
Tumor Necrosis Factor-α Receptor 1 Mediates Borna Disease Virus 1-Induced Changes in Peroxisomal and Mitochondrial Dynamics in Neurons


Abstract
1. Introduction
2. Results
2.1. Differences in Abundances of Peroxisomes, Mitochondria, Catalase, and SOD2 in Different Brain Regions of Non-Infected Wt Mice
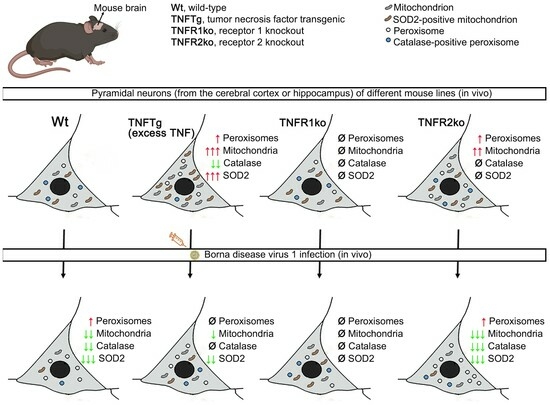
2.2. The Abundances of Peroxisomes, Mitochondria, Catalase, and SOD2 Differ in TNFTg Compared to Wt Mice
2.3. Similar Abundances of Peroxisomes, Mitochondria, Catalase, and SOD2 in Wt and TNFR1ko Mice, but a Higher Organelle Abundance with No Change in Antioxidant Enzymes in TNFR2ko Mice
2.4. Response of Wt Mice to BoDV1 Infection with Regard to Abundances of Peroxisomes, Mitochondria, Catalase, and SOD2
2.5. Response of TNFtg Mice to BoDV1 Infection with Regard to Abundances of Peroxisomes, Mitochondria, Catalase, and SOD2
2.6. Response of TNFR1- and TNFR2ko Mice to BoDV1 Infection with Regard to Abundances of Peroxisomes, Mitochondria, Catalase and SOD2
3. Discussion
3.1. Differences in the Abundances of Peroxisomes and Mitochondria as Well as Their Main Antioxidant Enzymes Catalase and SOD2 in the Brains of Wt, TNFTg, TNFR1ko, and TNFR2ko Mice
3.2. BoDV1 Induced Changes in the Abundances of Peroxisomes, Mitochondria, Catalase, and SOD2 in the Brains of Wt Mice
3.3. Differences in Responses after BoDV1 Infection of TNFTg, TNFR1ko, and TNFR2ko Mice in Comparison to Wt Mice with Regard to the Abundances of Peroxisomes and Mitochondria, Catalase, and SOD2
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Mouse Lines and Genotyping
4.2.2. Preparation of BoDV1 Suspension and Experimental Infection of Mice
4.2.3. Brain Tissue Perfusion and Preparation
4.2.4. IF Staining of Formalin-Fixed, Paraffin-Embedded (FFPE) Mouse Brain Tissue
4.2.5. Fluorescence Microscopic Morphometry of Peroxisomes and Mitochondria and Their Antioxidant Enzymes
4.2.6. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | activator protein-1 |
| ATP5B | adenosine triphosphate synthase F1 subunit beta |
| BD | Borna disease |
| BoDV1 | Borna disease virus 1 |
| InfTNFR1ko | BoDV1-infected TNFR1ko |
| InfTNFR2ko | BoDV1-infected TNFR2ko |
| InfTNFTg | BoDV1-infected TNFTg |
| InfWt | BoDV1-infected Wt |
| BSA | bovine serum albumin |
| CA | cornu ammonis |
| Cb | cerebellar cortex |
| CNS | central nervous system |
| Cx | cerebral cortex |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DG | dentate gyrus |
| DMEM | Dulbecco’s modified Eagle’s medium |
| dpi | days post-infection |
| H2O2 | hydrogen peroxide |
| IFN | interferon |
| IL | interleukin |
| IRFs | IFN regulatory factors |
| MAVS | mitochondrial antiviral signaling protein |
| NF-κB | nuclear factor kappa B |
| NR2B | N-methyl-D-aspartate 2b receptor subtype |
