1. Introduction
Pseudomonas aeruginosa is a multifunctional Gram-negative bacterium [
1]. It can cause inflammation, chronic persistent lung infection, and cystic fibrosis, and it has a high mortality rate [
2].
P. aeruginosa exhibits strong drug resistance because it limits the penetration rate of antibiotic molecules into cells by producing biological enzymes, reducing the permeability of the outer membrane, and promoting the efflux of antibiotics, so that it can quickly adapt to drugs [
3]. These resistance mechanisms are primarily regulated by cell density-dependent intercellular communication systems, also known as quorum sensing (QS).
QS is a chemical communication process used by bacteria to regulate their collective behavior [
4].
P. aeruginosa controls its virulence and biofilm formation through QS. Biofilms are regulated by the secretion and sensing of a series of signaling molecules referred to as autoinducers [
2]. Recent studies [
5] have reported that, compared with traditional antibiotics, quorum sensing inhibitors (QSIs) exert less selective pressure on
P. aeruginosa in terms of drug resistance; therefore, their own drug resistance evolution is also reduced [
6]; this is highly effective against Gram-negative bacteria. Therefore, the current mainstream research and development direction for inhibitors of
P. aeruginosa is to design inhibitors targeting QS systems.
Currently, there are several QSI studies on
P. aeruginosa, primarily focusing on las, rhl, and pqs systems [
7]. The key virulence regulatory receptor PqsR (also known as MvfR), an important target of the pq system, has recently attracted increasing attention from researchers. A series of studies on PqsR inhibitors have shown that PqsR inhibitors can effectively inhibit
P. aeruginosa biofilm formation and reduce the expression of virulence factor genes [
8,
9].
However, although there have been some structure–activity relationship (SAR) studies on inhibitors targeting the parent core of PqsR, most of these studies only explored the SAR of a few compounds of the same parent core, inferred the SAR based on experimental data, or verified the experimental results using docking and molecular dynamics simulations. Relevant systematic reviews are also only summaries of the SAR conclusions, and a set of hierarchical virtual screening criteria for PqsR inhibitors has not been established to guide the synthesis and virtual screening of PqsR inhibitors.
We found that there are currently multi-target ligand-based studies [
10,
11] in other drug design aspects, and this method is feasible and effective. Therefore, this study aimed to develop a set of virtual screening criteria for PqsR inhibitors based on a systematic study of the SAR of PqsR inhibitors. First, we constructed a database of PqsR inhibitors using systematic reviews, including training and testing sets. A suitable pharmacophore model was constructed based on the ligand level to identify the active groups and spatial distribution of the PqsR inhibitors. At the ligand–receptor level, molecular docking, molecular dynamics simulations, and other techniques were used to discover the core indicators of the key interactions between PqsR inhibitors and their receptors. Then, a systematic SAR-based virtual screening model was constructed; that is, the first-level screening of active compounds was based on the ligand level, and the second-level screening of potential candidate compounds was based on the ligand–receptor interaction level. Finally, our test set was screened using the existing screening methods, and the higher accuracy and superiority of multi-level screening were reflected through horizontal comparison.
2. Results and Discussion
2.1. Database Results
We obtained a total screening of ten articles [
2,
12,
13,
14,
15,
16,
17,
18,
19,
20] on PqsR inhibitors in
Table S1, and 283 compounds from these articles were built as a database in
Table S2. The training set pharmacophore, shown in
Table S3, consisted of the top 10% of the database’s activity (28 top-active compounds) and a decoy of 28 inactive compounds generated from the website
https://dude.docking.org/ [
21] (18 July 2023) in a 1:1 ratio. The molecular docking training set is shown in
Table S4 and consists of 20 active and 20 non-active compounds in the database. The molecular dynamics simulation training set is shown in
Table S5 and consists of five active compounds and five non-active compounds randomly selected from the database. The test set is shown in
Table S6 and consists of 74 active compounds in the top 30% of the database with better activity and 1600 non-active bait compounds.
