
Glioblastoma-Associated Mesenchymal Stem/Stromal Cells and Cancer-Associated Fibroblasts: Partners in Crime?

 ,
,
Abstract
1. Introduction
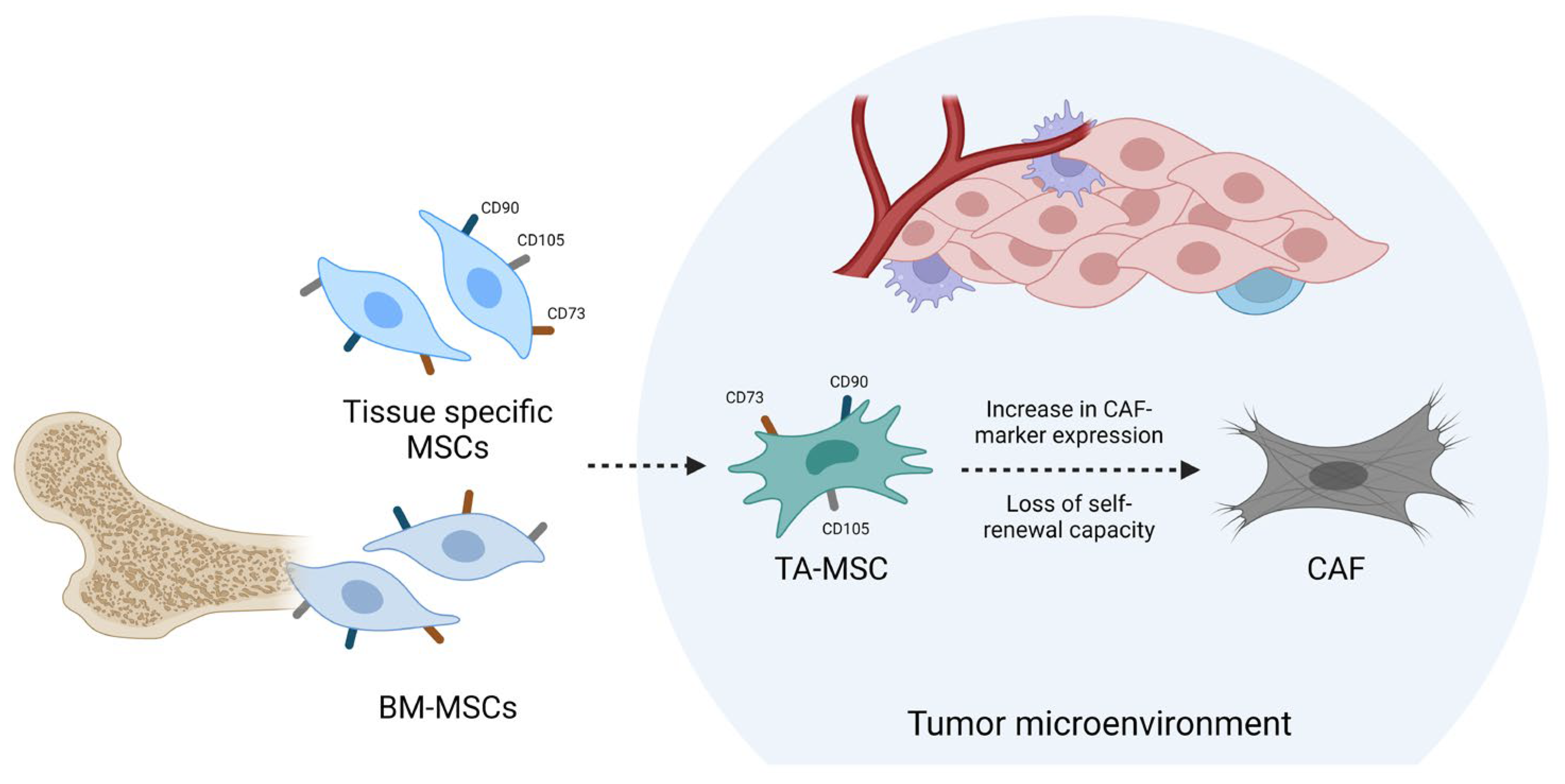
2. TA-MSCs as CAF Precursors
3. Homing of MSCs and Differentiation into GA-MSCs
4. GA-MSCs Modulate the Tumor Microenvironment
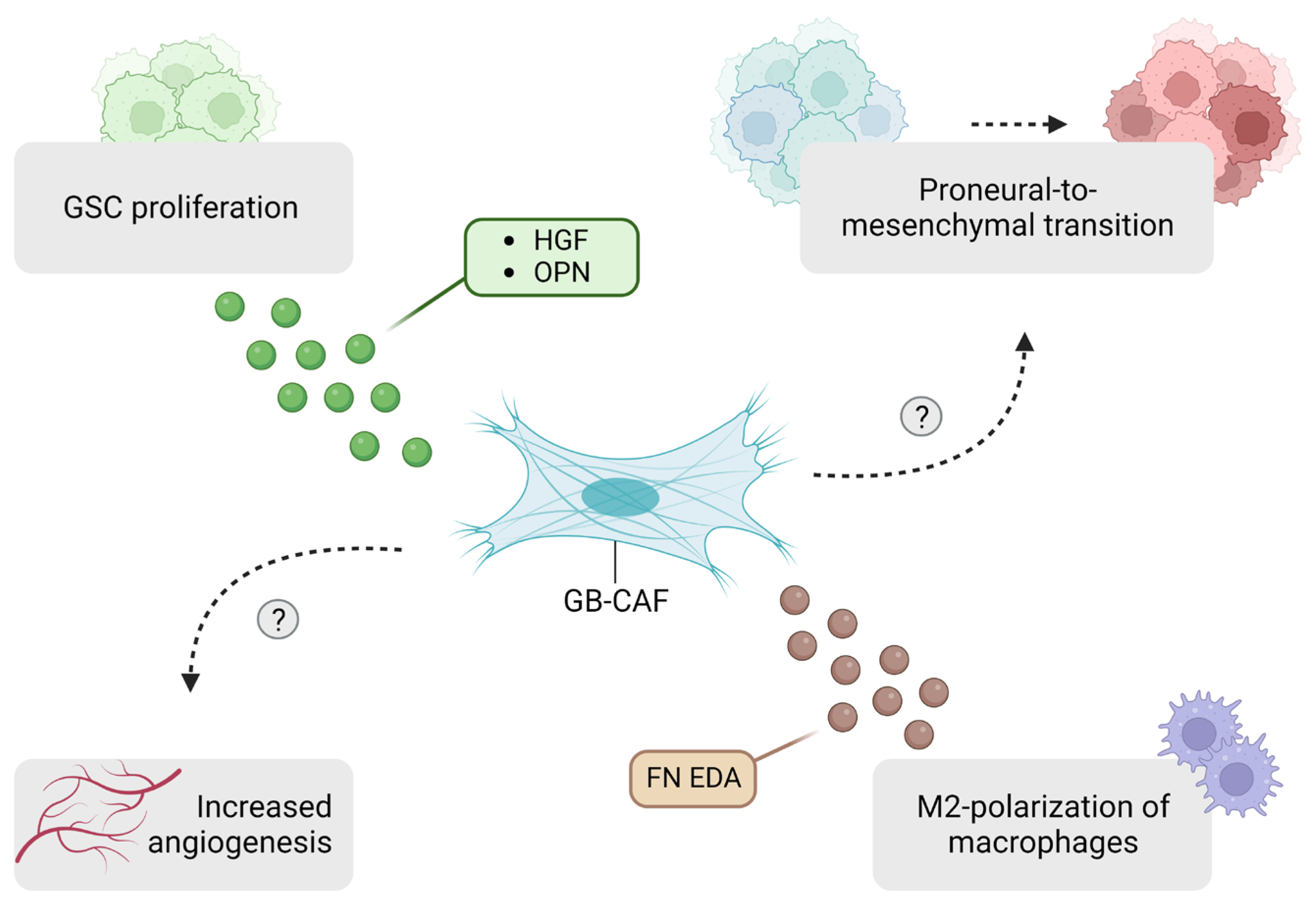
4.1. Tumor Growth and Invasion
4.2. Angiogenesis
4.3. GSC Crosstalk
4.4. Mitochondrial Transfer and Resistance to Therapy
4.5. Effects on the Immune Compartment
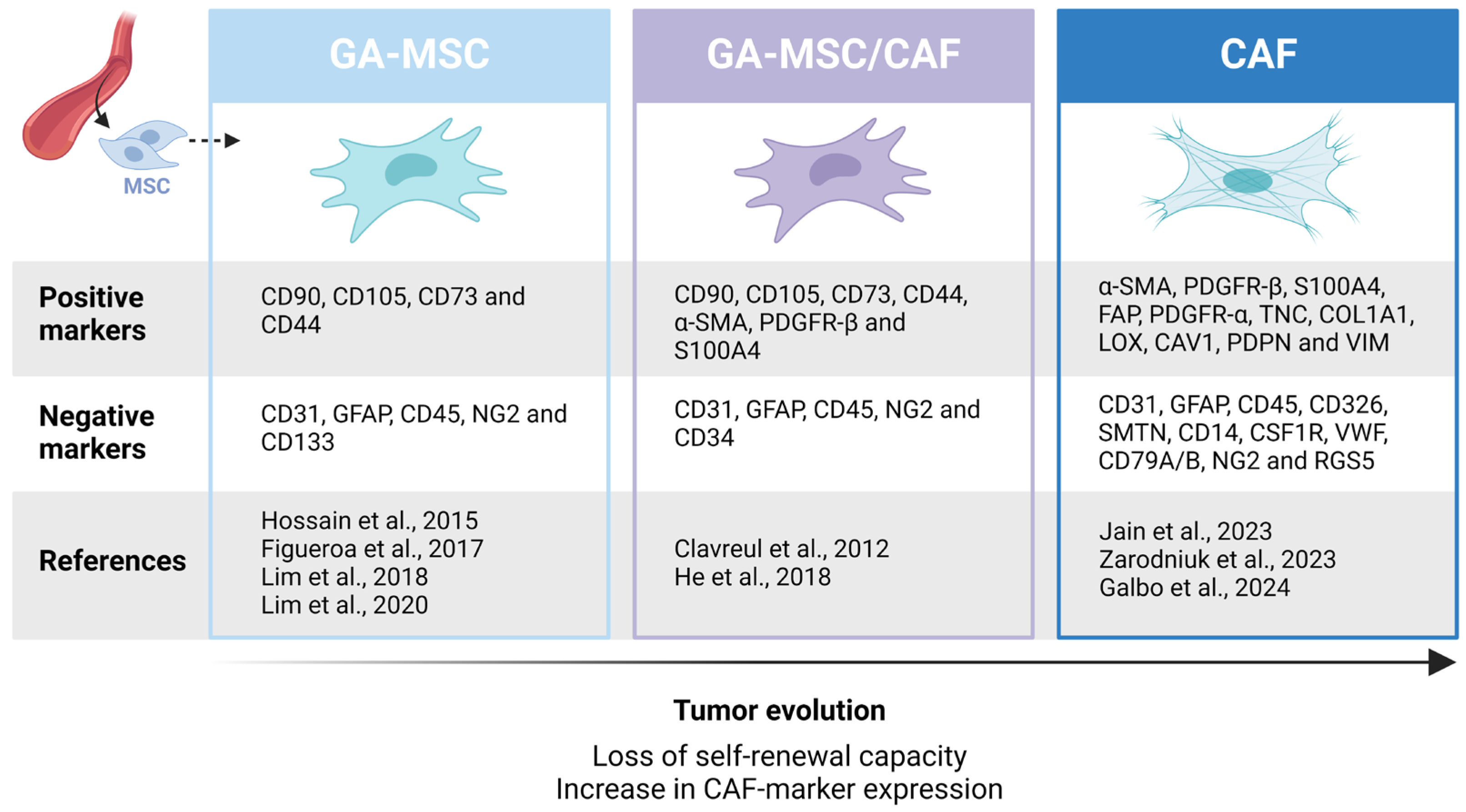
5. GA-MSC Subpopulations
6. Differentiation of GA-MSCs into CAFs
6.1. CAF-Marker Expression in GB
6.2. Identification of GB-Associated CAFs
6.3. GA-MSCs as CAF Precursors
6.4. A Shared Origin with Pericytes
7. CAF Subtypes in GB
8. Open Questions and Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| BBB | Blood-brain barrier |
| BM-MSC | Bone marrow-derived mesenchymal stem cell |
| CAF | Cancer-associated fibroblast |
| CCR2 | CC-chemokine receptor 2 |
| ECM | Extracellular matrix |
| EDA | Extra domain A |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| EVs | Extracellular vesicles |
| FAP | Fibroblast activation protein |
| FN | Fibronectin |
| GA-MSC | Glioblastoma-associated mesenchymal stem/stromal cell |
| GB | Glioblastoma |
| GSC | Glioblastoma stem cell |
| HA | Hyaluronic acid |
| HGF | Hepatocyte growth factor |
| IDH | Isocitrate dehydrogenase |
| IM | Immunomodulatory |
| LOX | Lysyl oxidase |
| MDSC | Myeloid-derived suppressor cell |
| MMP1 | Matrix metalloproteinase 1 |
| MR | Mechanoresponsive |
| MSC | Mesenchymal stem cell |
| NSC | Neural stem cell |
| OPN | Osteopontin |
| PDCL | Patient-derived cell line |
| RNA-seq | RNA sequencing |
| scRNA-seq | Single-cell RNA sequencing |
| SDF-1 | Stromal-derived factor-1 |
| SSL | Steady state-like |
| TAM | Tumor-associated macrophage |
| TA-MSC | Tumor-associated mesenchymal stem/stromal cell |
| TME | Tumor microenvironment |
| TMZ | Temozolomide |
| TNBC | Triple-negative breast cancer |
| TNT | Tunnelling nanotubes |
| VIM | Vimentin |
References
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular origin of glioblastoma and its implication in precision therapy. Cell. Mol. Immunol. 2018, 15, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Osborn, A.G.; Louis, D.N.; Poussaint, T.Y.; Linscott, L.L.; Salzman, K.L. The 2021 World Health Organization Classification of Tumors of the Central Nervous System: What Neuroradiologists Need to Know. Am. J. Neuroradiol. 2022, 43, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. CNS cancer: Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.D.; Malta, T.M.; Wade, T.E.; Sabedot, T.S.; et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 2022, 185, 2184–2199.e16. