
Transcriptomic Analyses Reveal Insights into the Shared Regulatory Network of Phenolic Compounds and Steviol Glycosides in Stevia rebaudiana

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. Phytochemical Evaluation and Antioxidant Activity
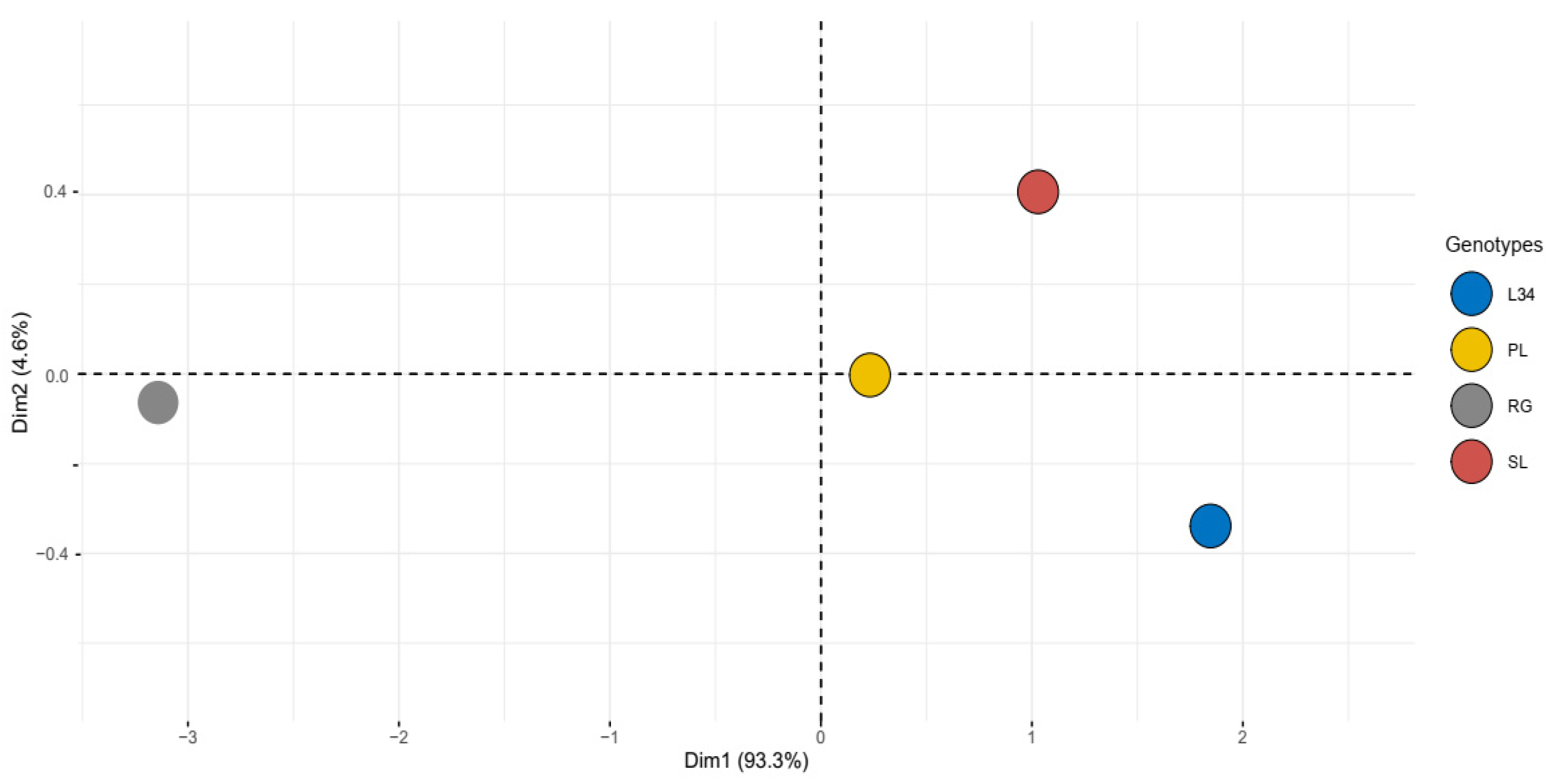
2.2. Transcriptomic Analyses
2.2.1. cDNA Sequencing and Mapping on S. rebaudiana Reference Transcriptome
2.2.2. Analysis and GO Enrichment of Differentially Expressed Genes
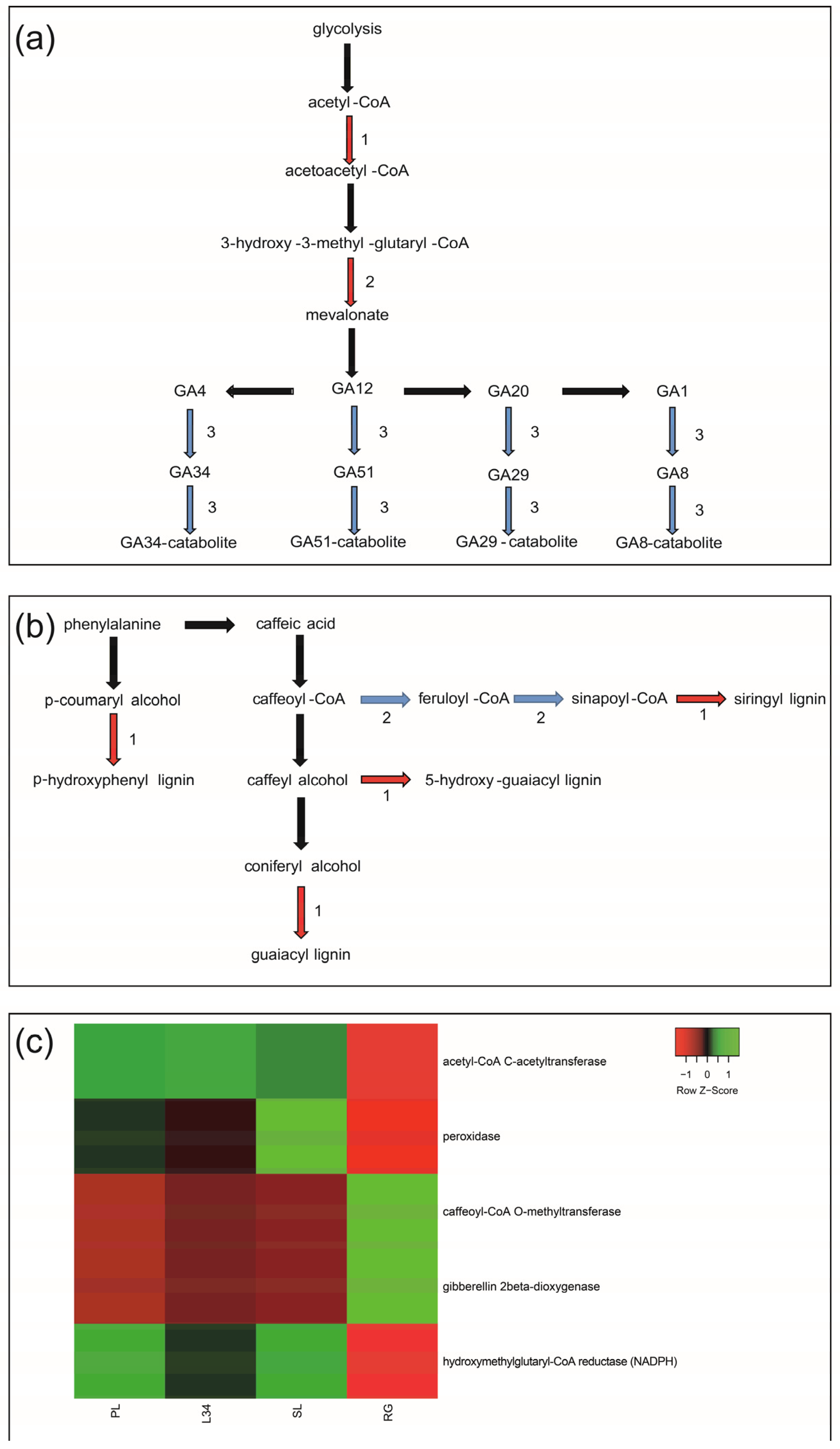
2.2.3. Functional Characterization of DEGs Related to Primary and Secondary Metabolism
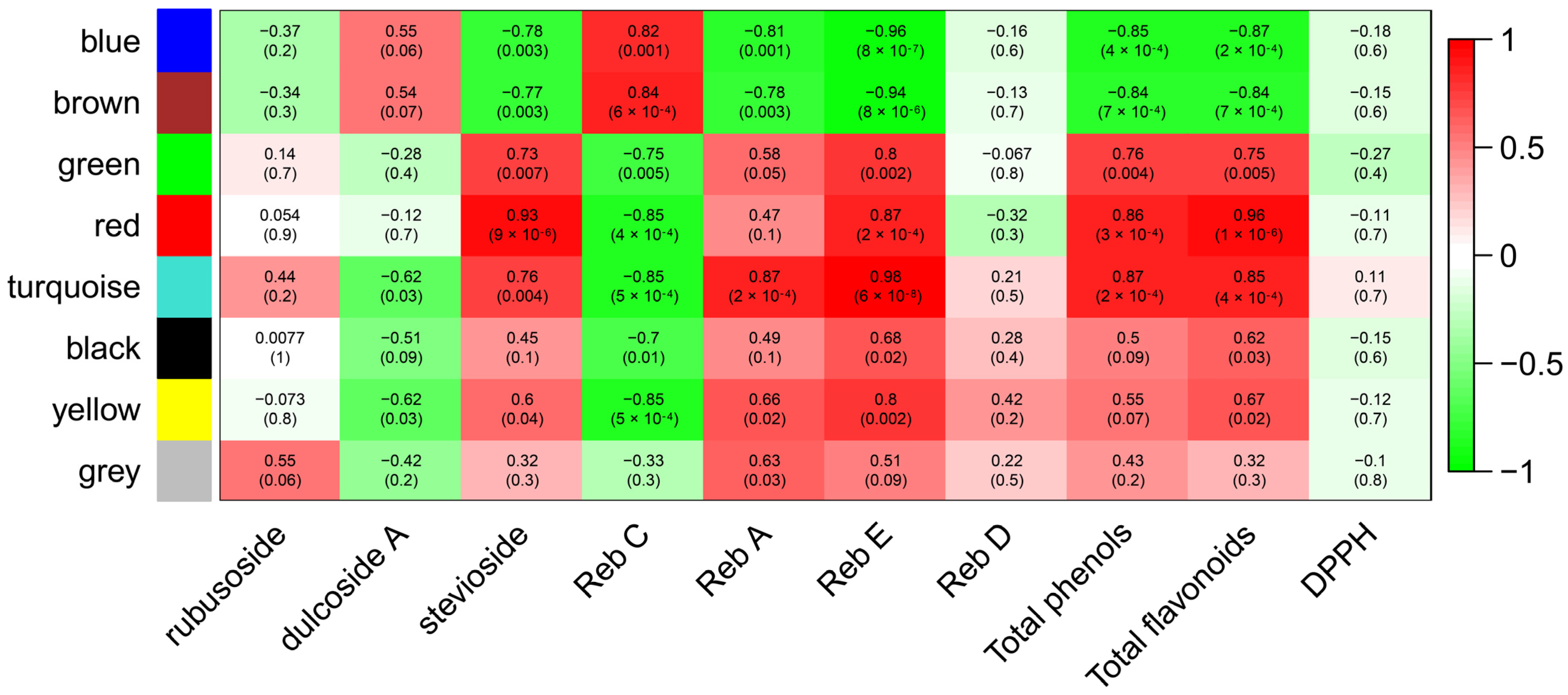
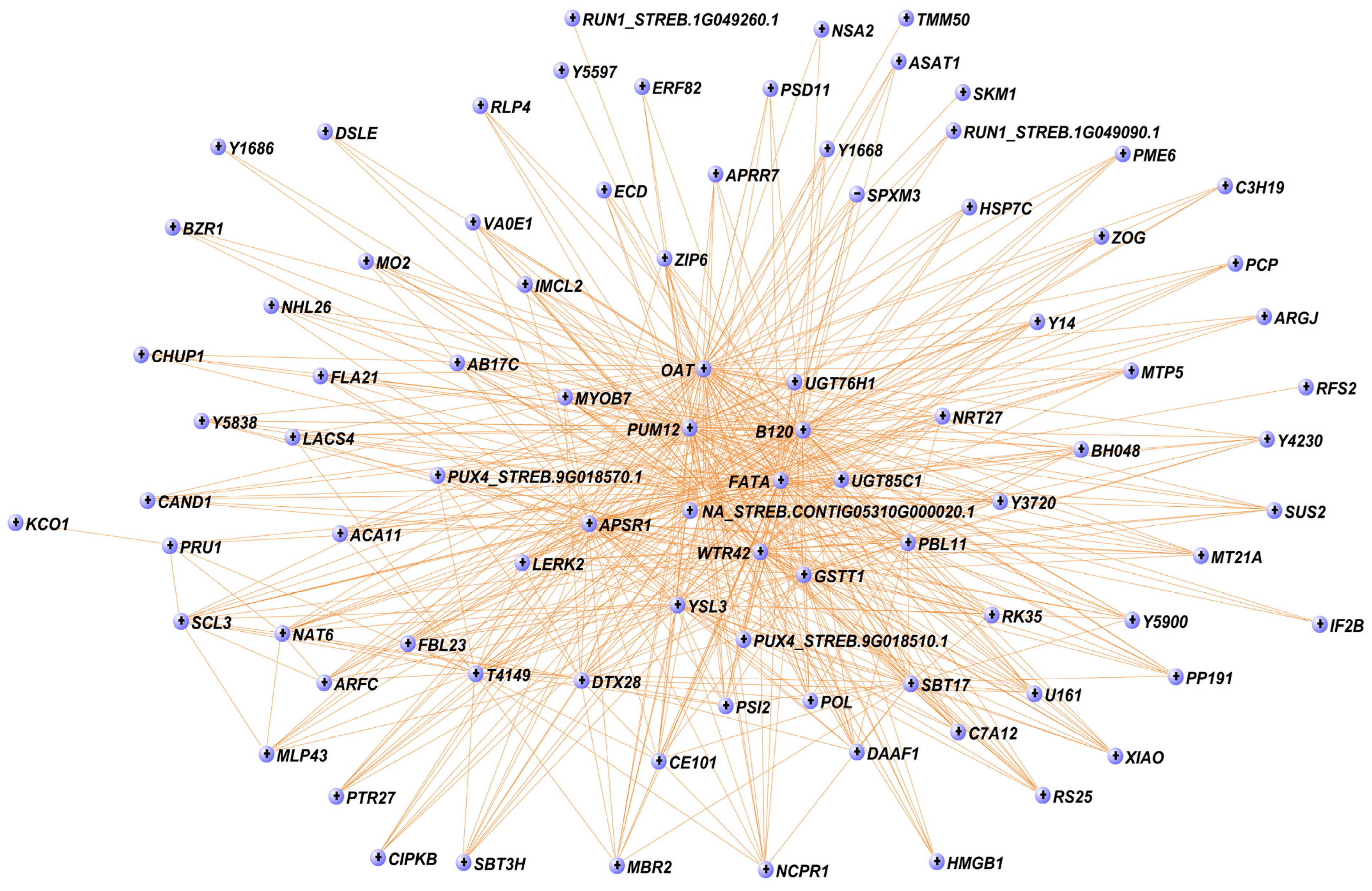
2.2.4. Analysis of Gene Networks and Correlation with Biochemical Traits
2.2.5. Characterization of Differentially Expressed UGTs
3. Discussion
3.1. Genes Putatively Involved in the Quantitative Variation of Steviol Glycosides and Phenolic Compounds in S. rebaudiana Leaf
3.2. Co-Expression and Phylogenetic Analyses Indicate Putative Functions of UGTs
4. Materials and Methods
4.1. Plant Material
4.2. Phytochemical Analyses
4.2.1. Sample Extraction
4.2.2. Spectrophotometric Assays
4.2.3. Steviol Glycoside Content and Composition
4.2.4. PCA Analysis
4.3. Transcriptomic Analyses
4.3.1. RNA Extraction
4.3.2. RNA Sequencing and Mapping Procedures
4.3.3. Differential Expression Analysis
4.3.4. Differentially Expressed Gene Characterization
4.3.5. Construction and Analysis of Co-Expression Networks
4.3.6. Construction of UGT Dendrogram
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ceunen, S.; Geuns, J.M.C. Steviol glycosides: Chemical diversity, metabolism, and function. J. Nat. Prod. 2013, 76, 1201–1228. [Google Scholar] [CrossRef] [PubMed]
- Tavarini, S.; Sgherri, C.; Ranieri, A.M.; Angelini, L.G. Effect of nitrogen fertilization and harvest time on steviol glycosides, flavonoid composition, and antioxidant properties in Stevia rebaudiana Bertoni. J. Agric. Food Chem. 2015, 63, 7041–7050. [Google Scholar] [CrossRef] [PubMed]
- Tavarini, S.; Passera, B.; Angelini, L.G. Crop and steviol glycoside improvement in Stevia by breeding. In Steviol Glycosides: Cultivation, Processing, Analysis and Applications in Food; The Royal Society of Chemistry: London, UK, 2018; pp. 1–31. [Google Scholar] [CrossRef]
- Yadav, A.K.; Singh, S.; Dhyani, D.; Ahuja, P.S.; Randhawa, H.; Puchalski, B.J.; Frick, M.; Goyal, A.; Despins, T.; Graf, R.J.; et al. A review on the improvement of stevia [Stevia rebaudiana (Bertoni)]. Can. J. Plant Sci. 2011, 91, 1–27. [Google Scholar] [CrossRef]
- Moraes, R.M.; Donega, M.A.; Cantrell, C.L.; Mello, S.C.; McChesney, J.D. Effect of harvest timing on leaf production and yield of diterpene glycosides in Stevia rebaudiana Bert: A specialty perennial crop for Mississippi. Ind. Crops Prod. 2013, 51, 385–389. [Google Scholar] [CrossRef]
- Chatsudthipong, V.; Muanprasat, C. Stevioside and related compounds: Therapeutic benefits beyond sweetness. Pharmacol. Ther. 2009, 121, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Puri, M.; Sharma, D. Antibacterial activity of stevioside towards food-borne pathogenic bacteria. Eng. Life Sci. 2011, 11, 326–329. [Google Scholar] [CrossRef]