2.2. Pharmacophore Results of the Training Set and Its Validation
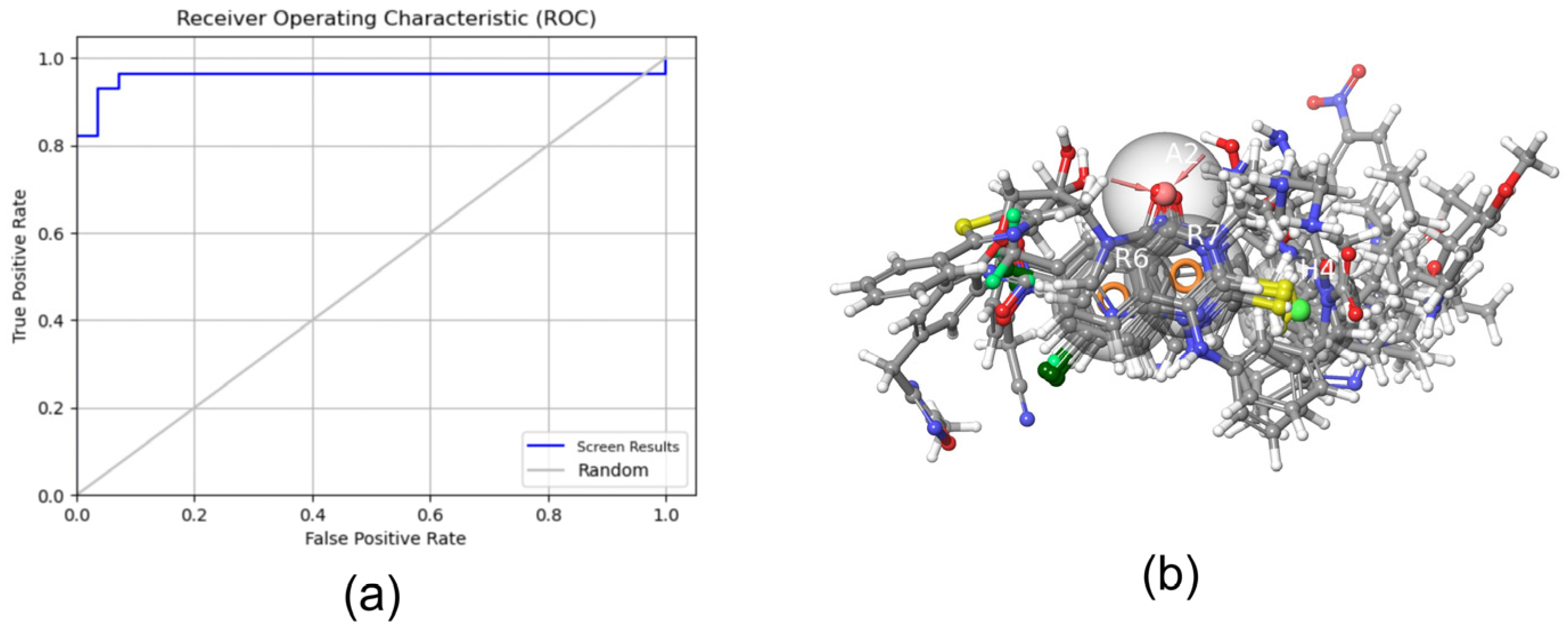
Using the
Develop Pharmacophore Model module to construct and validate the pharmacophore model (
Table 1), we determined that the ROC of the pharmacophore model AHRR_2 was 0.96 and its AUC was 0.74, both >0.7 (
Figure 1a). Therefore, it served as a candidate pharmacophore model. Further analysis of AHRR_2 showed that the model was composed of a hydrogen bond acceptor feature, two aromatic ring features, and one or two hydrophobic regions. This is also in accordance with the basic structure of quinazolinones and quinolones, which consist of an aromatic ring and a six-membered nitrogen heterocycle forming two aromatic groups. The carbonyl group in the heterocycle is a hydrogen-bond acceptor, whereas the long carbon chain is in the hydrophobic region. The active compounds in the pharmacophore training set were matched with the AHRR_2 pharmacophore (
Table 2 and
Figure 1b), and the good degree of matching indicated that the pharmacophore AHRR_2 can identify the key active groups and the spatial distribution of PqsR inhibitors. Therefore, we determined the following ligand-based screening criterion: the PqsR inhibitor should exactly match the four pharmacophore groups in AHRR_2.
2.3. Molecular Docking Results of the Training Set
The
Ligand docking module was used to construct and verify molecular docking criteria. Twenty active and twenty non-active compounds were analyzed in the training set.
