
CRISPR Manipulation of Age-Related Macular Degeneration Haplotypes in the Complement System: Potential Future Therapeutic Applications/Avenues


Abstract
1. Introduction
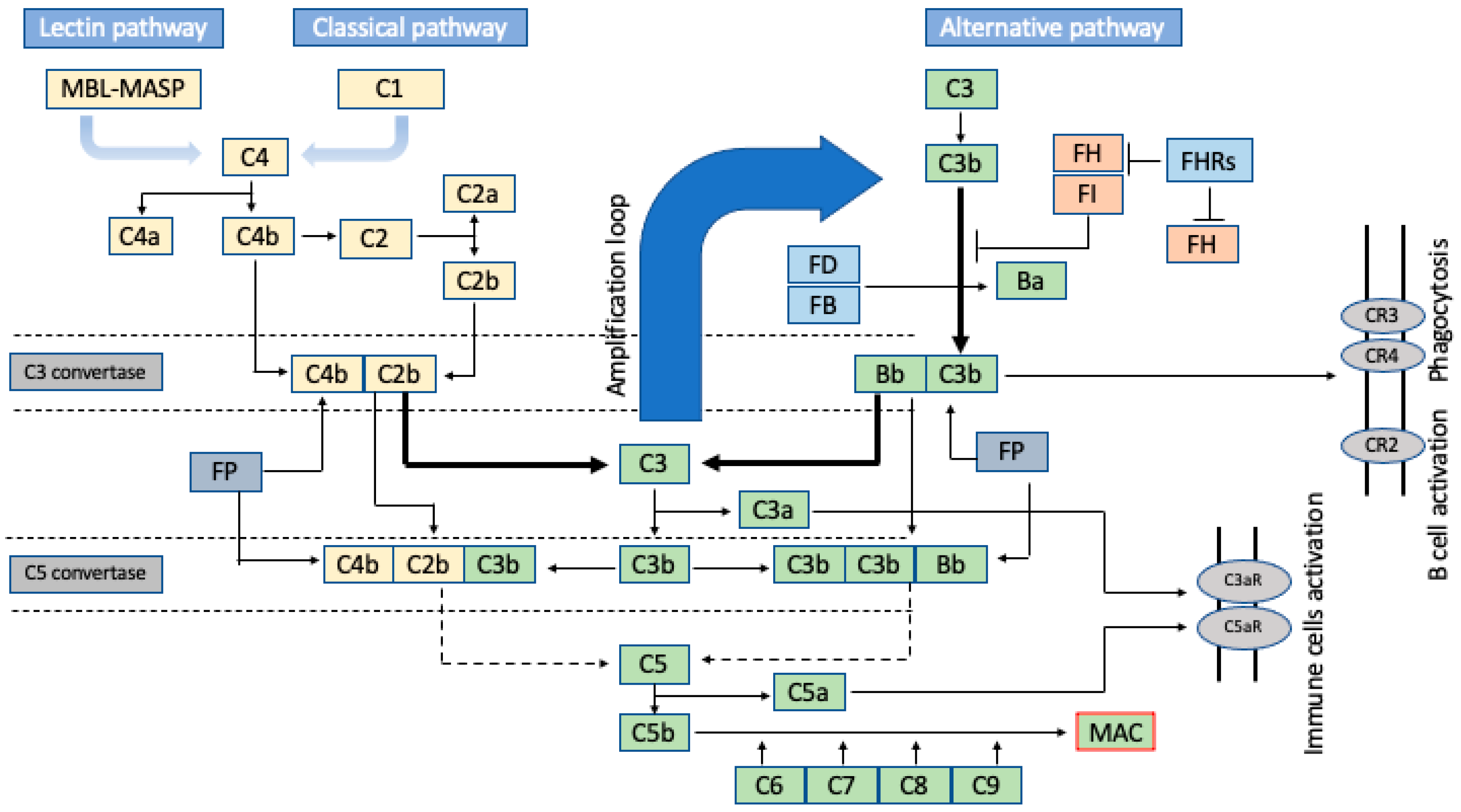
2. Brief Insight into the Amplification Nature of the Complement Cascade
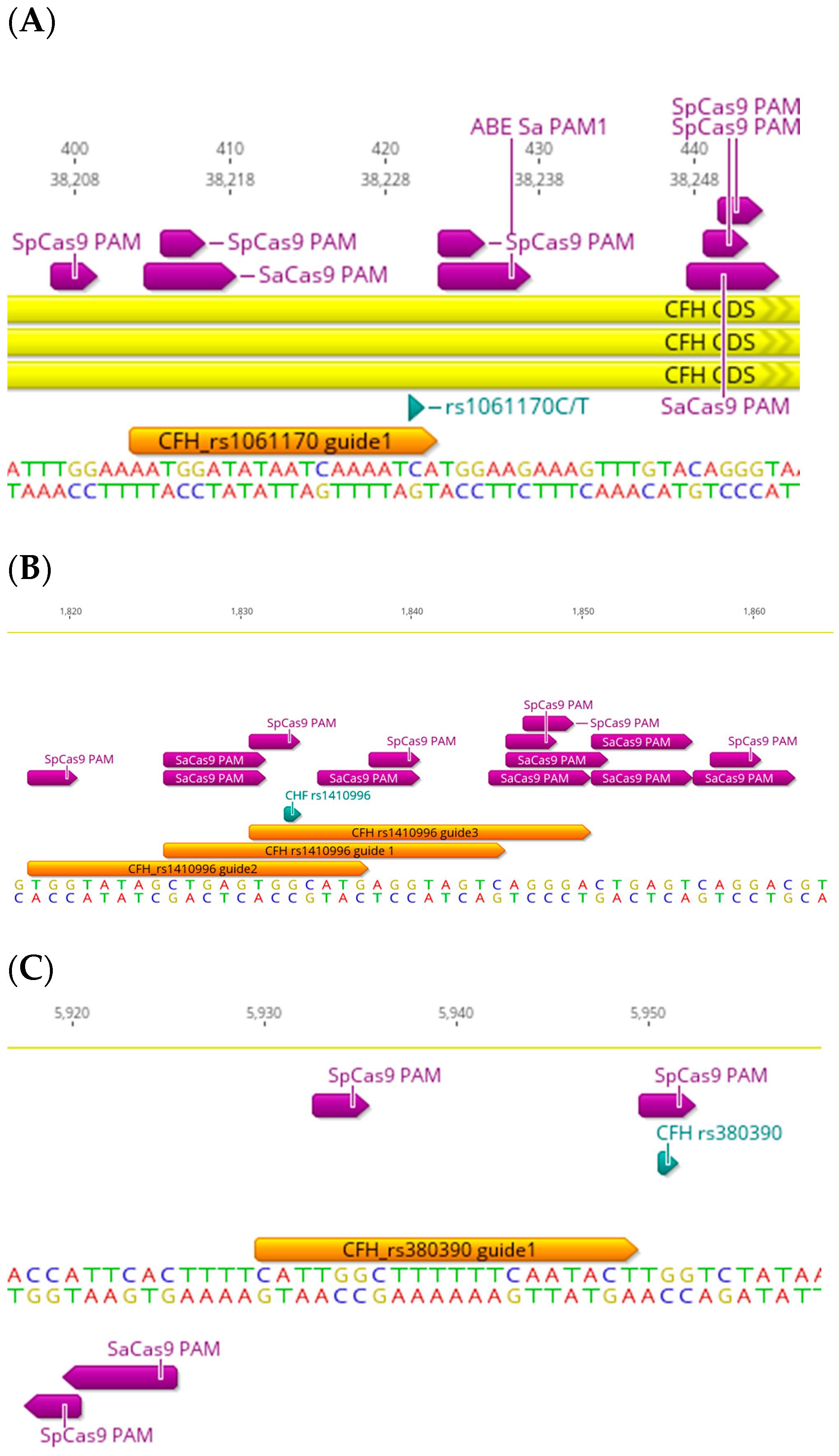
3. The CFH-CFHR Haplotypes
4. The CFB Haplotypes
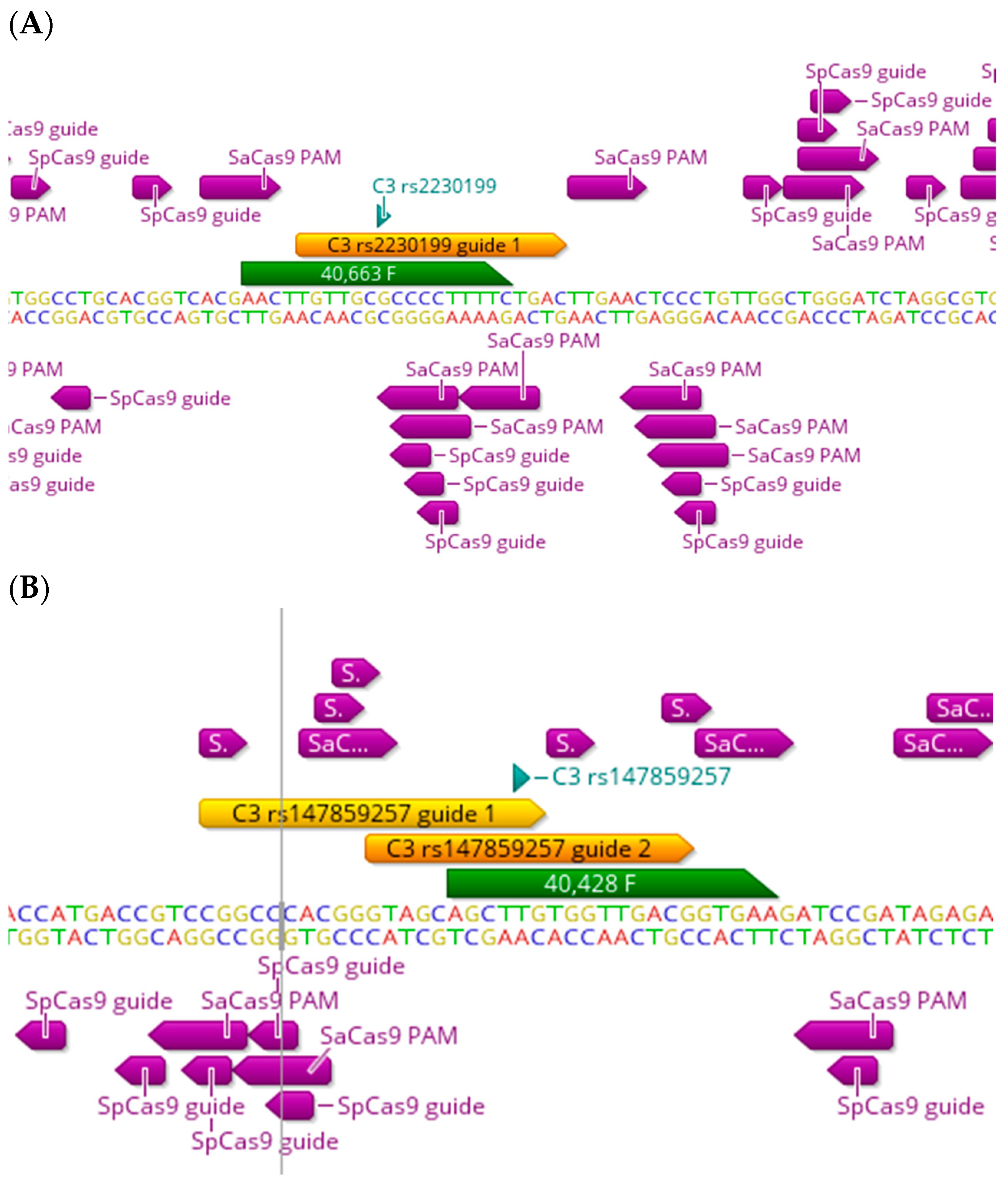
5. The C3 and Other Functional Haplotypes
6. Implications of Future CRISPR Therapeutics for AMD-Related SNPs
7. Conclusions and Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Jaskolka, M.C.; El-Husayni, S.; Duke, B.; Erlwein, A.; Myers, R.; Pennesi, M.E.; Pierce, E.A.; Michaels, L.A.; Shearman, M.; Zhang, K.; et al. Exploratory Safety Profile of EDIT-101, a First-in-Human CRISPR Gene Editing Therapy for related Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2022, 63, 2835-A0351. [Google Scholar]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.H.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 2002, 134, 411–431. [Google Scholar] [CrossRef]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Lo, M.W.; Woodruff, T.M. Complement: Bridging the innate and adaptive immune systems in sterile inflammation. J. Leukoc. Biol. 2020, 108, 339–351. [Google Scholar] [CrossRef]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflammation 2020, 17, 354. [Google Scholar] [CrossRef]
- Tenner, A.J.; Stevens, B.; Woodruff, T.M. New tricks for an ancient system: Physiological and pathological roles of complement in the CNS. Mol. Immunol. 2018, 102, 3–13. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Cortes, C.; Ferreira, V.P. Properdin: A multifaceted molecule involved in inflammation and diseases. Mol. Immunol. 2018, 102, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Hellwage, J.; Friese, M.A.; Hegasy, G.; Jokiranta, S.T.; Meri, S. Factor H and disease: A complement regulator affects vital body functions. Mol. Immunol. 1999, 36, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.W.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef]
- Fontaine, M.; Demares, M.J.; Koistinen, V.; Day, A.J.; Davrinche, C.; Sim, R.B.; Ripoche, J. Truncated forms of human complement factor H. Biochem. J. 1989, 258, 927–930. [Google Scholar] [CrossRef]
