
Bruton’s Tyrosine Kinase Inhibitors: Recent Updates

 ,
,
Abstract
1. Introduction
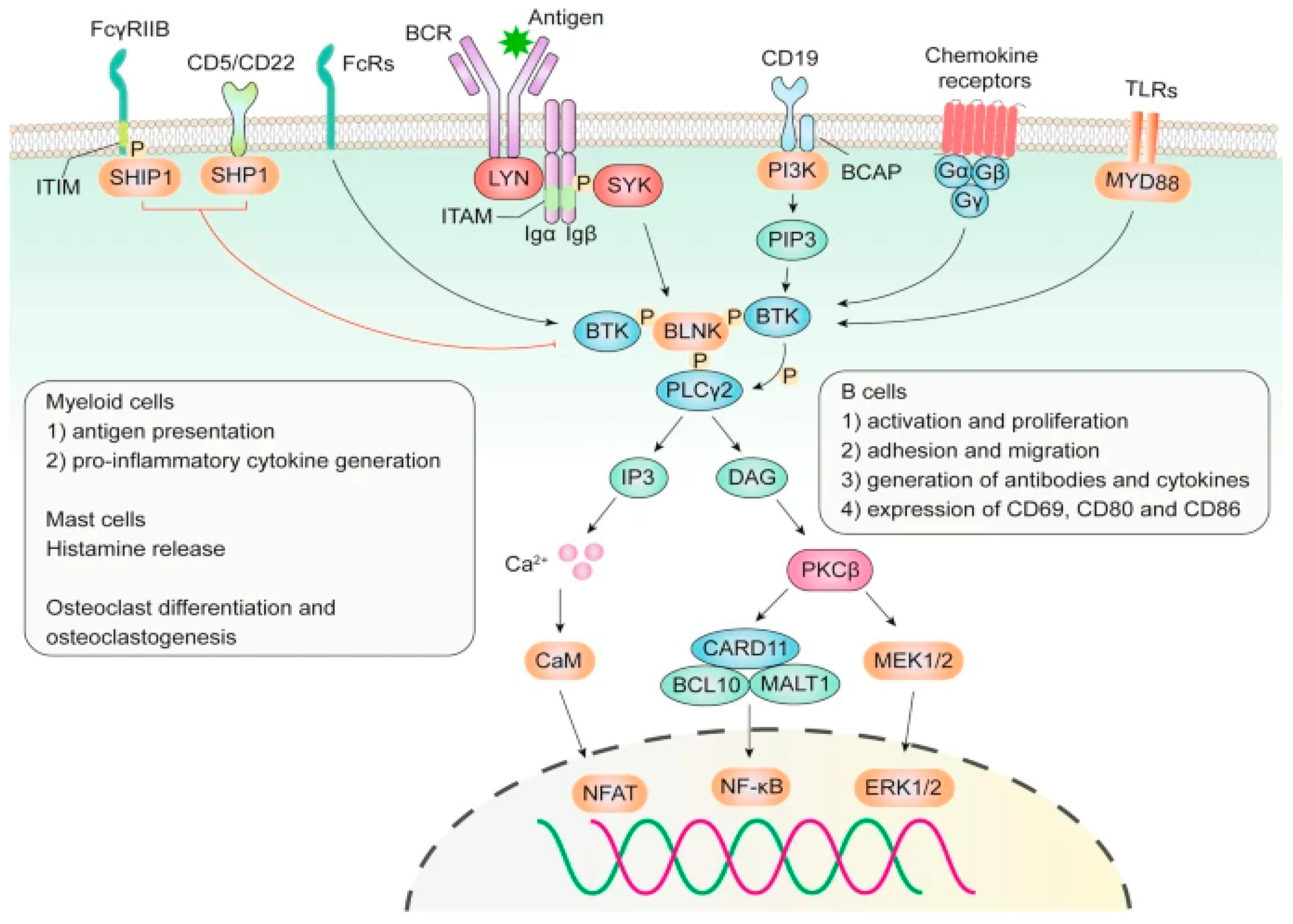
2. BCR Signaling Pathway Overview
2.1. BTK Receptor
2.2. BTK Inhibition
2.3. Adverse Effects and Off-Target Activity
3. Hematological Malignancies
3.1. Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma
3.1.1. Ibrutinib
3.1.2. Acalabrutinib
3.1.3. Zanubrutinib
3.1.4. Pirtobrutinib
3.2. Non-Hodgkin Lymphoma
3.2.1. Mantle Cell Lymphoma
Ibrutinib
Acalabrutinib
Zanubrutinib
Pirtobrutinib
3.2.2. Marginal Zone Lymphoma
Ibrutinib
Zanubrutinib
3.2.3. Waldenstrom’s Macroglobulinemia
Ibrutinib
4. Autoimmune Disorders
4.1. Rheumatoid Arthritis
4.2. Systemic Lupus Erythematosus
4.3. Multiple Sclerosis
5. Solid Tumors
6. Graft-versus-Host Disease
7. Future Directions
8. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tasso, B.; Spallarossa, A.; Russo, E.; Brullo, C. The Development of BTK Inhibitors: A Five-Year Update. Molecules 2021, 26, 7411. [Google Scholar] [CrossRef]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Alu, A.; Lei, H.; Han, X.; Wei, Y.; Wei, X. BTK inhibitors in the treatment of hematological malignancies and inflammatory diseases: Mechanisms and clinical studies. J. Hematol. Oncol. 2022, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Bruton Tyrosine Kinase Inhibitors: Present and Future. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef]
- Mano, H. Tec family of protein-tyrosine kinases: An overview of their structure and function. Cytokine Growth Factor Rev. 1999, 10, 267–280. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Wang, C.; Xu, Z.; Zhang, Y.; Liu, Y.; Zhao, G.; Ling, Y. Review of the development of BTK inhibitors in overcoming the clinical limitations of ibrutinib. Eur. J. Med. Chem. 2022, 229, 114009. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, A.; Lamanna, N. Managing toxicities of Bruton tyrosine kinase inhibitors. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Owen, C.; Robak, T.; Tedeschi, A.; Bairey, O.; Burger, J.A.; Hillmen, P.; Coutre, S.E.; Dearden, C.; Grosicki, S.; et al. Up to 8-year follow-up from RESONATE-2: First-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022, 6, 3440–3450. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Heerema, N.A.; Zhao, W.; Booth, A.M.; Ding, W.; Bartlett, N.L.; Brander, D.M.; Barr, P.M.; Rogers, K.; et al. Long-Term Results of Alliance A041202 Show Continued Advantage of Ibrutinib-Based Regimens Compared with Bendamustine Plus Rituximab (BR) Chemoimmunotherapy. Blood 2021, 138, 639. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Efficacy and safety in a 4-year follow-up of the ELEVATE-TN study comparing acalabrutinib with or without obinutuzumab versus obinutuzumab plus chlorambucil in treatment-naïve chronic lymphocytic leukemia. Leukemia 2022, 36, 1171–1175. [Google Scholar] [CrossRef]