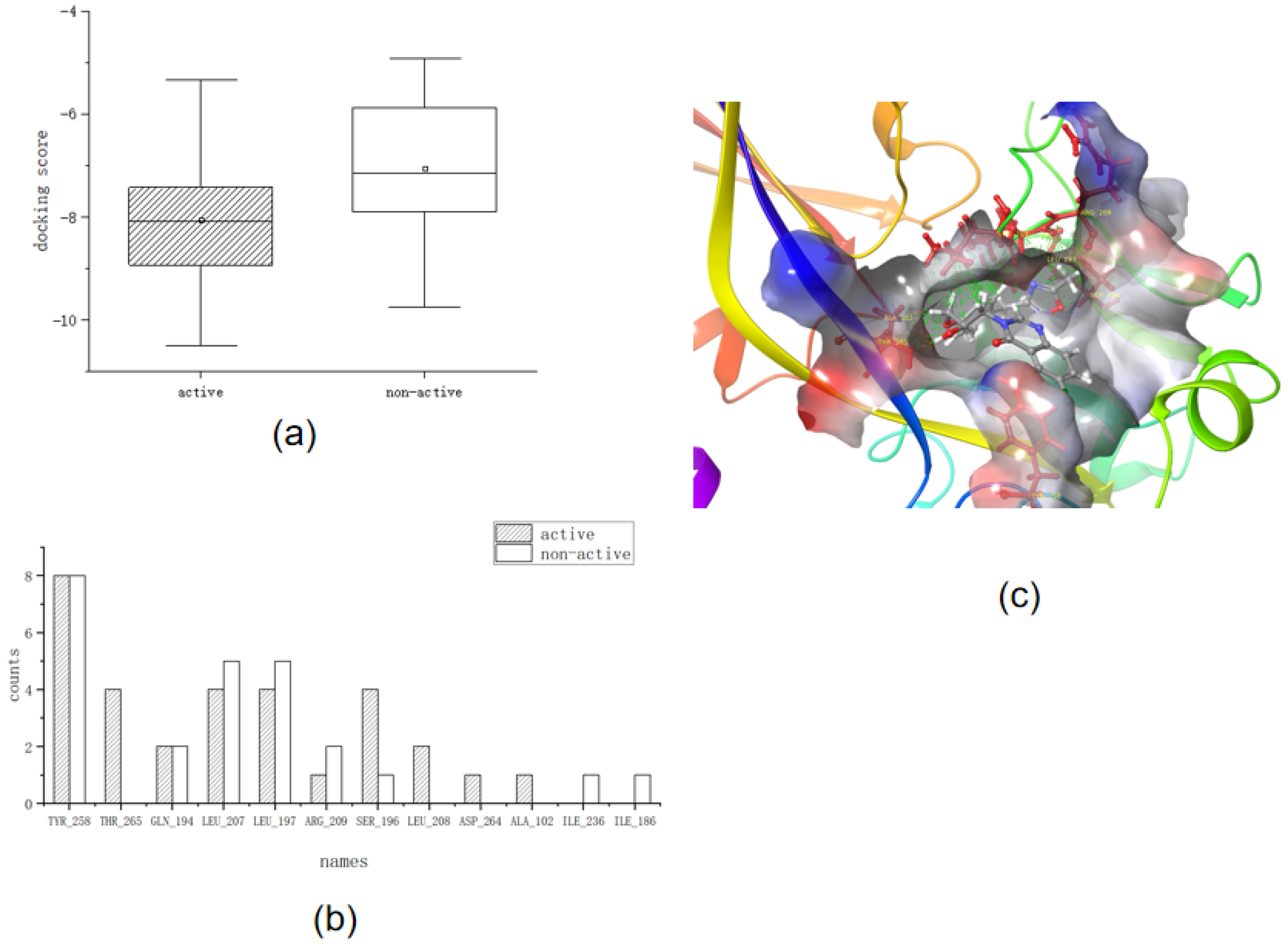
Table 3 and
Figure 2a show the docking scores for the molecular docking of the training set, and
Figure 2b,c show the key interactions of the training set.
First, we analyzed the docking scores. We determined that the main part of the docking score box plot ranged from −7.5 to −10 for active compounds, whereas the docking score box plot body ranged from −6 to −8 for non-active compounds. This indicated that the overall docking score of the active compounds was lower than that of the non-active compounds, which reflected a better interaction between the ligand and the receptor-docking conformation of the active compounds than that of the non-active compounds.
Second, we analyzed key interactions. Most of the active compounds interacted with TYR_258 through water bridges with the carbonyl group on the heterocycle. However, ARG_209, LEU_208, and others interact with hydrophobic groups such as hydrogen or methyl groups of long carbon chains. THR_265 is unique in that it reacts with long chains of nitrogen substituents close to the heterocyclic carbonyl group, such as the hydroxyl group of 16. From the frequency analysis table of amino acid residues, we determined that TYR_258, LEU_197, LEU_207, SER_196, and THR_265 were frequently involved in this interaction. The higher the frequency of the interaction, the more likely it was to be a key amino acid residue for ligand binding.
Thus, we determined the screening criteria in molecular docking to be as follows:
The docking score should be <−8.
The structure of the compound requires two aromatic rings that are directly connected and a hydrophobic long chain structure such as a carbon chain or benzene ring, which is inserted into the protein pocket for anchoring (the pocket is preferably hydrophobic as a whole).
The compound should interact with the six amino acid residues TYR_258, LEU_197, LEU_207, SER_196, THR_265, and LEU_208.
The hydrophobic group in the structure of the compound is close to ARG_209 (2.26 − 6.91 Å) or the compound interacts with ARG_209.
2.4. Molecular Dynamics Simulations and Binding Free Energy Results of the Training Set
The
Molecular Dynamics module in Desmond was used to perform molecular dynamics simulations, and the
MMGBSA module was used to calculate the binding free energy. Five active and five non-active compounds were randomly selected for molecular dynamics simulation.