| PBS | phosphate-buffered saline |
| PEX | peroxin |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| PPAR | peroxisome proliferator-activated receptor |
| RLRs | retinoic-acid-inducible gene I (RIG-I)-like receptors |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| TNFR | tumor necrosis factor-α receptor |
| TNFR1ko | TNF receptor 1 knockout |
| TNFR2ko | TNF receptor 2 knockout |
| TNFTg | TNF transgenic |
| TNF | tumor necrosis factor alpha |
| VLCFA | very-long-chain fatty acid |
| Wt | wild type |
References
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef]
- Tizard, I.; Ball, J.; Stoica, G.; Payne, S. The pathogenesis of bornaviral diseases in mammals. Anim. Health Res. Rev. 2016, 17, 92–109. [Google Scholar] [CrossRef]
- Ackermann, A.; Kugel, D.; Schneider, U.; Staeheli, P. Enhanced polymerase activity confers replication competence of Borna disease virus in mice. J. Gen. Virol. 2007, 88 Pt 11, 3130–3132. [Google Scholar] [CrossRef]
- Staeheli, P.; Sauder, C.; Hausmann, J.; Ehrensperger, F.; Schwemmle, M. Epidemiology of Borna disease virus. J. Gen. Virol. 2000, 81 Pt 9, 2123–2135. [Google Scholar] [CrossRef]
- Herden, C.; Briese, T.; Lipkin, W.I.; Richt, J.A. Bornaviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1124–1150. [Google Scholar]
- Shankar, V.; Kao, M.; Hamir, A.N.; Sheng, H.; Koprowski, H.; Dietzschold, B. Kinetics of virus spread and changes in levels of several cytokine mRNAs in the brain after intranasal infection of rats with Borna disease virus. J. Virol. 1992, 66, 992–998. [Google Scholar] [CrossRef]
- Stitz, L.; Bilzer, T.; Planz, O. The immunopathogenesis of Borna disease virus infection. Front. Biosci. 2002, 7, 541–555. [Google Scholar] [CrossRef]
- Kramer, K.; Schaudien, D.; Eisel, U.L.; Herzog, S.; Richt, J.A.; Baumgärtner, W.; Herden, C. TNF-overexpression in Borna disease virus-infected mouse brains triggers inflammatory reaction and epileptic seizures. PLoS ONE 2012, 7, e41476. [Google Scholar] [CrossRef]
- Schlottau, K.; Forth, L.; Angstwurm, K.; Höper, D.; Zecher, D.; Liesche, F.; Hoffmann, B.; Kegel, V.; Seehofer, D.; Platen, S.; et al. Fatal encephalitic Borna disease virus 1 in solid-organ transplant recipients. N. Engl. J. Med. 2018, 379, 1377–1379. [Google Scholar] [CrossRef] [PubMed]
- Niller, H.H.; Angstwurm, K.; Rubbenstroth, D.; Schlottau, K.; Ebinger, A.; Giese, S.; Wunderlich, S.; Banas, B.; Forth, L.F.; Hoffmann, D.; et al. Zoonotic spillover infections with Borna disease virus 1 leading to fatal human encephalitis, 1999–2019: An epidemiological investigation. Lancet Infect. Dis. 2020, 20, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Korn, K.; Coras, R.; Bobinger, T.; Herzog, S.M.; Lücking, H.; Stöhr, R.; Huttner, H.B.; Hartmann, A.; Ensser, A. Fatal encephalitis associated with Borna disease Virus 1. N. Engl. J. Med. 2018, 379, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- Nobach, D.; Müller, J.; Tappe, D.; Herden, C. Update on immunopathology of bornavirus infections in humans and animals. Adv. Virus Res. 2020, 107, 159–222. [Google Scholar] [CrossRef]