- Tam, C.S.; Brown, J.R.; Kahl, B.S.; Ghia, P.; Giannopoulos, K.; Jurczak, W.; Šimkovič, M.; Shadman, M.; Österborg, A.; Laurenti, L.; et al. Zanubrutinib versus bendamustine and rituximab in untreated chronic lymphocytic leukaemia and small lymphocytic lymphoma (SEQUOIA): A randomised, controlled, phase 3 trial. Lancet Oncol. 2022, 23, 1031–1043, Erratum in Lancet Oncol. 2023, 24, e106. [Google Scholar] [CrossRef]
- Mato, A.R.; Woyach, J.A.; Brown, J.R.; Ghia, P.; Patel, K.; Eyre, T.A.; Munir, T.; Lech-Maranda, E.; Lamanna, N.; Tam, C.S.; et al. Efficacy of pirtobrutinib in covalent BTK-inhibitor pre-treated relapsed / refractory CLL/SLL: Additional patients and extended follow-up from the phase 1/2 BRUIN study. Blood 2022, 140 (Suppl. 1), 2316–2320. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Yatabe, Y.; Suzuki, R.; Tobinai, K.; Matsuno, Y.; Ichinohasama, R.; Okamoto, M.; Yamaguchi, M.; Tamaru, J.; Uike, N.; Hashimoto, Y.; et al. Significance of cyclin D1 overexpression for the diagnosis of mantle cell lymphoma: A clinicopathologic comparison of cyclin D1-positive MCL and cyclin D1-negative MCL-like B-cell lymphoma. Blood 2000, 95, 2253–2261. [Google Scholar] [PubMed]
- Wang, M.; Rule, S.; Zinzani, P.L.; Goy, A.; Casasnovas, O.; Smith, S.D.; Damaj, G.; Doorduijn, J.; Lamy, T.; Morschhauser, F.; et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): A single-arm, multicentre, phase 2 trial. Lancet 2018, 391, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Romaguera, J.; Srour, S.A.; Lee, H.J.; Hagemeister, F.; Westin, J.; Fayad, L.; Samaniego, F.; Badillo, M.; Zhang, L.; et al. Four-year follow-up of a single arm, phase II clinical trial of ibrutinib with rituximab (IR) in patients with relapsed/refractory mantle cell lymphoma (MCL). Br. J. Haematol. 2018, 182, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhou, K.; Zou, D.; Zhou, J.; Hu, J.; Yang, H.; Zhang, H.; Ji, J.; Xu, W.; Jin, J.; et al. Treatment of Patients with Relapsed or Refractory Mantle-Cell Lymphoma with Zanubrutinib, a Selective Inhibitor of Bruton’s Tyrosine Kinase. Clin. Cancer Res. 2020, 26, 4216–4224. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Shah, N.; Jurczak, W.; Zinzani, P.L.; Eyre, T.A.; Cheah, C.Y.; Ujjani, C.S.; Koh, Y.; Izutsu, K.; Gerson, J.N.; et al. Efficacy of pirtobrutinib in covalent BTK-inhibitor pre-treated relapsed/refractory mantle cell lymphoma: Additional patients and extended follow-up from the phase 1/2 BRUIN study. Blood 2022, 140, 9368–9372. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Noy, A.; de Vos, S.; Coleman, M.; Martin, P.; Flowers, C.R.; Thieblemont, C.; Morschhauser, F.; Collins, G.P.; Ma, S.; Peles, S.; et al. Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: Long-term follow-up and biomarker analysis. Blood Adv. 2020, 4, 5773–5784. [Google Scholar] [CrossRef]