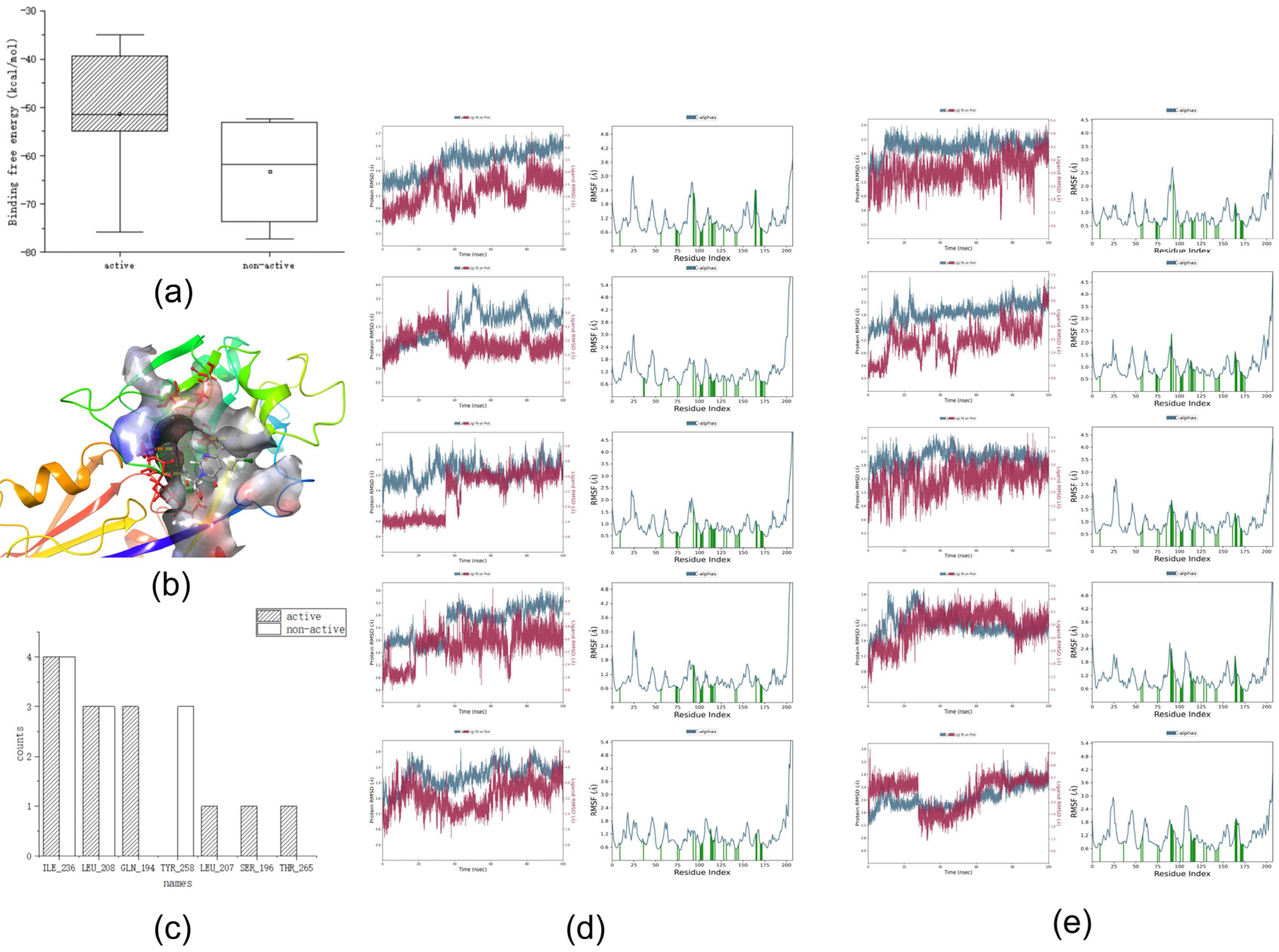
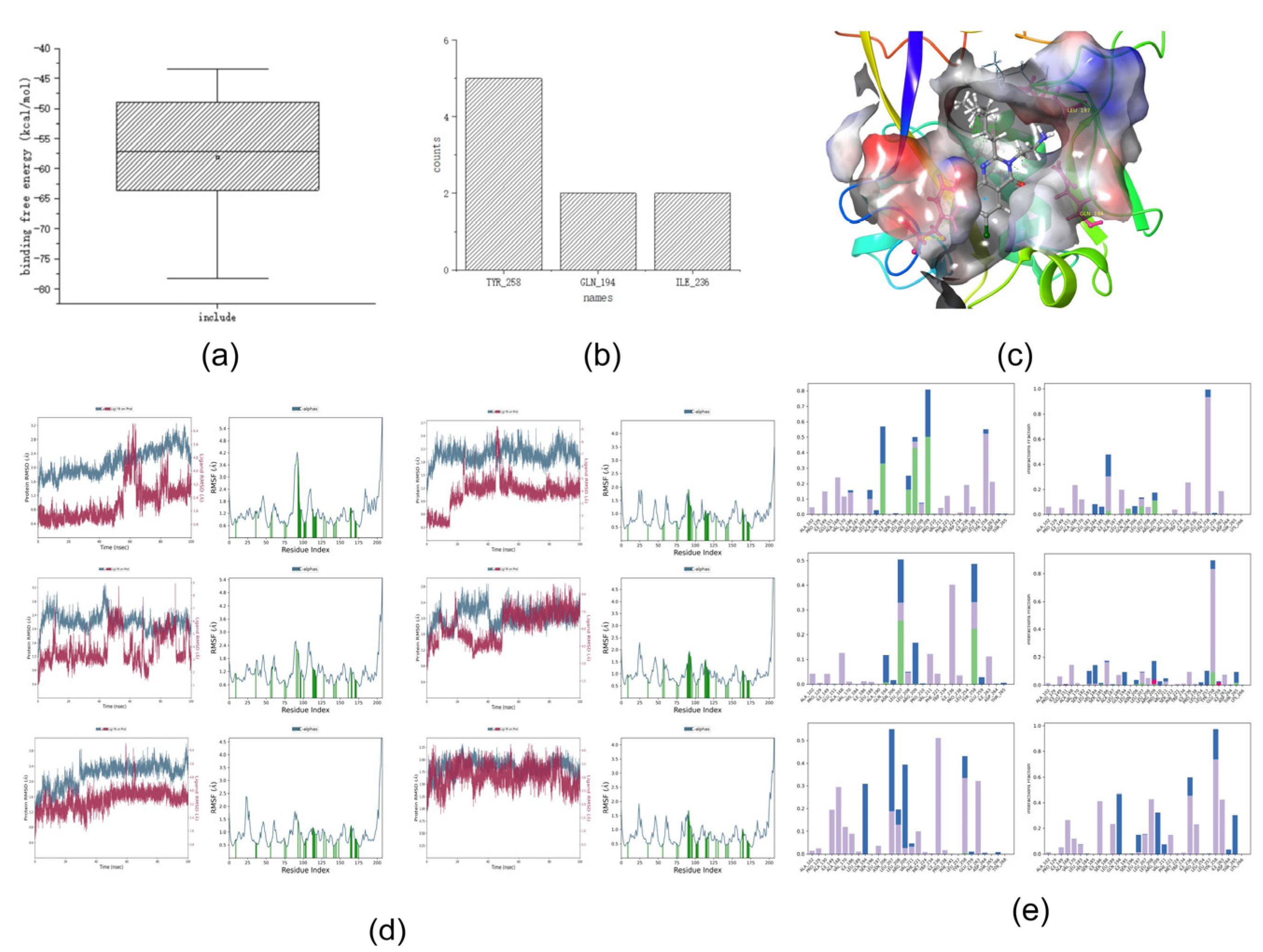
Figure 3a shows the binding free energies of the training set,
Figure 3b,c show the interactions in the dynamics, and
Figure 3d,e show the RMSD and RMSF values in the dynamic simulation.
First, we analyzed the RMSF and RMSD. Compounds with five activities in the training set exhibited large fluctuations in the early stage. Analysis of the three-dimensional (3D) structure map showed that if the hydrophobic tail was a long carbon chain, it would lead to large fluctuations in the RMSF and RMSD values of the compounds. This may be due to the heterogeneous interactions between the carbon chain and the surrounding hydrophobic groups, such as those in compound 106. However, when a benzene ring or another nitrogen-containing heterocyclic ring is added to the hydrophobic end, such as in compound 82, the initial fluctuation is small and stability can be achieved quickly. This may be because the benzene ring or nitrogen-containing hydrophobic heterocyclic ring is relatively structurally stable, and it is difficult to change the spatial structure position because of the surrounding water molecular force field. Simultaneously, we observed that the RMSF and RMSD values of the compound tended to be stable at approximately 80 ns; therefore, we focused on the analysis of the key force magnitude after stabilization (80–100 ns).
Second, we analyzed the dynamic interactions. From the active compounds, it can be concluded that TYR_258, ILE_236, LEU_208, and GLN_194 interacted with all the active compounds over a long time. This phenomenon may indicate that TYR_258, ILE_236, LEU_208, and GLN_194 are the key amino acids that interact with the compounds during the molecular dynamics simulation.
Finally, the binding free energies were analyzed. We determined that the binding free energy box plots of active compounds ranged from −40 to −55 kcal/mol in the main part, whereas those of non-active compounds ranged from −53 to −70 kcal/mol (
Figure 3). No significant differences were observed between the binding free energies of active and non-active sites. Because the binding free energy reflects the binding capacity, a lower value indicates that the compound is tightly bound to an amino acid residue. Therefore, considering the above analysis, we hypothesize that the binding free energy of an active compound should be <−40 kcal/mol.
After summarizing the above, we determined the following screening criteria for molecular dynamics:
The analysis of the binding free energy is best performed after stabilization (80–100 ns).
The compound should, if possible, interact continuously with the following four amino acid residues: TYR_258, ILE_236, LEU_208, and GLN_194.
The compound should, if possible, have a binding free energy (dG) of <−40 kcal/mol, and the main part should range from −40 to −55 kcal/mol.