- Shivanna, N.; Naika, M.; Khanum, F.; Kaul, V.K. Antioxidant, anti-diabetic and renal protective properties of Stevia rebaudiana. J. Diabetes Complicat. 2013, 27, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.Z.; Wu, K.; Xia, Y.; Fan, Y.; Shen, J.; Zhang, P.; Gu, J. Polyphenols from Stevia rebaudiana (Bertoni) leaves and their functional properties. J. Food Sci. 2020, 85, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, S.; Sharma, R.; Rajput, Y.S.; Mann, B.; Lata, K. Estimation of steviol glycosides in food matrices by high performance liquid chromatography. J. Food Sci. Technol. 2018, 55, 3325–3334. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, X.; Jia, X.; Zhu, L.; Yin, H. Comparative transcriptomic of Stevia rebaudiana provides insight into rebaudioside D and rebaudioside M biosynthesis. Plant Physiol. Biochem. 2021, 167, 541–549. [Google Scholar] [CrossRef]
- Yadav, S.K.; Guleria, P. Steviol glycosides from Stevia: Biosynthesis pathway review and their application in foods and medicine. Crit. Rev. Food Sci. Nutr. 2012, 52, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Brandle, J.E.; Telmer, P.G. Steviol glycoside biosynthesis. Phytochemistry 2007, 68, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Richman, A.; Swanson, A.; Humphrey, T.; Chapman, R.; McGarvey, B.; Pocs, R.; Brandle, J. Functional genomics uncovers three glucosyltransferases involved in the synthesis of the major sweet glucosides of Stevia rebaudiana. Plant J. 2005, 41, 56–67. [Google Scholar] [CrossRef]
- Chen, J.; Hou, K.; Qin, P.; Liu, H.; Yi, B.; Yang, W.; Wu, W. RNA-Seq for gene identification and transcript profiling of three Stevia rebaudiana genotypes. BMC Genom. 2014, 15, 571. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.; Carlsen, S.; Semmler, A.; Simón, E.; Mikkelsen, M.D.; Møller, B.L. Microbial production of next-generation stevia sweeteners. Microb. Cell Factories 2016, 15, 207. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Rodriguez-Mateos, A.; Corona, G.; Oruna-Concha, M.J.; Spencer, J.P.E. Polyphenols and human health: Prevention of disease and mechanisms of action. Nutrients 2010, 2, 1106–1131. [Google Scholar] [CrossRef]
- Arruda, H.S.; Neri-Numa, I.A.; Kido, L.A.; Júnior, M.R.M.; Pastore, G.M. Recent advances and possibilities for the use of plant phenolic compounds to manage ageing-related diseases. J. Funct. Foods 2020, 75, 104203. [Google Scholar] [CrossRef]
- Kinghorn, A.D. Stevia: The Genus Stevia; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- De, S.; Mondal, S.; Banerjee, S. Stevioside: Technology, Applications and Health; John Wiley Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Tavarini, S.; Angelini, L.G. Stevia rebaudiana Bertoni as a source of bioactive compounds: The effect of harvest time, experimental site and crop age on steviol glycoside content and antioxidant properties. J. Sci. Food Agric. 2013, 93, 2121–2129. [Google Scholar] [CrossRef]
- Karaköse, H.; Jaiswal, R.; Kuhnert, N. Characterization and quantification of hydroxycinnamate derivatives in Stevia rebaudiana leaves by LC-MSn. J. Agric. Food Chem. 2011, 59, 10143–10150. [Google Scholar] [CrossRef]