- Ripoche, J.; Day, A.J.; Harris, T.J.; Sim, R.B. The complete amino acid sequence of human complement factor H. Biochem. J. 1988, 249, 593–602. [Google Scholar] [CrossRef]
- Cserhalmi, M.; Papp, A.; Brandus, B.; Uzonyi, B.; Jozsi, M. Regulation of regulators: Role of the complement factor H-related proteins. Semin. Immunol. 2019, 45, 101341. [Google Scholar] [CrossRef]
- Edwards, A.O.; Ritter, R.; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Mitsunobu, H.; Teramoto, J.; Nishida, K.; Kondo, A. Beyond Native Cas9: Manipulating Genomic Information and Function. Trends Biotechnol. 2017, 35, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A.T to G.C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 169, 559. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Baral, R.; Singhal, P.; Loveluck, K.; Shen, S.; Sanchez, M.; Marco, E.; Gotta, G.M.; Maeder, M.L.; Kennedy, E.M.; et al. Characterization of Staphylococcus aureus Cas9: A smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015, 16, 257. [Google Scholar] [CrossRef]
- Maller, J.; George, S.; Purcell, S.; Fagerness, J.; Altshuler, D.; Daly, M.J.; Seddon, J.M. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat. Genet. 2006, 38, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Bortesi, L.; Zhu, C.; Zischewski, J.; Perez, L.; Bassie, L.; Nadi, R.; Forni, G.; Lade, S.B.; Soto, E.; Jin, X.; et al. Patterns of CRISPR/Cas9 activity in plants, animals and microbes. Plant Biotechnol. J. 2016, 14, 2203–2216. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Muckersie, E.; Robertson, M.; Forrester, J.V.; Xu, H.P. Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp. Eye Res. 2008, 87, 543–550. [Google Scholar] [CrossRef]
- Spencer, K.L.; Hauser, M.A.; Olson, L.M.; Schmidt, S.; Scott, W.K.; Gallins, P.; Agarwal, A.; Postel, E.A.; Pericak-Vance, M.A.; Haines, J.L. Protective effect of complement factor B and complement component 2 variants in age-related macular degeneration. Hum. Mol. Genet. 2007, 16, 1986–1992. [Google Scholar] [CrossRef]