- Opat, S.; Tedeschi, A.; Linton, K.; McKay, P.; Hu, B.; Chan, H.; Jin, J.; Sobieraj-Teague, M.; Zinzani, P.L.; Coleman, M.; et al. The MAGNOLIA Trial: Zanubrutinib, a Next-Generation Bruton Tyrosine Kinase Inhibitor, Demonstrates Safety and Efficacy in Relapsed/Refractory Marginal Zone Lymphoma. Clin. Cancer Res. 2021, 27, 6323–6332. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Treon, S.P.; Al-Katib, A.; Fonseca, R.; Greipp, P.R.; McMaster, M.L.; Morra, E.; Pangalis, G.A.; San Miguel, J.F.; Branagan, A.R.; et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: Consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin. Oncol. 2003, 30, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Tedeschi, A.; Trotman, J.; García-Sanz, R.; Macdonald, D.; Leblond, V.; Mahe, B.; Herbaux, C.; Tam, C.; Orsucci, L.; et al. iNNOVATE Study Group and the European Consortium for Waldenström’s Macroglobulinemia. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2018, 378, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C.; Chan, G.G.; Xu, L.; Tsakmaklis, N.; Kofides, A.; Demos, M.G.; Chen, J.; Liu, X.; Munshi, M.; Yang, G.; et al. Genomic evolution of ibrutinib-resistant clones in Waldenström macroglobulinaemia. Br. J. Haematol. 2020, 189, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Trotman, J.; Opat, S.; Gottlieb, D.; Simpson, D.; Marlton, P.; Cull, G.; Munoz, J.; Tedeschi, A.; Roberts, A.W.; Seymour, J.F.; et al. Zanubrutinib for the treatment of patients with Waldenström macroglobulinemia: 3 Years of follow-up. Blood 2020, 136, 2027–2037, Erratum in Blood 2021, 137, 1131. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 2020, 136, 2038–2050. [Google Scholar] [CrossRef]
- Owen, R.G.; McCarthy, H.; Rule, S.; D’Sa, S.; Thomas, S.K.; Tournilhac, O.; Forconi, F.; Kersten, M.J.; Zinzani, P.L.; Iyengar, S.; et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: A single-arm, multicentre, phase 2 study. Lancet Haematol. 2020, 7, e112–e121, Erratum in Lancet Haematol. 2021, 8, e249. [Google Scholar] [CrossRef]
- Sekiguchi, N.; Rai, S.; Munakata, W.; Suzuki, K.; Handa, H.; Shibayama, H.; Endo, T.; Terui, Y.; Iwaki, N.; Fukuhara, N.; et al. A multicenter, open-label, phase II study of tirabrutinib (ONO/GS-4059) in patients with Waldenström’s macroglobulinemia. Cancer Sci. 2020, 111, 3327–3337, Erratum in Cancer Sci. 2021, 112, 1669. [Google Scholar] [CrossRef]