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Antonio Chiocca, E.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro. Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Jiyong, L.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2016, 16, 35–52. [Google Scholar] [CrossRef]
- Frisbie, L.; Buckanovich, R.J.; Coffman, L. Carcinoma-Associated Mesenchymal Stem/Stromal Cells: Architects of the Pro-tumorigenic Tumor Microenvironment. Stem Cells 2022, 40, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Kim, S.; Oh, Y.; Suh, Y.; Kaushik, N.; Lee, J.H.; Lee, H.J.; Kim, M.J.; Park, M.J.; Kim, R.K.; et al. Crosstalk between GBM cells and mesenchymal stemlike cells promotes the invasiveness of GBM through the C5a/p38/ZEB1 axis. Neuro. Oncol. 2020, 22, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.G.; Joyce, C.; et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.H.; Do Shin, H.; Kim, S.H.; Mok, H.S.; Shim, J.K.; Lee, J.H.; Shin, H.J.; Huh, Y.M.; Kim, E.H.; Park, E.K.; et al. Increased in vivo angiogenic effect of glioma stromal mesenchymal stem-like cells on glioma cancer stem cells from patients with glioblastoma. Int. J. Oncol. 2013, 42, 1754–1762. [Google Scholar] [CrossRef]
- Nakhle, J.; Khattar, K.; Özkan, T.; Boughlita, A.; Abba Moussa, D.; Darlix, A.; Lorcy, F.; Rigau, V.; Bauchet, L.; Gerbal-Chaloin, S.; et al. Mitochondria Transfer from Mesenchymal Stem Cells Confers Chemoresistance to Glioblastoma Stem Cells through Metabolic Rewiring. Cancer Res. Commun. 2023, 3, 1041–1056. [Google Scholar] [CrossRef]
- Peng, Z.; Wu, Y.; Wang, J.; Gu, S.; Wang, Y.; Xue, B.; Fu, P.; Xiang, W. Development and validation of a glioma-associated mesenchymal stem cell-related gene prognostic index for predicting prognosis and guiding individualized therapy in glioma. Stem Cell Res. Ther. 2023, 14, 56. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 1795. [Google Scholar] [CrossRef] [PubMed]
- Coffman, L.G.; Pearson, A.T.; Frisbie, L.G.; Freeman, Z.; Christie, E.; Bowtell, D.D.; Buckanovich, R.J. Ovarian Carcinoma-Associated Mesenchymal Stem Cells Arise from Tissue-Specific Normal Stroma. Stem Cells 2019, 37, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Liu, Y.; Zhao, X.; Zhang, J.; Zheng, B.; Yuan, Z.R.; Zhang, L.; Qu, X.; Tischfield, J.A.; Shao, C.; et al. Tumor resident mesenchymal stromal cells endow naïve stromal cells with tumor-promoting properties. Oncogene 2014, 33, 4016–4020. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Rick, J.W.; Joshi, R.S.; Beniwal, A.; Spatz, J.; Gill, S.; Chang, A.C.; Choudhary, N.; Nguyen, A.T.; Sudhir, S.; et al. Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. J. Clin. Investig. 2023, 133, e147087. [Google Scholar] [CrossRef] [PubMed]