- Hallensleben, W.; Schwemmle, M.; Hausmann, J.; Stitz, L.; Volk, B.; Pagenstecher, A.; Staeheli, P. Borna disease virus-induced neurological disorder in mice: Infection of neonates results in immunopathology. J. Virol. 1998, 72, 4379–4386. [Google Scholar] [CrossRef]
- Hofer, M.; Hausmann, J.; Staeheli, P.; Pagenstecher, A. Cerebral expression of interleukin-12 induces neurological disease via differential pathways and recruits antigen-specific T cells in virus-infected mice. Am. J. Pathol. 2004, 165, 949–958. [Google Scholar] [CrossRef]
- Hirz, M. Pathogeneses of Epileptiform Convulsions in TNF-Transgenic Mice after Borna Disease Virus Infection: Role of Astroglia. Ph.D. Thesis, Institute of Veterinary Pathology, Justus-Liebig University, Giessen, Germany, 2017. [Google Scholar]
- Richt, J.A.; Grabner, A.; Herzog, S. Borna disease in horses. Vet. Clin. N. Am. Equine Pract. 2000, 16, 579–595. [Google Scholar] [CrossRef]
- Tappe, D.; Frank, C.; Offergeld, R.; Wagner-Wiening, C.; Stark, K.; Rubbenstroth, D.; Giese, S.; Lattwein, E.; Schwemmle, M.; Beer, M.; et al. Low prevalence of Borna disease virus 1 (BoDV-1) IgG antibodies in humans from areas endemic for animal Borna disease of Southern Germany. Sci. Rep. 2019, 9, 20154. [Google Scholar] [CrossRef]
- Kanda, T.; Tomonaga, K. Reverse genetics and artificial replication systems of Borna disease Virus 1. Viruses 2022, 14, 2236. [Google Scholar] [CrossRef]
- Neumann, B.; Hierl, A.; Wunderlich, S.; Meier, H.; Bauer, C.; Gerner, S.T.; Rieder, G.; Geis, T.; Kunkel, J.; Bauswein, M.; et al. Cerebrospinal fluid in Borna disease virus 1 (BoDV-1) encephalitis. J. Neurol. Sci. 2023, 446, 120568. [Google Scholar] [CrossRef]
- Staeheli, P.; Sentandreu, M.; Pagenstecher, A.; Hausmann, J. Alpha/beta interferon promotes transcription and inhibits replication of borna disease virus in persistently infected cells. J. Virol. 2001, 75, 8216–8223. [Google Scholar] [CrossRef] [PubMed]
- Szelechowski, M.; Bétourné, A.; Monnet, Y.; Ferré, C.A.; Thouard, A.; Foret, C.; Peyrin, J.M.; Hunot, S.; Gonzalez-Dunia, D. A viral peptide that targets mitochondria protects against neuronal degeneration in models of Parkinson’s disease. Nat. Commun. 2014, 5, 5181. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, M. Tumor necrosis factor alpha: A major cytokine of brain neuroinflammation. IntechOpen. 2019. Available online: https://www.intechopen.com/chapters/68123 (accessed on 20 December 2023).