- Cohen, S.; Tuckwell, K.; Katsumoto, T.R.; Zhao, R.; Galanter, J.; Lee, C.; Rae, J.; Toth, B.; Ramamoorthi, N.; Hackney, J.A.; et al. Fenebrutinib versus Placebo or Adalimumab in Rheumatoid Arthritis: A Randomized, Double-Blind, Phase II Trial (ANDES Study). Arthritis Rheumatol. 2020, 72, 1435–1446. [Google Scholar] [CrossRef]
- Rankin, A.L.; Seth, N.; Keegan, S.; Andreyeva, T.; Cook, T.A.; Edmonds, J.; Mathialagan, N.; Benson, M.J.; Syed, J.; Zhan, Y.; et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J. Immunol. 2013, 191, 4540–4550. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.A.; Glynn, E.; Garcia, S.J.; Panzenbeck, M.; Pelletier, J.; Dimock, J.; Seccareccia, E.; Bosanac, T.; Khalil, S.; Harcken, C.; et al. BTK inhibition ameliorates kidney disease in spontaneous lupus nephritis. Clin. Immunol. 2018, 197, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.; Furie, R.; Jones, N.S.; Guibord, P.; Galanter, J.; Lee, C.; McGregor, A.; Toth, B.; Rae, J.; Hwang, O.; et al. Efficacy, Safety, and Pharmacodynamic Effects of the Bruton’s Tyrosine Kinase Inhibitor Fenebrutinib (GDC-0853) in Systemic Lupus Erythematosus: Results of a Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2021, 73, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Dörner, T.; Pisetsky, D.S.; Sanchez-Guerrero, J.; Patel, A.C.; Parsons-Rich, D.; Le Bolay, C.; Drouin, E.E.; Kao, A.H.; Guehring, H.; et al. Efficacy and Safety of the Bruton’s Tyrosine Kinase Inhibitor Evobrutinib in Systemic Lupus Erythematosus: Results of a Phase II, Randomized, Double-Blind, Placebo-Controlled Dose-Ranging Trial. ACR Open Rheumatol. 2023, 5, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, G.; Berg, J.; Lindgren, P.; Fredrikson, S.; Jönsson, B. Costs and quality of life of patients with multiple sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2006, 77, 918–926. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Arnold, D.L.; Vermersch, P.; Bar-Or, A.; Fox, R.J.; Matta, A.; Turner, T.; Wallström, E.; Zhang, X.; Mareš, M.; et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: A phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef]
- Hong, D.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.J.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111. [Google Scholar] [CrossRef]
- Tempero, M.; Oh, D.Y.; Tabernero, J.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.T.; Starling, N.; Bachet, J.B.; Chang, H.M.; et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine for first-line treatment of patients with metastatic pancreatic adenocarcinoma: Phase III RESOLVE study. Ann. Oncol. 2021, 32, 600–608. [Google Scholar] [CrossRef]