- Kim, I.-S.; Yang, M.; Lee, O.-H.; Kang, S.-N. The antioxidant activity and the bioactive compound content of Stevia rebaudiana water extracts. LWT 2011, 44, 1328–1332. [Google Scholar] [CrossRef]
- Wölwer-Rieck, U. The leaves of Stevia rebaudiana (Bertoni), their constituents and the analyses thereof: A review. J. Agric. Food Chem. 2012, 60, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Lemus-Mondaca, R.; Vega-Gálvez, A.; Rojas, P.; Stucken, K.; Delporte, C.; Valenzuela-Barra, G.; Jagus, R.J.; Agüero, M.V.; Pasten, A. Antioxidant, antimicrobial and anti-inflammatory potential of Stevia rebaudiana leaves: Effect of different drying methods. J. Appl. Res. Med. Aromat. Plants 2018, 11, 37–46. [Google Scholar] [CrossRef]
- Pacifico, S.; Piccolella, S.; Nocera, P.; Tranquillo, E.; Poggetto, F.D.; Catauro, M. New insights into phenol and polyphenol composition of Stevia rebaudiana leaves. J. Pharm. Biomed. Anal. 2019, 163, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Tapas, A.; Sakarkar, D.; Kakde, R. Flavonoids as nutraceuticals: A review. Trop. J. Pharm. Res. 2008, 7, 1089–1099. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Chapple, C. The genetics of lignin biosynthesis: Connecting genotype to phenotype. Annu. Rev. Genet. 2010, 44, 337–363. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.A.; Zhao, Q.; Gallego-Giraldo, L.I.N.A.; Dai, X.; Zhao, P.X.; Dixon, R.A. Genome-wide analysis of phenylpropanoid defence pathways. Mol. Plant Pathol. 2010, 11, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, S. Biosynthesis and regulation of phenylpropanoids in plants. Crit. Rev. Plant Sci. 2017, 36, 257–290. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Biosynthesis of flavonoids and effects of stress. Curr. Opin. Plant Biol. 2002, 5, 218–223. [Google Scholar] [CrossRef]
- Du, H.; Huang, Y.; Tang, Y. Genetic and metabolic engineering of isoflavonoid biosynthesis. Appl. Microbiol. Biotechnol. 2010, 86, 1293–1312. [Google Scholar] [CrossRef]
- Plumb, G.W.; De Pascual-Teresa, S.; Santos-Buelga, C.; Cheynier, V.; Williamson, G. Antioxidant properties of catechins and proanthocyanidins: Effect of polymerisation, galloylation and glycosylation. Free Radic. Res. 1998, 29, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, J.; Huss, B.; Creach, A.; Hawkins, S.; Neutelings, G. Glycosylation is a major regulator of phenylpropanoid availability and biological activity in plants. Front. Plant Sci. 2016, 7, 735. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.-Q.; Sun, Y.; Guo, T.; Shi, C.-L.; Zhang, Y.-M.; Kan, Y.; Xiang, Y.-H.; Zhang, H.; Yang, Y.-B.; Li, Y.-C.; et al. UDP-glucosyltransferase regulates grain size and abiotic stress tolerance associated with metabolic flux redirection in rice. Nat. Commun. 2020, 11, 2629. [Google Scholar] [CrossRef] [PubMed]
- Brandle, J.; Richman, A.; Swanson, A.; Chapman, B. Leaf Ests from Stevia rebaudiana: A resource for gene discovery in diterpene synthesis. Plant Mol. Biol. 2002, 50, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Kahrizi, D.; Ghari, S.M.; Ghaheri, M.; Fallah, F.; Ghorbani, T.; Agha, A.B.A.; Kazemi, E.; Ansarypour, Z. Effect of KH2PO4 on gene expression, morphological and biochemical characteristics of Stevia rebaudiana Bertoni under in vitro conditions. Cell. Mol. Biol. 2017, 63, 107–111. [Google Scholar] [CrossRef]