- Galbo, P.M.; Madsen, A.T.; Liu, Y.; Peng, M.; Wei, Y.; Ciesielski, M.J.; Fenstermaker, R.A.; Graff, S.; Montagna, C.; Segall, J.E.; et al. Functional Contribution and Clinical Implication of Cancer-Associated Fibroblasts in Glioblastoma. Clin. Cancer Res. 2024. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef]
- Khan, F.; Pang, L.; Dunterman, M.; Lesniak, M.S.; Heimberger, A.B.; Chen, P. Macrophages and microglia in glioblastoma: Heterogeneity, plasticity, and therapy. J. Clin. Investig. 2023, 133, e163446. [Google Scholar] [CrossRef]
- Zarodniuk, M.; Steele, A.; Lu, X.; Li, J.; Datta, M. CNS tumor stroma transcriptomics identify perivascular fibroblasts as predictors of immunotherapy resistance in glioblastoma patients. NPJ Genomic Med. 2023, 8, 35. [Google Scholar] [CrossRef]
- Schaffenrath, J.; Wyss, T.; He, L.; Rushing, E.J.; Delorenzi, M.; Vasella, F.; Regli, L.; Neidert, M.C.; Keller, A. Blood-brain barrier alterations in human brain tumors revealed by genome-wide transcriptomic profiling. Neuro. Oncol. 2021, 23, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, M.; Lu, W.; Tang, J.; Deng, L.; Wen, Q.; Huang, M.; Deng, R.; Ye, G.; Ye, W.; et al. Targeting FAPα-expressing tumor-associated mesenchymal stromal cells inhibits triple-negative breast cancer pulmonary metastasis. Cancer Lett. 2021, 503, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Salati, M.; Sorgentoni, G.; Barbisan, F.; Orciani, M. Characterization of tumor-derived mesenchymal stem cells potentially differentiating into cancer-associated fibroblasts in lung cancer. Clin. Transl. Oncol. 2018, 20, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Ito, Y.; Kawano, Y.; Kurozumi, K.; Kobune, M.; Tsuda, H.; Bizen, A.; Honmou, O.; Niitsu, Y.; Hamada, H. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004, 11, 1155–1164. [Google Scholar] [CrossRef]
- Nakamizo, A.; Marini, F.; Amano, T.; Khan, A.; Studeny, M.; Gumin, J.; Chen, J.; Hentschel, S.; Vecil, G.; Dembinski, J.; et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005, 65, 3307–3318. [Google Scholar] [CrossRef]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [Google Scholar] [CrossRef]
- Li, M.; Zeng, L.; Liu, S.; Dangelmajer, S.; Kahlert, U.D.; Huang, H.; Han, Y.; Chi, X.; Zhu, M.; Lei, T. Transforming growth factor-β promotes homing and therapeutic efficacy of human mesenchymal stem cells to glioblastoma. J. Neuropathol. Exp. Neurol. 2019, 78, 315–325. [Google Scholar] [CrossRef]