- Kim, D.W.; Tan, E.; Zhou, J.M.; Schell, M.J.; Martinez, M.; Yu, J.; Carballido, E.; Mehta, R.; Strosberg, J.; Imanirad, I.; et al. A phase 1/2 trial of ibrutinib in combination with pembrolizumab in patients with mismatch repair proficient metastatic colorectal cancer. Br. J. Cancer 2021, 124, 1803–1808. [Google Scholar] [CrossRef]
- Al-Toubah, T.; Schell, M.J.; Cives, M.; Zhou, J.M.; Soares, H.P.; Strosberg, J.R. A Phase II Study of Ibrutinib in Advanced Neuroendocrine Neoplasms. Neuroendocrinology 2020, 110, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Al-Kadhimi, Z.; Gul, Z.; Chen, W.; Smith, D.; Abidi, M.; Deol, A.; Ayash, L.; Lum, L.; Waller, E.K.; Ratanatharathorn, V.; et al. High incidence of severe acute graft-versus-host disease with tacrolimus and mycophenolate mofetil in a large cohort of related and unrelated allogeneic transplantation patients. Biol. Blood Marrow Transplant. 2014, 20, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Finke, J.; Bethge, W.A.; Schmoor, C.; Ottinger, H.D.; Stelljes, M.; Zander, A.R.; Volin, L.; Ruutu, T.; Heim, D.A.; Schwerdtfeger, R.; et al. Standard graft-versus-host disease prophylaxis with or without anti-T-cell globulin in haematopoietic cell transplantation from matched unrelated donors: A randomised, open-label, multicentre phase 3 trial. Lancet Oncol. 2009, 10, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Solano, C.; Wolschke, C.; Bandini, G.; Patriarca, F.; Pini, M.; Nagler, A.; Selleri, C.; Risitano, A.; Messina, G.; et al. Antilymphocyte Globulin for Prevention of Chronic Graft-versus-Host Disease. N. Engl. J. Med. 2016, 374, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Walker, I.; Panzarella, T.; Couban, S.; Couture, F.; Devins, G.; Elemary, M.; Gallagher, G.; Kerr, H.; Kuruvilla, J.; Lee, S.J.; et al. Pretreatment with anti-thymocyte globulin versus no anti-thymocyte globulin in patients with haematological malignancies undergoing haemopoietic cell transplantation from unrelated donors: A randomised, controlled, open-label, phase 3, multicentre trial. Lancet Oncol. 2016, 17, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Foell, J.; Schulte, J.H.; Pfirstinger, B.; Troeger, A.; Wolff, D.; Edinger, M.; Hofmann, P.; Aslanidis, C.; Lang, P.; Holler, E.; et al. Haploidentical CD3 or α/β T-cell depleted HSCT in advanced stage sickle cell disease. Bone Marrow Transplant. 2019, 54, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Lum, S.H.; Sobh, A.; Carruthers, K.; Nademi, Z.; Watson, H.; McNaughton, P.; Selvarajah, S.; Deyà-Martínez, A.; Abinun, M.; Flood, T.; et al. Improved survival and graft function in ex vivo T-cell depleted haploidentical hematopoietic cell transplantation for primary immunodeficiency. Bone Marrow Transplant. 2021, 56, 1200–1204, Erratum in Bone Marrow Transplant. 2021, 56, 1483–1484. [Google Scholar] [CrossRef]