- Ghaheri, M.; Kahrizi, D.; Bahrami, G.; Mohammadi-Motlagh, H.-R. Study of gene expression and steviol glycosides accumulation in Stevia rebaudiana Bertoni under various mannitol concentrations. Mol. Biol. Rep. 2019, 46, 7–16. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Vangelisti, A.; Guidi, L.; Cavallini, A.; Natali, L.; Piccolo, E.L.; Landi, M.; Lorenzini, G.; Malorgio, F.; Massai, R.; Nali, C.; et al. Red versus green leaves: Transcriptomic comparison of foliar senescence between two Prunus cerasifera genotypes. Sci. Rep. 2020, 10, 1959. [Google Scholar] [CrossRef]
- Xu, X.; Yuan, H.; Yu, X.; Huang, S.; Sun, Y.; Zhang, T.; Liu, Q.; Tong, H.; Zhang, Y.; Wang, Y.; et al. The chromosome-level Stevia genome provides insights into steviol glycoside biosynthesis. Hortic. Res. 2021, 8, 129. [Google Scholar] [CrossRef]
- Clemente, C.; Angelini, L.G.; Ascrizzi, R.; Tavarini, S. Stevia rebaudiana (Bertoni) as a multifunctional and sustainable crop for the mediterranean climate. Agriculture 2021, 11, 123. [Google Scholar] [CrossRef]
- Singh, G.; Singh, G.; Singh, P.; Parmar, R.; Paul, N.; Vashist, R.; Swarnkar, M.K.; Kumar, A.; Singh, S.; Singh, A.K.; et al. Molecular dissection of transcriptional reprogramming of steviol glycosides synthesis in leaf tissue during developmental phase transitions in Stevia rebaudiana Bert. Sci. Rep. 2017, 7, 11835. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, X.; Zhang, T.; Yang, Y.; Tong, H.; Yuan, H. Comparative transcriptome analysis provides insights into steviol glycoside synthesis in stevia (Stevia rebaudiana Bertoni) leaves under nitrogen deficiency. Plant Cell Rep. 2021, 40, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Q.; Lyu, C.; Chen, J.; Xiao, R.; Chen, J.; Yang, Y.; Zhang, H.; Hou, K.; Wu, W. Characterizing glycosyltransferases by a combination of sequencing platforms applied to the leaf tissues of Stevia rebaudiana. BMC Genom. 2020, 21, 794. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Pajoro, A.; Angenent, G.C. Regulation of transcription in plants: Mechanisms controlling developmental switches. Nat. Rev. Genet. 2010, 11, 830–842. [Google Scholar] [CrossRef]
- Mastrangelo, A.M.; Marone, D.; Laidò, G.; De Leonardis, A.M.; De Vita, P. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012, 185–186, 40–49. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Zhu, F.; Fan, T.; Zhang, J.; Lo, C. Alternative splicing is a Sorghum bicolor defense response to fungal infection. Planta 2020, 251, 14. [Google Scholar] [CrossRef]
- Lam, P.Y.; Wang, L.; Lo, C.; Zhu, F.-Y. Alternative splicing and its roles in plant metabolism. Int. J. Mol. Sci. 2022, 23, 7355. [Google Scholar] [CrossRef]