- Aghi, M.; Cohen, K.S.; Klein, R.J.; Scadden, D.T.; Chiocca, E.A. Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res. 2006, 66, 9054–9064. [Google Scholar] [CrossRef]
- Ho, I.A.W.; Chan, K.Y.W.; Ng, W.H.; Guo, C.M.; Hui, K.M.; Cheang, P.; Lam, P.Y.P. Matrix metalloproteinase 1 is necessary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells 2009, 27, 1366–1375. [Google Scholar] [CrossRef]
- Vogel, S.; Peters, C.; Etminan, N.; Börger, V.; Schimanski, A.; Sabel, M.C.; Sorg, R.V. Migration of mesenchymal stem cells towards glioblastoma cells depends on hepatocyte-growth factor and is enhanced by aminolaevulinic acid-mediated photodynamic treatment. Biochem. Biophys. Res. Commun. 2013, 431, 428–432. [Google Scholar] [CrossRef]
- Pietrobono, D.; Giacomelli, C.; Marchetti, L.; Martini, C.; Trincavelli, M.L. High adenosine extracellular levels induce glioblastoma aggressive traits modulating the mesenchymal stromal cell secretome. Int. J. Mol. Sci. 2020, 21, 7706. [Google Scholar] [CrossRef]
- Lou, Q.; Zhao, M.; Xu, Q.; Xie, S.; Liang, Y.; Chen, J.; Yuan, L.; Wang, L.; Jiang, L.; Mou, L.; et al. Retinoic Acid Inhibits Tumor-Associated Mesenchymal Stromal Cell Transformation in Melanoma. Front. Cell Dev. Biol. 2021, 9, 658757. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; You, C.; Zhao, D. Long non-coding RNA UCA1/miR-182/PFKFB2 axis modulates glioblastoma-associated stromal cells-mediated glycolysis and invasion of glioma cells. Biochem. Biophys. Res. Commun. 2018, 500, 569–576. [Google Scholar] [CrossRef]
- Novak, M.; Krajnc, M.K.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-mediated signaling is involved in invasion of glioblastoma cells in its microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef]
- Lim, E.J.; Suh, Y.; Yoo, K.C.; Lee, J.H.; Kim, I.G.; Kim, M.J.; Chang, J.H.; Kang, S.G.; Lee, S.J. Tumor-associated mesenchymal stem-like cells provide extracellular signaling cue for invasiveness of glioblastoma cells. Oncotarget 2017, 8, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Suh, Y.; Kim, S.; Kang, S.G.; Lee, S.J. Force-mediated proinvasive matrix remodeling driven by tumor-associated mesenchymal stem-like cells in glioblastoma. BMB Rep. 2018, 51, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lim, E.; Yoo, K.; Zhao, Y.; Kang, J.; Lim, E.; Shin, I.; Kang, S.; Lim, H.W.; Lee, S. Glioblastoma-educated mesenchymal stem-like cells promote glioblastoma infiltration via extracellular matrix remodelling in the tumour microenvironment. Clin. Transl. Med. 2022, 12, e997. [Google Scholar] [CrossRef]