- Neto, J.M.; Viturino, M.G.; Ananina, G.; Bajano, F.F.; Costa, S.; Roque, A.B.; Borges, G.F.; Franchi, R.; Rim, P.H.; Medina, F.M.; et al. Association of genetic variants rs641153 (CFB), rs2230199 (C3), and rs1410996 (CFH) with age-related macular degeneration in a Brazilian population. Exp. Biol. Med. 2021, 246, 2290–2296. [Google Scholar] [CrossRef]
- Nakata, I.; Yamashiro, K.; Yamada, R.; Gotoh, N.; Nakanishi, H.; Hayashi, H.; Akagi-Kurashige, Y.; Tsujikawa, A.; Otani, A.; Saito, M.; et al. Significance of C2/CFB Variants in Age-Related Macular Degeneration and Polypoidal Choroidal Vasculopathy in a Japanese Population. Invesig. Ophthalmol. Vis. Sci. 2012, 53, 794–798. [Google Scholar] [CrossRef]
- Zhan, X.; Larson, D.E.; Wang, C.; Koboldt, D.C.; Sergeev, Y.V.; Fulton, R.S.; Fulton, L.L.; Fronick, C.C.; Branham, K.E.; Bragg-Gresham, J.; et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat. Genet. 2013, 45, 1375–1379. [Google Scholar] [CrossRef]
- Helgason, H.; Sulem, P.; Duvvari, M.R.; Luo, H.R.; Thorleifsson, G.; Stefansson, H.; Jonsdottir, I.; Masson, G.; Gudbjartsson, D.F.; Walters, G.B.; et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat. Genet. 2013, 45, 1371–1374. [Google Scholar] [CrossRef]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, P.J.; Lay, E.; Seilly, D.J.; Buchberger, A.; Schwaeble, W.; Khadake, J. Further studies of the down-regulation by Factor I of the C(3)b feedback cycle using endotoxin as a soluble activator and red cells as a source of CR1 on sera of different complotype. Clin. Exp. Immunol. 2016, 183, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Lay, E.; Nutland, S.; Smith, J.E.; Hiles, I.; Smith, R.A.; Seilly, D.J.; Buchberger, A.; Schwaeble, W.; Lachmann, P.J. Complotype affects the extent of down-regulation by Factor I of the C3b feedback cycle in vitro. Clin. Exp. Immunol. 2015, 181, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Bessho, H.; Honda, S.; Negi, A. Additional evidence to support the role of a common variant near the complement factor I gene in susceptibility to age-related macular degeneration. Eur. J. Hum. Genet. 2010, 18, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Fagerness, J.A.; Maller, J.B.; Neale, B.M.; Reynolds, R.C.; Daly, M.J.; Seddon, J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bhangale, T.R.; Fagerness, J.; Ripke, S.; Thorleifsson, G.; Tan, P.L.; Souied, E.H.; Richardson, A.J.; Merriam, J.E.; Buitendijk, G.H.; et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum. Mol. Genet. 2011, 20, 3699–3709. [Google Scholar] [CrossRef]