- Li, W.; Lin, W.-D.; Ray, P.; Lan, P.; Schmidt, W. Genome-wide detection of condition-sensitive alternative splicing in Arabidopsis roots. Plant Physiol. 2013, 162, 1750–1763. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Zhang, H.; Wang, L.; Zhu, Z.; Gao, J.; Li, C.; Zhu, Y. High temperature inhibits the accumulation of storage materials by inducing alternative splicing of OsbZIP58 during filling stage in rice. Plant Cell Environ. 2020, 43, 1879–1896. [Google Scholar] [CrossRef]
- Albaqami, M. The Splicing Factor SR45 Negatively Regulates Anthocyanin Accumulation under High-Light Stress in Arabidopsis thaliana. Life 2023, 13, 1386. [Google Scholar] [CrossRef] [PubMed]
- Mehrtens, F.; Kranz, H.; Bednarek, P.; Weisshaar, B. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005, 138, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Jahns, O.; Keck, M.; Tohge, T.; Niehaus, K.; Fernie, A.R.; Weisshaar, B. Analysis of production of flavonol glycosides-dependent flavonol glycoside accumulation in Arabidopsis thaliana plants reveals MYB11-, MYB12- and MYB111-independent flavonol glycoside accumulation. New Phytol. 2010, 188, 985–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xu, H.; Jiang, S.; Zhang, Z.; Lu, N.; Qiu, H.; Qu, C.; Wang, Y.; Wu, S.; Chen, X. MYB12 and MYB22 play essential roles in proanthocyanidin and flavonol synthesis in red-fleshed apple (Malus sieversii f. niedzwetzkyana). Plant J. 2017, 90, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Whetten, R.; Sederoff, R. Lignin biosynthesis. Plant Cell 1995, 7, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Qian, Q.; Huang, Z.; Wang, Y.; Li, M.; Hong, L.; Zeng, D.; Gu, M.; Chu, C.; Cheng, Z. GOLD HULL AND INTERNODE2 encodes a primarily multifunctional cinnamyl-alcohol dehydrogenase in rice. Plant Physiol. 2006, 140, 972–983. [Google Scholar] [CrossRef]
- Schilmiller, A.L.; Stout, J.; Weng, J.; Humphreys, J.; Ruegger, M.O.; Chapple, C. Mutations in the cinnamate 4-hydroxylase gene impact metabolism, growth and development in Arabidopsis. Plant J. 2009, 60, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gu, M.; Lai, Z.; Fan, B.; Shi, K.; Zhou, Y.-H.; Yu, J.-Q.; Chen, Z. Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiol. 2010, 153, 1526–1538. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Kim, J.I.; Tobimatsu, Y.; Ciesielski, P.N.; Anderson, N.A.; Ximenes, E.; Maeda, J.; Ralph, J.; Donohoe, B.S.; Ladisch, M.; et al. Disruption of Mediator rescues the stunted growth of a lignin-deficient Arabidopsis mutant. Nature 2014, 509, 376–380. [Google Scholar] [CrossRef]
- Villalobos-González, L.; Peña-Neira, A.; Ibáñez, F.; Pastenes, C. Long-term effects of abscisic acid (ABA) on the grape berry phenylpropanoid pathway: Gene expression and metabolite content. Plant Physiol. Biochem. 2016, 105, 213–223. [Google Scholar] [CrossRef]
- Wei, K.; Ruan, L.; Wang, L.; Cheng, H. Auxin-induced adventitious root formation in nodal cuttings of Camellia sinensis. Int. J. Mol. Sci. 2019, 20, 4817. [Google Scholar] [CrossRef]
- Li, B.; Fan, R.; Guo, S.; Wang, P.; Zhu, X.; Fan, Y.; Chen, Y.; He, K.; Kumar, A.; Shi, J.; et al. The Arabidopsis MYB transcription factor, MYB111 modulates salt responses by regulating flavonoid biosynthesis. Environ. Exp. Bot. 2019, 166, 103807. [Google Scholar] [CrossRef]