- Coy, S.; Wang, S.; Stopka, S.A.; Lin, J.R.; Yapp, C.; Ritch, C.C.; Salhi, L.; Baker, G.J.; Rashid, R.; Baquer, G.; et al. Single cell spatial analysis reveals the topology of immunomodulatory purinergic signaling in glioblastoma. Nat. Commun. 2022, 13, 4814. [Google Scholar] [CrossRef]
- Das, S.; Marsden, P.A. Angiogenesis in Glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef]
- Clavreul, A.; Guette, C.; Faguer, R.; Tétaud, C.; Boissard, A.; Lemaire, L.; Rousseau, A.; Avril, T.; Henry, C.; Coqueret, O.; et al. Glioblastoma-associated stromal cells (GASCs) from histologically normal surgical margins have a myofibroblast phenotype and angiogenic properties. J. Pathol. 2014, 233, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T.; et al. Exosomes from Glioma-Associated Mesenchymal Stem Cells Increase the Tumorigenicity of Glioma Stem-like Cells via Transfer of miR-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.S.; Lou, E. Tunneling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep. 2019, 2, e1185. [Google Scholar] [CrossRef]
- Venkataramani, V.; Schneider, M.; Giordano, F.A.; Kuner, T.; Wick, W.; Herrlinger, U.; Winkler, F. Disconnecting multicellular networks in brain tumours. Nat. Rev. Cancer 2022, 22, 481–491. [Google Scholar] [CrossRef]
- Salaud, C.; Alvarez-Arenas, A.; Geraldo, F.; Belmonte-Beitia, J.; Calvo, G.F.; Gratas, C.; Pecqueur, C.; Garnier, D.; Pérez-Garcià, V.; Vallette, F.M.; et al. Mitochondria transfer from tumor-activated stromal cells (TASC) to primary Glioblastoma cells. Biochem. Biophys. Res. Commun. 2020, 533, 139–147. [Google Scholar] [CrossRef]
- Xue, B.; Xiang, W.; Zhang, Q.; Wang, H.; Zhou, Y.; Tian, H.; Abdelmaksou, A.; Xue, J.; Sun, M.; Yi, D.; et al. CD90low glioma-associated mesenchymal stromal/stem cells promote temozolomide resistance by activating FOXS1-mediated epithelial-mesenchymal transition in glioma cells. Stem Cell Res. Ther. 2021, 12, 394. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tu, M.; Wang, L. Pan-Cancer Analysis Predicts FOXS1 as a Key Target in Prognosis and Tumor Immunotherapy. Int. J. Gen. Med. 2022, 15, 2171–2185. [Google Scholar] [CrossRef]
- Shahar, T.; Rozovski, U.; Hess, K.R.; Hossain, A.; Gumin, J.; Gao, F.; Fuller, G.N.; Goodman, L.; Sulman, E.P.; Lang, F.F. Percentage of mesenchymal stem cells in high-grade glioma tumor samples correlates with patient survival. Neuro. Oncol. 2017, 19, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Svensson, A.; Ramos-Moreno, T.; Eberstål, S.; Scheding, S.; Bengzon, J. Identification of two distinct mesenchymal stromal cell populations in human malignant glioma. J. Neurooncol. 2017, 131, 245–254. [Google Scholar] [CrossRef]