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef]
- Probert, L.; Akassoglou, K.; Kassiotis, G.; Pasparakis, M.; Alexopoulou, L.; Kollias, G. TNF-α transgenic and knockout models of CNS inflammation and degeneration. J. Neuroimmunol. 1997, 72, 137–141. [Google Scholar] [CrossRef]
- Jean Beltran, P.M.; Cook, K.C.; Hashimoto, Y.; Galitzine, C.; Murray, L.A.; Vitek, O.; Cristea, I.M. Infection-induced peroxisome biogenesis is a metabolic strategy for Herpesvirus replication. Cell Host Microbe 2018, 24, 526–541.e7. [Google Scholar] [CrossRef]
- Elesela, S.; Lukacs, N.W. Role of mitochondria in viral infections. Life 2021, 11, 232. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 8, 1126. [Google Scholar] [CrossRef]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Terlecky, S.R.; Terlecky, L.J.; Giordano, C.R. Peroxisomes, oxidative stress, and inflammation. World J. Biol. Chem. 2012, 3, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Trompier, D.; Vejux, A.; Zarrouk, A.; Gondcaille, C.; Geillon, F.; Nury, T.; Savary, S.; Lizard, G. Brain peroxisomes. Biochimie 2014, 98, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Del Río, L.A. Peroxisomes as a cellular source of reactive nitrogen species signal molecules. Arch. Biochem. Biophys. 2011, 506, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.V.; Chernov, N.N.; Saprin, A.N. Involvement of thio-, peroxi-, and glutaredoxins in cellular redox-dependent processes. Biochemistry 2008, 73, 1493–1510. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Marques, M.; Ribeiro, D. Peroxisomes and innate immunity: Antiviral response and beyond. Int. J. Mol. Sci. 2019, 20, 3795. [Google Scholar] [CrossRef]
- Poenisch, M.; Burger, N.; Staeheli, P.; Bauer, G.; Schneider, U. Protein X of Borna disease virus inhibits apoptosis and promotes viral persistence in the central nervous systems of newborn-infected rats. J. Virol. 2009, 83, 4297–4307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jin, D.H.; Jung, Y.W.; Ko, B.H.; Moon, I.S. Immunoblot analyses on the differential distribution of NR2A and NR2B subunits in the adult rat brain. Mol. Cells 1997, 7, 749–754. [Google Scholar] [CrossRef]
- Watanabe, M.; Inoue, Y.; Sakimura, K.; Mishina, M. Distinct distributions of five N-methyl-D-aspartate receptor channel subunit mRNAs in the forebrain. J. Comp. Neurol. 1993, 338, 377–390. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and non-signaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef]
- Raziyeva, K.; Kim, Y.; Zharkinbekov, Z.; Kassymbek, K.; Jimi, S.; Saparov, A. Immunology of acute and chronic wound healing. Biomolecules 2021, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, F.; Hayashi, H. Significance of catalase in peroxisomal fatty acyl-CoA beta-oxidation. Biochim. Biophys. Acta 1987, 921, 142–150. Available online: https://pubmed.ncbi.nlm.nih.gov/2887206/ (accessed on 20 December 2023).
- Vijayan, V.; Srinu, T.; Karnati, S.; Garikapati, V.; Linke, M.; Kamalyan, L.; Mali, S.R.; Sudan, K.; Kollas, A.; Schmid, T.; et al. A new immunomodulatory role for peroxisomes in macrophages activated by the TLR4 ligand lipopolysaccharide. J. Immunol. 2017, 198, 2414–2425. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef]
- Lu, Q.; Zong, W.; Zhang, M.; Chen, Z.; Yang, Z. The overlooked transformation mechanisms of VLCFAs: Peroxisomal β-oxidation. Agriculture 2022, 12, 947. Available online: https://www.mdpi.com/2077-0472/12/7/947 (accessed on 20 December 2023). [CrossRef]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef]