- Shelikhova, L.; Ilushina, M.; Shekhovtsova, Z.; Shasheleva, D.; Khismatullina, R.; Kurnikova, E.; Pershin, D.; Balashov, D.; Radygina, S.; Trakhtman, P.; et al. αβ T Cell-Depleted Haploidentical Hematopoietic Stem Cell Transplantation without Antithymocyte Globulin in Children with Chemorefractory Acute Myelogenous Leukemia. Biol. Blood Marrow Transplant. 2019, 25, e179–e182. [Google Scholar] [CrossRef]
- Pavletic, S.Z.; Carter, S.L.; Kernan, N.A.; Henslee-Downey, J.; Mendizabal, A.M.; Papadopoulos, E.; Gingrich, R.; Casper, J.; Yanovich, S.; Weisdorf, D.; et al. Influence of T-cell depletion on chronic graft-versus-host disease: Results of a multicenter randomized trial in unrelated marrow donor transplantation. Blood 2005, 106, 3308–3313. [Google Scholar] [CrossRef]
- Baron, F.; Mohty, M.; Blaise, D.; Socié, G.; Labopin, M.; Esteve, J.; Ciceri, F.; Giebel, S.; Gorin, N.C.; Savani, B.N.; et al. Anti-thymocyte globulin as graft-versus-host disease prevention in the setting of allogeneic peripheral blood stem cell transplantation: A review from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica 2017, 102, 224–234. [Google Scholar] [CrossRef]
- Shinners, N.P.; Carlesso, G.; Castro, I.; Hoek, K.L.; Corn, R.A.; Woodland, R.T.; Scott, M.L.; Wang, D.; Khan, W.N. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J. Immunol. 2007, 179, 3872–3880, Erratum in J. Immunol. 2007, 179, 6369. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef]
- Miklos, D.; Cutler, C.S.; Arora, M.; Waller, E.K.; Jagasia, M.; Pusic, I.; Flowers, M.E.; Logan, A.C.; Nakamura, R.; Blazar, B.R.; et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 2017, 130, 2243–2250. [Google Scholar] [CrossRef]
- Waller, E.K.; Miklos, D.; Cutler, C.; Arora, M.; Jagasia, M.H.; Pusic, I.; Flowers, M.E.D.; Logan, A.C.; Nakamura, R.; Chang, S.; et al. Ibrutinib for Chronic Graft-versus-Host Disease After Failure of Prior Therapy: 1-Year Update of a Phase 1b/2 Study. Biol. Blood Marrow Transplant. 2019, 25, 2002–2007. [Google Scholar] [CrossRef]
- Carpenter, P.A.; Kang, H.J.; Yoo, K.H.; Zecca, M.; Cho, B.; Lucchini, G.; Nemecek, E.R.; Schultz, K.R.; Stepensky, P.; Chaudhury, S.; et al. Ibrutinib Treatment of Pediatric Chronic Graft-versus-Host Disease: Primary Results from the Phase 1/2 iMAGINE Study. Transplant. Cell Ther. 2022, 28, 771.e1–771.e10. [Google Scholar] [CrossRef]