- Ding, K.; Pei, T.; Bai, Z.; Jia, Y.; Ma, P.; Liang, Z. SmMYB36, a novel R2R3-MYB transcription factor, enhances tanshinone accumulation and decreases phenolic acid content in Salvia miltiorrhiza hairy roots. Sci. Rep. 2017, 7, 5104. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Luo, T.; Guo, X.; Zou, X.; Zhou, D.; Afrin, S.; Li, G.; Zhang, Y.; Zhang, R.; Luo, Z. PgMYB2, a MeJA-responsive transcription factor, positively regulates the dammarenediol synthase gene expression in Panax Ginseng. Int. J. Mol. Sci. 2019, 20, 2219. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-G.; Mou, F.-J.; Li, K.-Z. De novo RNA sequencing and analysis reveal the putative genes involved in diterpenoid biosynthesis in Aconitum vilmorinianum roots. 3 Biotech 2021, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Guillaumie, S.; Mzid, R.; Méchin, V.; Léon, C.; Hichri, I.; Destrac-Irvine, A.; Trossat-Magnin, C.; Delrot, S.; Gomès, P.; Martel, L.; et al. The grapevine transcription factor WRKY2 influences the lignin pathway and xylem development in tobacco. Plant Mol. Biol. 2010, 72, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Avci, U.; Nakashima, J.; Hahn, M.G.; Chen, F.; Dixon, R.A. Mutation of WRKY transcription factors initiates pith secondary wall formation and increases stem biomass in dicotyledonous plants. Proc. Natl. Acad. Sci. USA 2010, 107, 22338–22343. [Google Scholar] [CrossRef] [PubMed]
- Schluttenhofer, C.; Yuan, L. Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 2015, 167, 295–306. [Google Scholar] [CrossRef]
- Yogendra, K.N.; Kumar, A.; Sarkar, K.; Li, Y.; Pushpa, D.; Mosa, K.A.; Duggavathi, R.; Kushalappa, A.C. Transcription factor StWRKY1 regulates phenylpropanoid metabolites conferring late blight resistance in potato. J. Exp. Bot. 2015, 66, 7377–7389. [Google Scholar] [CrossRef]
- Ulmasov, T.; Hagen, G.; Guilfoyle, T.J. ARF1, a transcription factor that binds to auxin response elements. Science 1997, 276, 1865–1868. [Google Scholar] [CrossRef]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Y.; Wu, C.; Yang, G.; Li, Y.; Zheng, C. Genome-wide analysis of CCCH zinc finger family in Arabidopsis and rice. BMC Genom. 2008, 9, 44. [Google Scholar] [CrossRef]
- Griffiths, S.; Dunford, R.P.; Coupland, G.; Laurie, D.A. The evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiol. 2003, 131, 1855–1867. [Google Scholar] [CrossRef] [PubMed]
- Galego, L.; Almeida, J. Role of DIVARICATA in the control of dorsoventral asymmetry in Antirrhinum flowers. Genes Dev. 2002, 16, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Bundock, P.; Hooykaas, P. An Arabidopsis hAT-like transposase is essential for plant development. Nature 2005, 436, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.Y.; Ohta, M.; Usui, A.; Shinshi, H.; Ohme-Takagi, M. Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box–mediated gene expression. Plant Cell 2000, 12, 393–404. [Google Scholar] [PubMed]