2.5. Pharmacophore Results of the Test Set
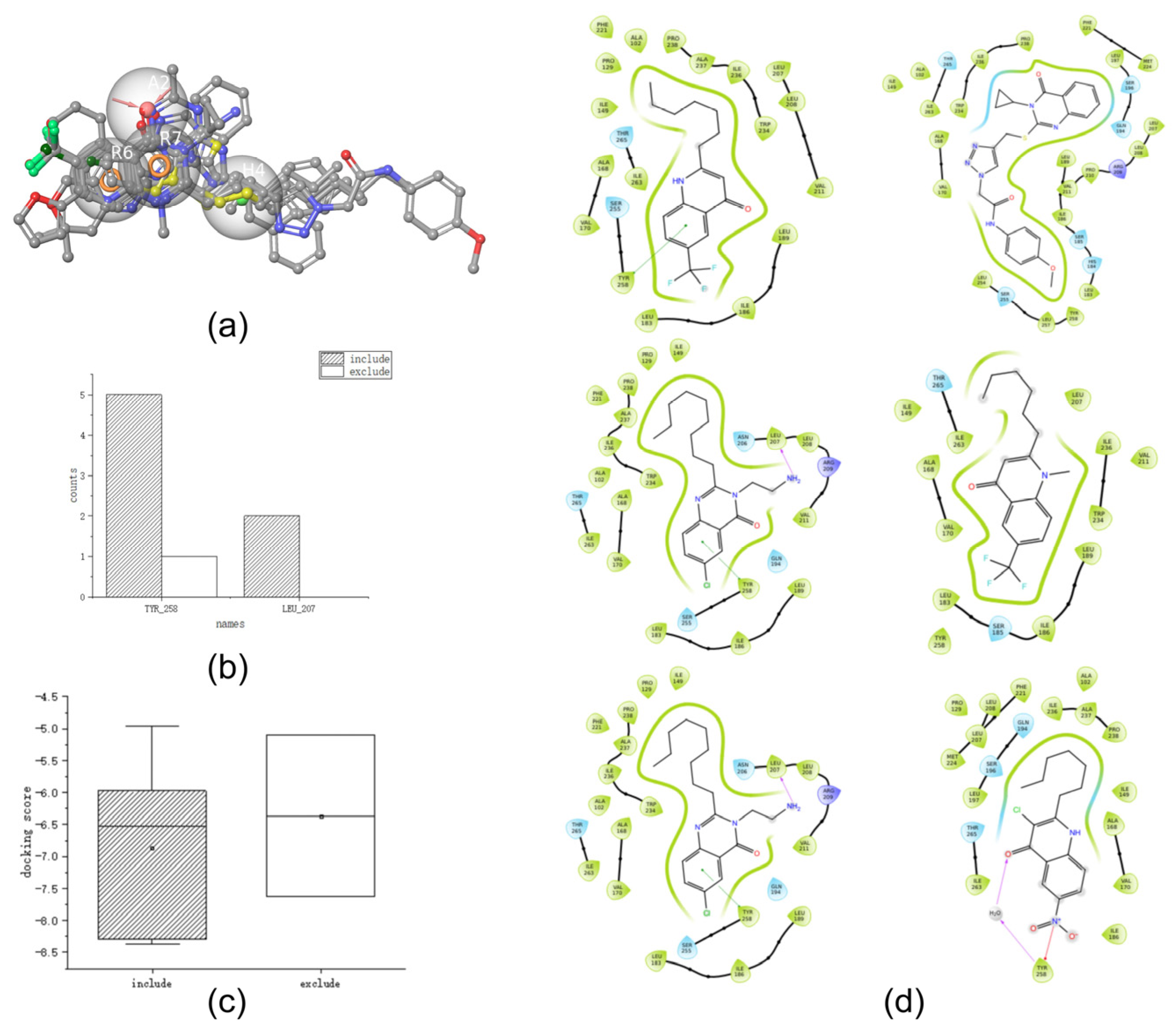
After importing the test set into the pharmacophore, the top ten compounds were analyzed from high to low pharmacophore scores (
Table 4). Following the analysis, all results were in line with the conclusions obtained from the training set; that is, all four pharmacophore groups in AHRR_2 were matched and the 3D structure diagram was constructed (
Figure 4a), which is in line with the conclusions drawn in
Section 2.2. Therefore, all ten compounds were retained for further verification.
2.6. Molecular Docking Results of the Test Set
Based on the pharmacophore results above, we proceeded with further validation using the docking scores and key interactions.
Table 5 and
Figure 4b,c show the docking scores and key docking interaction diagrams, and
Figure 4d shows the 2D docking diagram of the active compounds of the test set with protein amino acid residues. Based on the docking score analysis, the docking scores of
232,
56, and
107 were all <−8 kcal/mol, which was consistent with the docking score conclusion drawn in
Section 2.3. Key docking interaction analysis showed that compounds
232,
56,
107, and
253 interacted with TYR_258. Furthermore, compounds
183 and
280 interacted with LEU_207, and the hydrophobic groups on these compounds were close to or interacted with ARG_209. These results are consistent with those obtained in
Section 2.3 for key interactions between PqsR inhibitors and PqsR receptors. Together with the docking scores and docking interaction analyses described above, these six compounds fit the conclusions of molecular docking in the training set. Therefore, these six compounds were retained for further validation. Four other compounds were excluded because they either failed to interact with the key amino acid residues (TYR_258, LEU_197, LEU_207, SER_196, THR_265, and LEU_208) or had docking scores of >−8 kcal/mol.