- Miklos, D.B.; Abu Zaid, M.; Cooney, J.P.; Albring, J.C.; Flowers, M.; Skarbnik, A.P.; Yakoub-Agha, I.; Ko, B.S.; Bruno, B.; Waller, E.K.; et al. Ibrutinib for First-Line Treatment of Chronic Graft-Versus-Host Disease: Results From the Randomized Phase III iNTEGRATE Study. J. Clin. Oncol. 2023, 41, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Flinn, I.W.; Awan, F.T.; Eradat, H.; Brander, D.; Tees, M.; Parikh, S.A.; Phillips, T.J.; Ghori, R.; Reddy, N.M.; et al. Efficacy and safety of nemtabrutinib, a wild-type and C481S-mutated Bruton tyrosine kinase inhibitor for B-Cell malignancies: Updated analysis of the open-label phase 1/2 dose-expansion bellwave-001 study. Blood 2022, 140, 7004–7006. [Google Scholar] [CrossRef]
- Mato, A.R.; Woyach, J.A.; Brown, J.R.; Ghia, P.; Patel, K.; Eyre, T.A.; Munir, T.; Lech-Maranda, E.; Lamanna, N.; Tam, C.S.; et al. Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023, 389, 33–44. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| BTK Inhibitor | Year of FDA Approval | Generation | BTK Inhibition | Indication |
|---|---|---|---|---|
| Ibrutinib (Imbruvica) | 2013 | First | Irreversible | CLL/SLL, GVHD, MCL, MZL, WM |
| Acalabrutinib (Calquence) | 2017 | Second | Irreversible | CLL/SLL, MCL |
| Zanubrutinib (Brukinsa) | 2019 | Second | Irreversible | CLL/SLL, MCL, MZL, WM |
| Pirtobrutinib (Jaypirca) | 2023 | Third | Reversible | CLL/SLL, MCL |
| Adverse Effect | Ibrutinib | Acalabrutinib | Zanubrutinib | Pirtobrutinib |
|---|---|---|---|---|
| Arthalgias | 16% to 24% | 8% to 16% | - | ≤12% |
| Atrial fibrillation | ≤8% | ≤5% | ≤5% | - |
| Bleeding events | 48% | 8% to 20% | 24% to 42% | 11% |
| Diarrhea | 28% to 59% | 18% to 35% | 14% to 22% | 19% |
| Headache | 12% to 21% | 22% to 39% | 8% to 18% | - |
| Hypertension | 11% to 16% | 3% | 14% to 19% | - |
| Infections | 10% to 11% | 56% to 65% | - | - |
| Lymphocytosis | 66% | 16% to 26% | 24% | 34% |
| Neutropenia | ≥30% | 23% to 48% | - | - |
| Rash | 12% to 25% | 9% to 25% | 20% to 29% | 14% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fares, A.; Carracedo Uribe, C.; Martinez, D.; Rehman, T.; Silva Rondon, C.; Sandoval-Sus, J. Bruton’s Tyrosine Kinase Inhibitors: Recent Updates. Int. J. Mol. Sci. 2024, 25, 2208. https://doi.org/10.3390/ijms25042208
Fares A, Carracedo Uribe C, Martinez D, Rehman T, Silva Rondon C, Sandoval-Sus J. Bruton’s Tyrosine Kinase Inhibitors: Recent Updates. International Journal of Molecular Sciences. 2024; 25(4):2208. https://doi.org/10.3390/ijms25042208
Chicago/Turabian StyleFares, Amneh, Carlos Carracedo Uribe, Diana Martinez, Tauseef Rehman, Carlos Silva Rondon, and Jose Sandoval-Sus. 2024. "Bruton’s Tyrosine Kinase Inhibitors: Recent Updates" International Journal of Molecular Sciences 25, no. 4: 2208. https://doi.org/10.3390/ijms25042208
APA StyleFares, A., Carracedo Uribe, C., Martinez, D., Rehman, T., Silva Rondon, C., & Sandoval-Sus, J. (2024). Bruton’s Tyrosine Kinase Inhibitors: Recent Updates. International Journal of Molecular Sciences, 25(4), 2208. https://doi.org/10.3390/ijms25042208