi.org/10.3390/ijms25052560
APA StyleBork, T., Hernando-Erhard, C., Liang, W., Tian, Z., Yamahara, K., & Huber, T. B. (2024). Cisplatin Nephrotoxicity Is Critically Mediated by the Availability of BECLIN1. International Journal of Molecular Sciences, 25(5), 2560. https://doi.org/10.3390/ijms25052560