- Roczkowsky, A.; Doan, M.A.L.; Hlavay, B.; Mamik, M.K.; Branton, W.G.; McKenzie, B.A.; Saito, L.B.; Schmitt, L.; Eitzen, G.; Di Cara, F.; et al. Peroxisome injury in multiple sclerosis: Protective effects of 4-phenylbutyrate in CNS-associated macrophages. J. Neurosci. 2022, 42, 7152–7165. [Google Scholar] [CrossRef]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Marin Mathieu, N.; Sieck, G.C. TNFα induces mitochondrial fragmentation and biogenesis in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 320, L137–L151. [Google Scholar] [CrossRef] [PubMed]
- Davizon-Castillo, P.; McMahon, B.; Aguila, S.; Bark, D.; Ashworth, K.; Allawzi, A.; Campbell, R.A.; Montenont, E.; Nemkov, T.; D’Alessandro, A.; et al. TNF-α-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019, 134, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Kastl, L.; Sauer, S.W.; Ruppert, T.; Beissbarth, T.; Becker, M.S.; Süss, D.; Krammer, P.H.; Gülow, K. TNF-α mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-κB activation in liver cells. FEBS Lett. 2014, 588, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Gupta Vallur, P.; Phaëton, R.; Mythreye, K.; Hempel, N. Insights into the dichotomous regulation of SOD2 in cancer. Antioxidants 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Hou, S.; Malik-Soni, N.; Xu, Z.; Kumar, A.; Rachubinski, R.A.; Frappier, L.; Hobman, T.C. Flavivirus infection impairs peroxisome biogenesis and early antiviral signaling. J. Virol. 2015, 89, 12349–12361. [Google Scholar] [CrossRef]
- Tanner, L.B.; Chng, C.; Guan, X.L.; Lei, Z.; Rozen, S.G.; Wenk, M.R. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J. Lipid Res. 2014, 55, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Lazarow, P.B. Viruses exploiting peroxisomes. Curr. Opin. Microbiol. 2011, 14, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lei, Y.; Liu, X.; Wang, X.; Liu, Z.; Li, D.; Zheng, P.; Zhang, L.; Chen, S.; Xie, P. Glutamate and lipid metabolic perturbation in the hippocampi of asymptomatic borna disease virus-infected horses. PLoS ONE 2014, 9, e99752. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.C.; Moreno, J.A.; Jean Beltran, P.M.; Cristea, I.M. Peroxisome plasticity at the virus-host interface. Trends Microbiol. 2019, 27, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Knoblach, B.; Ishida, R.; Hobman, T.C.; Rachubinski, R.A. Peroxisomes exhibit compromised structure and matrix protein content in SARS-CoV-2-infected cells. Mol. Biol. Cell 2021, 32, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Coyaud, E.; Ranadheera, C.; Cheng, D.; Gonçalves, J.; Dyakov, B.J.A.; Laurent, E.M.N.; St-Germain, J.; Pelletier, L.; Gingras, A.C.; Brumell, J.H.; et al. Global interactomics uncovers extensive organellar targeting by Zika virus. Mol. Cell Proteom. 2018, 17, 2242–2255. [Google Scholar] [CrossRef]
- Refolo, G.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Ciccosanti, F. Mitochondrial interactome: A focus on antiviral signaling pathways. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Burtscher, J.; Cappellano, G.; Omori, A.; Koshiba, T.; Millet, G.P. Mitochondria: In the cross fire of SARS-CoV-2 and immunity. iScience 2020, 23, 101631. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Hossain, M.G.; Akter, S.; Ohsaki, E.; Ueda, K. Impact of the interaction of Hepatitis B virus with mitochondria and associated proteins. Viruses 2020, 12, 175. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Javed, F.; Manzoor, S. HCV non-structural NS4A protein of genotype 3a induces mitochondria mediated death by activating Bax and the caspase cascade. Microb. Pathog. 