- Sobrin, L.; Ripke, S.; Yu, Y.; Fagerness, J.; Bhangale, T.R.; Tan, P.L.; Souied, E.H.; Buitendijk, G.H.S.; Merriam, J.E.; Richardson, A.J.; et al. Heritability and Genome-Wide Association Study to Assess Genetic Differences between Advanced Age-related Macular Degeneration Subtypes. Ophthalmology 2012, 119, 1874–1885. [Google Scholar] [CrossRef]
- Ennis, S.; Gibson, J.; Cree, A.J.; Collins, A.; Lotery, A.J. Support for the involvement of complement factor I in age-related macular degeneration. Eur. J. Hum. Genet. 2010, 18, 15–16. [Google Scholar] [CrossRef]
- Cipriani, V.; Matharu, B.K.; Khan, J.C.; Shahid, H.; Hayward, C.; Wright, A.F.; Armbrecht, A.M.; Dhillon, B.; Harding, S.P.; Bishop, P.N.; et al. No evidence of association between complement factor I genetic variant rs10033900 and age-related macular degeneration. Eur. J. Hum. Genet. 2012, 20, 1–2. [Google Scholar] [CrossRef]
- Geerlings, M.J.; de Jong, E.K.; den Hollander, A.I. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- de Jong, S.; Gagliardi, G.; Garanto, A.; de Breuk, A.; Lechanteur, Y.T.E.; Katti, S.; van den Heuvel, L.P.; Volokhina, E.B.; den Hollander, A.I. Implications of genetic variation in the complement system in age-related macular degeneration. Prog. Retin. Eye Res. 2021, 84, 100952. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.A.; Edwards, A.O.; Ryu, E.; Tosakulwong, N.; Baratz, K.H.; Brown, W.L.; Charbel Issa, P.; Scholl, H.P.; Pollok-Kopp, B.; Schmid-Kubista, K.E.; et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum. Mol. Genet. 2010, 19, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Smailhodzic, D.; Klaver, C.C.; Klevering, B.J.; Boon, C.J.; Groenewoud, J.M.; Kirchhof, B.; Daha, M.R.; den Hollander, A.I.; Hoyng, C.B. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Ophthalmology 2012, 119, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Ristau, T.; Paun, C.; Ersoy, L.; Hahn, M.; Lechanteur, Y.; Hoyng, C.; de Jong, E.K.; Daha, M.R.; Kirchhof, B.; den Hollander, A.I.; et al. Impact of the Common Genetic Associations of Age-Related Macular Degeneration upon Systemic Complement Component C3d Levels. PLoS ONE 2014, 9, e93459. [Google Scholar] [CrossRef] [PubMed]