- Onai, K.; Ishiura, M. PHYTOCLOCK 1 encoding a novel GARP protein essential for the Arabidopsis circadian clock. Genes Cells 2005, 10, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Jakoby, M.; Weisshaar, B.; Dröge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; DePamphilis, C.W.; Ma, H. Phylogenetic analysis of the plant-specific zinc finger-homeobox and mini zinc finger gene families. J. Integr. Plant Biol. 2008, 50, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.K.; Jackson, R.G.; Bowles, D.J. Identification and characterisation of Arabidopsis glycosyltransferases capable of glucosylating coniferyl aldehyde and sinapyl aldehyde. FEBS Lett. 2005, 579, 2802–2806. [Google Scholar] [CrossRef]
- Caputi, L.; Malnoy, M.; Goremykin, V.; Nikiforova, S.; Martens, S. A genome-wide phylogenetic reconstruction of family 1 UDP-glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. 2012, 69, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W. The UDP-glycosyltransferase (UGT) superfamily expressed in humans, insects and plants: Animal plant arms-race and co-evolution. Biochem. Pharmacol. 2016, 99, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Honke, K.; Fukuda, M.; Narimatsu, H.; Yamaguchi, Y.; Angata, T. Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 331–336. [Google Scholar]
- Basharat, S.; Huang, Z.; Gong, M.; Lv, X.; Ahmed, A.; Hussain, I.; Li, J.; Du, G.; Liu, L. A review on current conventional and biotechnical approaches to enhance biosynthesis of steviol glycosides in Stevia rebaudiana. Chin. J. Chem. Eng. 2021, 30, 92–104. [Google Scholar] [CrossRef]
- Dewanto, V.; Wu, X.; Liu, R.H. Processed sweet corn has higher antioxidant activity. J. Agric. Food Chem. 2002, 50, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Tang, M.; Wu, J. The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chem. 1999, 64, 555–559. [Google Scholar] [CrossRef]
- Zimmermann, B.F.; Woelwer-Rieck, U.; Papagiannopoulos, M. Separation of steviol glycosides by hydrophilic liquid interaction chromatography. Food Anal. Methods 2012, 5, 266–271. [Google Scholar] [CrossRef]
- Logemann, J.; Schell, J.; Willmitzer, L. Improved method for the isolation of RNA from plant tissues. Anal. Biochem. 1987, 163, 16–20. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 February 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Simoni, S.; Clemente, C.; Usai, G.; Vangelisti, A.; Natali, L.; Tavarini, S.; Angelini, L.G.; Cavallini, A.; Mascagni, F.; Giordani, T. Characterisation of LTR-Retrotransposons of Stevia rebaudiana and Their Use for the Analysis of Genetic Variability. Int. J. Mol. Sci. 2022, 23, 6220. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Torre, S.; Tattini, M.; Brunetti, C.; Guidi, L.; Gori, A.; Marzano, C.; Landi, M.; Sebastiani, F. De novo assembly and comparative transcriptome analyses of red and green morphs of sweet basil grown in full sunlight. PLoS ONE 2016, 11, e0160370. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Mellor, J.; Wu, J.; DeLisi, C. VisANT: An online visualization and analysis tool for biological interaction data. BMC Bioinform. 2004, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Yao, S.; Dai, X.; Yin, Q.; Liu, Y.; Jiang, X.; Wu, Y.; Qian, Y.; Pang, Y.; Gao, L.; et al. Identification of UDP-glycosyltransferases involved in the biosynthesis of astringent taste compounds in tea (Camellia sinensis). J. Exp. Bot. 2016, 67, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Dai, X.; Gao, L.; Guo, L.; Zhuang, J.; Liu, Y.; Ma, X.; Wang, R.; Xia, T.; Wang, Y. Functional analysis of an uridine diphosphate glycosyltransferase involved in the biosynthesis of polyphenolic glucoside in tea plants (Camellia sinensis). J. Agric. Food Chem. 2017, 65, 10993–11001. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Total SGs (g 100 g−1 DW) | Total Phenols (mg GAE g−1 DW) | Total Flavonoids (mg CE g−1 DW) | DPPH (mmol TE g −1 DW) |