2018, 124, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Chen, M.; Gu, W.; Wan, J.; Cheng, Z.; Shen, K.; Zhang, W.; He, J.; Wang, Y.; Deng, X. The molecular mechanism of cardiac injury in SARS-CoV-2 infection: Focus on mitochondrial dysfunction. J. Infect. Public Health 2023, 16, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, E.; Vanhorebeek, I.; Grabenbauer, M.; Borgers, M.; Declercq, P.E.; Fahimi, H.D.; Baes, M. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am. J. Pathol. 2001, 159, 1477–1494. [Google Scholar] [CrossRef]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Hennessy, C.; McKernan, D.P. Anti-viral pattern recognition receptors as therapeutic targets. Cells 2021, 10, 2258. [Google Scholar] [CrossRef]
- Odendall, C.; Kagan, J.C. Activation and pathogenic manipulation of the sensors of the innate immune system. Microbes Infect. 2017, 19, 229–237. [Google Scholar] [CrossRef]
- Chathuranga, K.; Weerawardhana, A.; Dodantenna, N.; Lee, J.S. Regulation of antiviral innate immune signaling and viral evasion following viral genome sensing. Exp. Mol. Med. 2021, 53, 1647–1668. [Google Scholar] [CrossRef]
- Ferré, C.A.; Davezac, N.; Thouard, A.; Peyrin, J.M.; Belenguer, P.; Miquel, M.C.; Gonzalez-Dunia, D.; Szelechowski, M. Manipulation of the N-terminal sequence of the Borna disease virus X protein improves its mitochondrial targeting and neuroprotective potential. FASEB J. 2016, 30, 1523–1533. [Google Scholar] [CrossRef]
- Zhai, A.; Qian, J.; Kao, W.; Li, A.; Li, Y.; He, J.; Zhang, Q.; Song, W.; Fu, Y.; Wu, J.; et al. Borna disease virus encoded phosphoprotein inhibits host innate immunity by regulating miR-155. Antivir. Res. 2013, 98, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lei, Y.; Deng, J.; Zhou, C.; Zhang, Y.; Li, W.; Huang, H.; Cheng, S.; Zhang, H.; Zhang, L.; et al. Human but Not Laboratory Borna Disease Virus Inhibits Proliferation and Induces Apoptosis in Human Oligodendrocytes In Vitro. PLoS ONE 2013, 8, e66623. [Google Scholar] [CrossRef] [PubMed]
- Koepke, J.I.; Nakrieko, K.A.; Wood, C.S.; Boucher, K.K.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Restoration of peroxisomal catalase import in a model of human cellular aging. Traffic 2007, 8, 1590–1600. [Google Scholar] [CrossRef]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell. 2011, 22, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, V.; Mohand-Said, S.; Hanoteau, N.; Fuchs, C.; Pfizenmaier, K.; Eisel, U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: Opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002, 22, RC216. [Google Scholar] [CrossRef] [PubMed]
- Suvannavejh, G.C.; Lee, H.O.; Padilla, J.; Dal Canto, M.C.; Barrett, T.A.; Miller, S.D. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35-55)-induced experimental autoimmune encephalomyelitis. Cell Immunol. 2000, 205, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Priatel, J.J.; Teh, S.-J.; The, H.-S. TNF receptor type 2 (p75) functions as a costimulator for antigen-driven T-cell responses in vivo. J. Immunol. 2006, 176, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Rothe, J.; Lesslauer, W.; Lötscher, H.; Lang, Y.; Koebel, P.; Köntgen, F.; Althage, A.; Zinkernagel, R.; Steinmetz, M.; Bluethmann, H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 26, 798–802. [Google Scholar] [CrossRef]
- Erickson, S.L.; de Sauvage, F.J.; Kikly, K.; Carver-Moore, K.; Pitts-Meek, S.; Gillett, N.; Sheehan, K.C.; Schreiber, R.D.; Goeddel, D.V.; Moore, M.W. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994, 372, 560–563. [Google Scholar] [CrossRef]