- Zhao, H.; Donna, M.P. Tumor-Promoting Phenotype of CD90hi Prostate Cancer-Associated Fibroblasts. Prostate 2009, 69, 991–1000. [Google Scholar] [CrossRef]
- Clavreul, A.; Etcheverry, A.; Tétaud, C.; Rousseau, A.; Avril, T.; Henry, C.; Mosser, J.; Menei, P. Identification of two glioblastoma-associated stromal cell subtypes with different carcinogenic properties in histologically normal surgical margins. J. Neurooncol. 2015, 122, 1–10. [Google Scholar] [CrossRef]
- Trylcova, J.; Busek, P.; Smetana, K.; Balaziova, E.; Dvorankova, B.; Mifkova, A.; Sedo, A. Effect of cancer-associated fibroblasts on the migration of glioma cells in vitro. Tumor Biol. 2015, 36, 5873–5879. [Google Scholar] [CrossRef] [PubMed]
- Balaziova, E.; Vymola, P.; Hrabal, P.; Mateu, R.; Zubal, M.; Tomas, R.; Netuka, D.; Kramar, F.; Zemanova, Z.; Svobodova, K.; et al. Fibroblast activation protein expressing mesenchymal cells promote glioblastoma angiogenesis. Cancers 2021, 13, 3304. [Google Scholar] [CrossRef]
- Busek, P.; Balaziova, E.; Matrasova, I.; Hilser, M.; Tomas, R.; Syrucek, M.; Zemanova, Z.; Krepela, E.; Belacek, J.; Sedo, A. Fibroblast activation protein alpha is expressed by transformed and stromal cells and is associated with mesenchymal features in glioblastoma. Tumor Biol. 2016, 37, 13961–13971. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, G.; Kiyokawa, J.; Tirmizi, Z.; Richardson, L.G.; Ning, J.; Das, S.; Martuza, R.L.; Stemmer-Rachamimov, A.; Rabkin, S.D.; et al. Characterization and oncolytic virus targeting of FAP-expressing tumor-associated pericytes in glioblastoma. Acta Neuropathol. Commun. 2020, 8, 221. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, V.G.; Trinh, D.L.; Aslanpour, S.; Hughes, M.; Livingstone, D.; Jin, D.; Ahn, B.Y.; Blough, M.D.; Cairncross, J.G.; Chan, J.A.; et al. Single-cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter- and intratumor heterogeneity. Cancer Cell 2022, 40, 379–392.e9. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Wick, N.; Hantusch, B.; Novatchkova, M.; Taubenschmid, J.; Hämmerle, M.; Esk, C.; Bagley, J.A.; et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 2019, 565, 505–510. [Google Scholar] [CrossRef]
- Goodpaster, T.; Legesse-Miller, A.; Hameed, M.R.; Aisner, S.C.; Randolph-Habecker, J.; Coller, H.A. An immunohistochemical method for identifying fibroblasts in formalin-fixed, paraffin-embedded tissue. J. Histochem. Cytochem. 2008, 56, 347–358. [Google Scholar] [CrossRef]
- Davidson, C.; Nel, S.; Heerden, M.B.V.; Heerden, W.V. Immunohistochemical Expression of Fibroblast Marker TE-7 in Human Dental Pulp Cells. J. Med. Lab. Sci. Technol. S. Afr. 2019, 1, 6–11. [Google Scholar]