2.7. Molecular Dynamics Simulations and Binding Free Energy Results of the Test Set
Based on the molecular docking results, further validation was performed from molecular dynamics simulations and binding free energies.
Table 6 and
Figure 5a show the binding free energy results of the test set, and
Figure 5b,c show the persistent interactions between the compounds and amino acid residues in the molecular dynamics simulations.
Figure 5d shows the RMSD and RMSF values.
Figure 5e shows the long-term interaction between compounds and amino acid residues.
First, we analyzed the RMSD and RMSF. The values of these six compounds tended to be stable at 80 ns and 100 ns, which is in line with the conclusion of
Section 2.4. Second, we analyzed dynamic interactions, as shown in
Table 6 (note: when the interaction fraction between the amino acid residues and the compound is >0.5, it is considered to have a long-term interaction, indicated by “√”). We determined that six compounds interacted with three amino acid residues, TYR_258, GLN_194, and ILE_236, for a long time. For example, compounds 280 and 56 had a persistent interaction with GLN_194, and 183 had a persistent interaction with ILE_236, which is consistent with the conclusions of
Section 2.4. Finally, we analyzed the binding free energy and determined that the binding free energy dG of the above six compounds was <−40 kcal/mol, which was in line with the conclusion of
Section 2.4 regarding the active binding free energy. Collectively, the results of the molecular dynamics simulations and binding free energies suggested that all six of the above candidates were active compounds.
In this study, a multidimensional virtual screening model for PqsR inhibitors was constructed using the training set, and multidimensional hierarchical screening was performed on the test set based on the evaluation criteria of the virtual screening model, including pharmacophore screening based on ligand conformation. Based on the screening of molecular docking of receptor–ligand interactions, molecular dynamics simulations, and binding free energy, six candidate compounds were selected. These six compounds were all active, indicating that the multidimensional virtual screening model constructed in this study can reliably identify active PqsR inhibitors.
2.8. Comparison of Results with Existing Screening Criteria
To further demonstrate the superiority of the multidimensional screening model, we selected two articles that searched for PqsR quorum sensing inhibitors using computer software [
20,
21]. The difference is that these two studies only considered a single-dimension aspect, that is, molecular docking and molecular dynamics simulations of compounds and binding free energy calculations based on receptors only. Among them, Vieira [
22] et al. suggested that the two residues Ile_186 and Tyr_258 may be the key to the activation mechanism of MvfR (PqsR). Shandil [
20] et al. suggested that the higher level of pqs inhibition may be due to the hydrogen bond interaction with GLN_194, LEU_207, THR_265, and SER_196; the pi sulfur bond with PHE_221; and the hydrophobic interaction with TYR_258 and LEU_197. Therefore, we used the above conclusions as screening criteria and verified them using the test set in this study.
First, we downloaded two protein crystal structures (PDBID: 4JVI and 6B8A) selected from the two studies, preprocessed the molecules in the test set, and performed molecular docking with the two crystal structures. The compounds were ranked from high to low based on the absolute value of the docking scores, and the compounds that were consistent with the conclusions in the literature were retained. The top ten compounds were analyzed and the screening accuracy was calculated (
Table 7).
The results showed that only seven of the top ten compounds were active after protein structure 4JVI docking, including three false-positive results, with an accuracy of 57.1%. After docking the protein structure 6B8A, the hit rates of the top ten compounds were not high enough (55.6% and 71.4%, respectively) (
Table 7). In contrast, the ligand-based pharmacophores followed by receptor-based docking screening used in this study identified six active compounds out of the top ten docking scores, and no false positives were indicated. This reflects the advantages of multidimensional screening.
3. Discussion
In this study, a database was constructed based on existing studies on the reported inhibitors of PqsR. Based on this database, a pharmacophore model of the PqsR inhibitor was established, and key interactions between the PqsR inhibitor and its receptor were investigated using docking and molecular dynamics techniques. Finally, a set of multidimensional virtual screening criteria considering both the structural characteristics of the ligand and the key interaction characteristics of the receptor and ligand was successfully developed.
As an important aspect of the pq system, the role of PqsR should not be underestimated. Once HHQ/PQS activates PqsR, it induces its own biosynthesis with an exponential increase in the concentration of signaling molecules [
23]. Some studies have shown that PqsR can also bind to and directly control various sites expressed throughout the genome of
P. aeruginosa, including major regulators such as LasR and RhlR, as well as genes involved in protein translation, secretion, and response to oxidative stress [
8]. Therefore, it is important to identify PqsR inhibitors as a potential strategy to reverse
P. aeruginosa resistance. However, currently, the discovery and screening strategies for inhibitors are primarily traditional structure derivation and activity screening, which are of great application value to accelerate the discovery of inhibitors.