|---|---|---|---|---|
| SL | 29.59 ± 1.74 a | 118.45 ± 2.82 a | 87.14 ± 4.41 b | 0.749 ± 0.049 ab |
| L34 | 28.26 ± 2.1 a | 130.20 ± 3.56 a | 118.43 ± 3.66 a | 0.879 ± 0.044 a |
| PL | 24.85 ± 2.31 a | 89.56 ± 6.58 b | 91.13 ± 4.38 b | 0.659 ± 0.062 b |
| RG | 9.23 ± 0.79 b | 41.98 ± 0.38 c | 33.96 ± 2.44 c | 0.196 ± 0.003 c |
| Significance | *** | *** | *** | *** |
| Genotype | Rubusoside | Dulcoside A | Stevioside | Reb C | Reb A | Reb E | Reb M | Reb D |
|---|---|---|---|---|---|---|---|---|
| SL | 1.59 ± 0.004 a | 1.19 ± 0.11 b | 9.07 ± 0.37 b | 2.60 ± 0.15 b | 12.68 ± 1.03 a | 0.58 ± 0.001 b | tr | 1.80 ± 0.08 b |
| L34 | 0.48 ± 0.03 b | 2.13 ± 0.14 a | 17.14 ± 1.79 a | 1.58 ± 0.11 c | 6.32 ± 0.25 c | 0.62 ± 0.004 a | tr | nd |
| PL | 0.15 ± 0.01 d | 1.24 ± 0.05 b | 10.24 ± 1.25 b | 1.42 ± 0.06 c | 9.07 ± 0.90 b | 0.57 ± 0.001 c | tr | 2.17 ± 0.03 a |
| RG | 0.27 ± 0.001 c | 2.21 ± 0.07 a | 0.18 ± 0.06 c | 4.76 ± 0.54 a | 0.79 ± 0.07 d | nd | nd | 1.01 ± 0.05 c |
| Significance | *** | *** | *** | *** | *** | *** | ns | *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simoni, S.; Vangelisti, A.; Clemente, C.; Usai, G.; Santin, M.; Ventimiglia, M.; Mascagni, F.; Natali, L.; Angelini, L.G.; Cavallini, A.; et al. Transcriptomic Analyses Reveal Insights into the Shared Regulatory Network of Phenolic Compounds and Steviol Glycosides in Stevia rebaudiana. Int. J. Mol. Sci. 2024, 25, 2136. https://doi.org/10.3390/ijms25042136
Simoni S, Vangelisti A, Clemente C, Usai G, Santin M, Ventimiglia M, Mascagni F, Natali L, Angelini LG, Cavallini A, et al. Transcriptomic Analyses Reveal Insights into the Shared Regulatory Network of Phenolic Compounds and Steviol Glycosides in Stevia rebaudiana. International Journal of Molecular Sciences. 2024; 25(4):2136. https://doi.org/10.3390/ijms25042136
Chicago/Turabian StyleSimoni, Samuel, Alberto Vangelisti, Clarissa Clemente, Gabriele Usai, Marco Santin, Maria Ventimiglia, Flavia Mascagni, Lucia Natali, Luciana G. Angelini, Andrea Cavallini, and et al. 2024. "Transcriptomic Analyses Reveal Insights into the Shared Regulatory Network of Phenolic Compounds and Steviol Glycosides in Stevia rebaudiana" International Journal of Molecular Sciences 25, no. 4: 2136. https://doi.org/10.3390/ijms25042136
APA StyleSimoni, S., Vangelisti, A., Clemente, C., Usai, G., Santin, M., Ventimiglia, M., Mascagni, F., Natali, L., Angelini, L. G., Cavallini, A., Tavarini, S., & Giordani, T. (2024). Transcriptomic Analyses Reveal Insights into the Shared Regulatory Network of Phenolic Compounds and Steviol Glycosides in Stevia rebaudiana. International Journal of Molecular Sciences, 25(4), 2136. https://doi.org/10.3390/ijms25042136