- Zhao, J.; Yang, S.; Lv, C.; Liu, Y. Cancer associated fibroblasts suppressed ferroptosis in glioblastoma via upregulating lncRNA DLEU1. Am. J. Physiol. Physiol. 2023, 324, C1039–C1052. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef] [PubMed]
- Oliver, L.; Álvarez-Arenas, A.; Salaud, C.; Jiménez-Sanchez, J.; Calvo, G.F.; Belmonte-Beitia, J.; Blandin, S.; Vidal, L.; Pérez, V.; Heymann, D.; et al. A Simple 3D Cell Culture Method for Studying the Interactions between Human Mesenchymal Stromal/Stem Cells and Patients Derived Glioblastoma. Cancers 2023, 15, 1304. [Google Scholar] [CrossRef]
- Garnier, D.; Ratcliffe, E.; Briand, J.; Cartron, P.F.; Oliver, L.; Vallette, F.M. The Activation of Mesenchymal Stem Cells by Glioblastoma Microvesicles Alters Their Exosomal Secretion of miR-100-5p, miR-9-5p and let-7d-5p. Biomedicines 2022, 10, 112. [Google Scholar] [CrossRef]
- Clavreul, A.; Etcheverry, A.; Chassevent, A.; Quillien, V.; Avril, T.; Jourdan, M.L.; Michalak, S.; François, P.; Carré, J.L.; Mosser, J.; et al. Isolation of a new cell population in the glioblastoma microenvironment. J. Neurooncol. 2012, 106, 493–504. [Google Scholar] [CrossRef]
- Madar, S.; Goldstein, I.; Rotter, V. “Cancer associated fibroblasts”-more than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef]
- Yi, D.; Xiang, W.; Zhang, Q.; Cen, Y.; Su, Q.; Zhang, F.; Lu, Y.; Zhao, H.; Fu, P. Human Glioblastoma-Derived Mesenchymal Stem Cell to Pericytes Transition and Angiogenic Capacity in Glioblastoma Microenvironment. Cell. Physiol. Biochem. 2018, 46, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, T.; Hildebrandt, J.; Nuebling, G.; Sostak, P.; Straube, A. Glioblastoma-dependent differentiation and angiogenic potential of human mesenchymal stem cells in vitro. J. Neurooncol. 2011, 105, 57–65. [Google Scholar] [CrossRef]
- Ochs, K.; Sahm, F.; Opitz, C.A.; Lanz, T.V.; Oezen, I.; Couraud, P.O.; von Deimling, A.; Wick, W.; Platten, M. Immature mesenchymal stem cell-like pericytes as mediators of immunosuppression in human malignant glioma. J. Neuroimmunol. 2013, 265, 106–116. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Salhia, B. Cancer-Associated Fibroblast Subpopulations With Diverse and Dynamic Roles in the Tumor Microenvironment. Mol. Cancer Res. 2022, 20, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.S.; Januszyk, M.; Delitto, D.; Chang, H.Y.; Norton, J.A.; Longaker, M.T.; Foster, D.S.; Januszyk, M.; Delitto, D.; Yost, K.E.; et al. Multiomic analysis reveals conservation of cancer- associated fibroblast phenotypes across species and tissue of origin ll Article Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin. Cancer Cell 2022, 40, 1392–1406.e7. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef]