- Lores-Motta, L.; Paun, C.C.; Corominas, J.; Pauper, M.; Geerlings, M.J.; Altay, L.; Schick, T.; Daha, M.R.; Fauser, S.; Hoyng, C.B.; et al. Genome-Wide Association Study Reveals Variants in CFH and CFHR4 Associated with Systemic Complement Activation: Implications in Age-Related Macular Degeneration. Ophthalmology 2018, 125, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Mckeigue, P.M.; Skerka, C.; Hayward, C.; Rudan, I.; Vitart, V.; Polasek, O.; Armbrecht, A.M.; Yates, J.R.W.; Vatavuk, Z.; et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum. Mol. Genet. 2013, 22, 4857–4869. [Google Scholar] [CrossRef] [PubMed]
- Hakobyan, S.; Harris, C.L.; Tortajada, A.; Goicochea de Jorge, E.; Garcia-Layana, A.; Fernandez-Robredo, P.; Rodriguez de Cordoba, S.; Morgan, B.P. Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: Application to assessing risk of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1983–1990. [Google Scholar] [CrossRef]
- Reynolds, R.; Hartnett, M.E.; Atkinson, J.P.; Giclas, P.C.; Rosner, B.; Seddon, J.M. Plasma complement components and activation fragments: Associations with age-related macular degeneration genotypes and phenotypes. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5818–5827. [Google Scholar] [CrossRef]
- Sharma, N.K.; Sharma, S.K.; Gupta, A.; Prabhakar, S.; Singh, R.; Anand, A. Predictive model for earlier diagnosis of suspected age-related macular degeneration patients. DNA Cell Biol. 2013, 32, 549–555. [Google Scholar] [CrossRef]
- Silva, A.S.; Teixeira, A.G.; Bavia, L.; Lin, F.; Velletri, R.; Belfort, R.; Isaac, L. Plasma levels of complement proteins from the alternative pathway in patients with age-related macular degeneration are independent of Complement Factor H Tyr(402)His polymorphism. Mol. Vis. 2012, 18, 2288–2299. [Google Scholar]