Virtual screening is an effective method to identify candidate compounds; however, the use of a single technology (single ligand-based or single key interaction-based) often leads to an increase in false positives or false negatives. For example, Ji et al. [
24] used 5′ -O-(N-acylsulfonyl) adenosine (acyl-AMS) and related compounds to mimic homologous and tightly bound acyl-AMP intermediates to inhibit such enzymes by designing inhibitors related to the target PqsA upstream of the PQS system. This operation has its own rationality, but it cannot control the activity of the newly designed compound because of the lack of structure–activity relationship analysis. “Vinyl sulfonamide analogues 6 and 7 were excluded due to a lack of biochemical potency”, the article states. In contrast, salicyl-AMS (3), salicyl-AMSN (4), and benzoyl-AMS are inhibitors of quinolone production but are much less potent [
24]. To further improve the accuracy of virtual screening, systematic integration of pharmacophore modeling, molecular docking, molecular dynamics, and other methods is required to construct multidimensional virtual screening criteria. For example, Luo et al. [
25] applied Pharmacome modeling, virtual screening, molecular docking, ADMET, and molecular dynamics simulations to identify potential PD-L1 inhibitors in a marine natural product library. In this study, pharmacophore-based virtual screening was performed using a marine natural product library of 52765 compounds according to the generated pharmacophore model. Twelve compounds conforming to all the pharmacophore properties were generated, and molecular docking was performed. Finally, the selected compounds, 51320 and 37080, were analyzed using molecular dynamics simulations, which confirmed that the compound and protein binding sites were stable. In addition, high dG binding values were observed for MM/GBSA, as calculated from individual trajectories, indicating the long-term stability of the selected protein–ligand complexes. Compound 51320 exhibited low toxicity and good activity. The data and results obtained also reflect the feasibility and accuracy of virtual screening based on various SAR analysis techniques.
At present, some studies on the structure–activity relationship and virtual screening of PqsR inhibitors exist. For example, Zender et al. [
26] further synthesized compounds based on 2-aminoxediazole as the backbone, evaluated their activity in Escherichia coli, and analyzed their structure–activity relationship. Based on the aminopyridine derivatives of fragments, Zender et al. [
27] determined the activity of the fragment compounds and screened new inhibitors with aminopyridine as the backbone using the fragment growth method. Vieira et al. [
22] performed molecular docking, molecular dynamics simulation, and MM/GBSA free energy calculations on 64 known drugs and protein pockets to confirm docking prediction and clarify interaction modes; the ROC and AUC were measured to be approximately 0.5. The last five compounds were identified as promising PqsR inhibitors. The above studies fully prove the feasibility of traditional compound screening based on structure–activity relationships, fragment growth, and computer-assisted virtual screening for drug development; however, they are only based on a single dimension of screening: whether it is an activity assay based on a certain skeleton or a simple virtual screening based on receptors, certain limitations exist. It is reasonable to consider that if the evaluation index of virtual screening considers both the structural characteristics of the ligand and the key ligand–receptor interactions, the prediction accuracy will be improved.
Therefore, in this study, we used a ligand-based pharmacophore model, receptor–ligand molecular docking, and molecular dynamics simulation techniques to analyze the structure–activity relationships of compounds to construct a multidimensional virtual screening model and standard for PqsR inhibitors. The first level of ligand-based pharmacophore screening concluded that compounds should cover two aromatic rings, a hydrogen bond acceptor, and two hydrophobic groups; The second level of screening based on molecular docking and molecular dynamics simulation of the receptor concluded that the docking score should be <−8 kcal/mol, the compound should interact continuously with four amino acid residues as much as possible (TYR_258, ILE_236, LEU_208, and GLN_194), and the dG of the compound should be <−40 kcal/mol. This test set was used for further horizontal verification. By comparing the results of the test set screened according to the screening criteria of a single dimension with the results of multidimensional screening in this study, we determined that the accuracy of multidimensional screening was higher. Therefore, we believe that this virtual screening model can be applied for the subsequent development of PqsR inhibitors for P. aeruginosa.
However, the results of the present study require further improvement. First, the sample size was insufficiently large. Only ten studies were included in this review, and the total number of compounds in these studies was not more than 300. Further expansion of the number of compounds is required. Second, the methods used to measure the activity data and the strains used were different in different studies, and the activity indices were not the same. Some studies have used IC50 values, whereas others have used MIC values. Therefore, at present, our evaluation model primarily focuses on qualitative indicators, that is, whether the candidate compounds are PqsR inhibitors, and cannot make further quantitative judgments. Despite the limitations of this study, our research group is committed to studying drug-resistant bacteria. Based on the multidimensional virtual screening model of PqsR inhibitors constructed in this study, we will continue to perform the strategy of “virtual screening-activity validation-optimization model” to find new PqsR inhibitors. In this process, the database of PqsR inhibitors was continuously expanded and the evaluation criteria for pharmacophores, molecular docking, and molecular dynamics were optimized to continuously improve the prediction accuracy of the multidimensional virtual screening model of PqsR inhibitors.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}