- Luo, H.; Xia, X.; Huang, L.B.; An, H.; Cao, M.; Kim, G.D.; Chen, H.N.; Zhang, W.H.; Shu, Y.; Kong, X.; et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 2022, 13, 6619. [Google Scholar] [CrossRef] [PubMed]
- Akturk, G.; Sweeney, R.; Remark, R.; Merad, M.; Gnjatic, S. Multiplexed Immunohistochemical Consecutive Staining on Single Slide (MICSSS): Multiplexed Chromogenic IHC Assay for High-Dimensional Tissue Analysis. Methods Mol. Biol. 2020, 2055, 497–519. [Google Scholar] [CrossRef] [PubMed]
- Kinkhabwala, A.; Herbel, C.; Pankratz, J.; Yushchenko, D.A.; Rüberg, S.; Praveen, P.; Reiß, S.; Rodriguez, F.C.; Schäfer, D.; Kollet, J.; et al. MACSima imaging cyclic staining (MICS) technology reveals combinatorial target pairs for CAR T cell treatment of solid tumors. Sci. Rep. 2022, 12, 1911. [Google Scholar] [CrossRef] [PubMed]
- Yeo, A.T.; Rawal, S.; Delcuze, B.; Christofides, A.; Atayde, A.; Strauss, L.; Balaj, L.; Rogers, V.A.; Uhlmann, E.J.; Varma, H.; et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat. Immunol. 2022, 23, 971–984. [Google Scholar] [CrossRef]
- Chen, Z.; Soni, N.; Pinero, G.; Giotti, B.; Eddins, D.J.; Lindblad, K.E.; Ross, J.L.; Puigdelloses Vallcorba, M.; Joshi, T.; Angione, A.; et al. Monocyte depletion enhances neutrophil influx and proneural to mesenchymal transition in glioblastoma. Nat. Commun. 2023, 14, 1839. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lootens, T.; Roman, B.I.; Stevens, C.V.; De Wever, O.; Raedt, R. Glioblastoma-Associated Mesenchymal Stem/Stromal Cells and Cancer-Associated Fibroblasts: Partners in Crime? Int. J. Mol. Sci. 2024, 25, 2285. https://doi.org/10.3390/ijms25042285
Lootens T, Roman BI, Stevens CV, De Wever O, Raedt R. Glioblastoma-Associated Mesenchymal Stem/Stromal Cells and Cancer-Associated Fibroblasts: Partners in Crime? International Journal of Molecular Sciences. 2024; 25(4):2285. https://doi.org/10.3390/ijms25042285
Chicago/Turabian StyleLootens, Thibault, Bart I. Roman, Christian V. Stevens, Olivier De Wever, and Robrecht Raedt. 2024. "Glioblastoma-Associated Mesenchymal Stem/Stromal Cells and Cancer-Associated Fibroblasts: Partners in Crime?" International Journal of Molecular Sciences 25, no. 4: 2285. https://doi.org/10.3390/ijms25042285
APA StyleLootens, T., Roman, B. I., Stevens, C. V., De Wever, O., & Raedt, R. (2024). Glioblastoma-Associated Mesenchymal Stem/Stromal Cells and Cancer-Associated Fibroblasts: Partners in Crime? International Journal of Molecular Sciences, 25(4), 2285. https://doi.org/10.3390/ijms25042285