- Schaudien, D. Experimental Infection of TNF-Alpha Transgenic Mice with the Borna Disease Virus—Influence of Cytokine Profile, Neurodegeneration and Neuroprotection. Ph.D. Thesis, Institute for Pathology, School of Veterinary Medicine, Hannover, Germany, 2007. [Google Scholar]
- Grant, P.; Ahlemeyer, B.; Karnati, S.; Berg, T.; Stelzig, I.; Nenicu, A.; Kuchelmeister, K.; Crane, D.I.; Baumgart-Vogt, E. The biogenesis protein PEX14 is an optimal marker for the identification and localization of peroxisomes in different cell types, tissues, and species in morphological studies. Histochem. Cell Biol. 2013, 140, 423–442. [Google Scholar] [CrossRef] [PubMed]
- Ahlemeyer, B.; Neubert, I.; Kovacs, W.J.; Baumgart-Vogt, E. Differential expression of peroxisomal matrix and membrane proteins during postnatal development of mouse brain. J. Comp. Neurol. 2002, 505, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Lee, I.; Chung, H.S.; Bae, G.U.; Chang, M.; Song, E.; Kim, M.J. ATP5B regulates mitochondrial fission and fusion in mammalian cells. Anim. Cell Syst. 2016, 20, 157–164. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Lines | ||||
|---|---|---|---|---|
| Wt vs. InfWt | TNFTg vs. InfTNFTg | TNFR1ko vs. InfTNFR1ko | TNFR2ko vs. InfTNFR2ko | |
| PEX14 | ||||
| GrDG | * | n.s. | n.s. | n.s. |
| PyCA | * | n.s. | n.s. | * |
| PyCx | * | n.s. | n.s. | * |
| GrCb | ** | n.s. | n.s. | n.s. |
| PuCb | * | n.s. | n.s. | * |
| ATP5B | ||||
| GrDG | n.s. | ** | n.s. | **** |
| PyCA | * | * | n.s. | ** |
| PyCx | n.s. | *** | * | *** |
| GrCb | * | n.s. | n.s. | * |
| PuCb | ** | * | n.s. | *** |
| Catalase | ||||
| GrDG | **** | n.s. | n.s. | ** |
| PyCA | **** | n.s. | n.s. | **** |
| PyCx | **** | n.s. | n.s. | *** |
| GrCb | ** | * | ** | **** |
| PuCb | *** | ** | n.s. | **** |
| SOD2 | ||||
| GrDG | n.s. | *** | n.s. | ** |
| PyCA | * | * | n.s. | * |
| PyCx | * | ** | n.s. | ** |
| GrCb | * | n.s. | n.s. | * |
| PuCb | * | * | n.s. | *** |
| Target Antigen | Host | Source; Catalogue Number | Dilution |
|---|---|---|---|
| PEX14 | Rb | Kindly donated by Denis I. Crane, Griffith University, Brisbane, Australia | 1:10,000 |
| Catalase | Rb | Kindly donated by Denis I. Crane, Griffith University, Brisbane, Australia | 1:5000 |
| ATP5B | Rb | Sigma-Aldrich, Deisenhofen, Germany; APA001520 | 1:2000 |
| SOD2 | Rb | Abcam, Cambridge, United Kingdom; ab13533 | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osei, D.; Baumgart-Vogt, E.; Ahlemeyer, B.; Herden, C. Tumor Necrosis Factor-α Receptor 1 Mediates Borna Disease Virus 1-Induced Changes in Peroxisomal and Mitochondrial Dynamics in Neurons. Int. J. Mol. Sci. 2024, 25, 1849. https://doi.org/10.3390/ijms25031849
Osei D, Baumgart-Vogt E, Ahlemeyer B, Herden C. Tumor Necrosis Factor-α Receptor 1 Mediates Borna Disease Virus 1-Induced Changes in Peroxisomal and Mitochondrial Dynamics in Neurons. International Journal of Molecular Sciences. 2024; 25(3):1849. https://doi.org/10.3390/ijms25031849
Chicago/Turabian StyleOsei, Dominic, Eveline Baumgart-Vogt, Barbara Ahlemeyer, and Christiane Herden. 2024. "Tumor Necrosis Factor-α Receptor 1 Mediates Borna Disease Virus 1-Induced Changes in Peroxisomal and Mitochondrial Dynamics in Neurons" International Journal of Molecular Sciences 25, no. 3: 1849. https://doi.org/10.3390/ijms25031849
APA StyleOsei, D., Baumgart-Vogt, E., Ahlemeyer, B., & Herden, C. (2024). Tumor Necrosis Factor-α Receptor 1 Mediates Borna Disease Virus 1-Induced Changes in Peroxisomal and Mitochondrial Dynamics in Neurons. International Journal of Molecular Sciences, 25(3), 1849. https://doi.org/10.3390/ijms25031849