- Cipriani, V.; Lores-Motta, L.; He, F.; Fathalla, D.; Tilakaratna, V.; McHarg, S.; Bayatti, N.; Acar, I.E.; Hoyng, C.B.; Fauser, S.; et al. Increased circulating levels of Factor H-Related Protein 4 are strongly associated with age-related macular degeneration. Nat. Commun. 2020, 11, 778. [Google Scholar] [CrossRef]
- Warwick, A.; Khandhadia, S.; Ennis, S.; Lotery, A. Age-Related Macular Degeneration: A Disease of Systemic or Local Complement Dysregulation? J. Clin. Med. 2014, 3, 1234–1257. [Google Scholar] [CrossRef]
- Doench, J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018, 19, 67–80. [Google Scholar] [CrossRef]
- Pulecio, J.; Verma, N.; Mejia-Ramirez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Horvath, P. A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2017, 2, 17092. [Google Scholar] [CrossRef]
- Fellmann, C.; Gowen, B.G.; Lin, P.C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Kelsh, R.N.; Richardson, R.J. New advances in CRISPR/Cas-mediated precise gene-editing techniques. Dis. Model. Mech. 2023, 16, dmm049874. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 2020, 18, 2401–2415. [Google Scholar] [CrossRef]
- Liu, W.; Li, L.; Jiang, J.; Wu, M.; Lin, P. Applications and challenges of CRISPR-Cas gene-editing to disease treatment in clinics. Precis. Clin. Med. 2021, 4, 179–191. [Google Scholar] [CrossRef]
- Salman, A.; Kantor, A.; McClements, M.E.; Marfany, G.; Trigueros, S.; MacLaren, R.E. Non-Viral Delivery of CRISPR/Cas Cargo to the Retina Using Nanoparticles: Current Possibilities, Challenges, and Limitations. Pharmaceutics 2022, 14, 1842. [Google Scholar] [CrossRef] [PubMed]
- Taha, E.A.; Lee, J.; Hotta, A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J. Control Release 2022, 342, 345–361. [Google Scholar] [CrossRef]
- Shinwari, Z.K.; Tanveer, F.; Khalil, A.T. Ethical Issues Regarding CRISPR-mediated Genome Editing. Cris. Cas. Syst. Emerg. Technol. Appl. 2018, 26, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, R.; Offen, D. Single-Base Resolution: Increasing the Specificity of the CRISPR-Cas System in Gene Editing. Mol. Ther. 2021, 29, 937–948. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP | Gene | SaCas9 PAM | SpCas9 PAM | xCas9 PAM | Nme2 Cas9 PAM | CJCas9 PAM | SNP (Intron/Exon) | Consequence |
|---|---|---|---|---|---|---|---|---|
| rs1410996 G>A | CFH | Y | Y | (NG)Y, (GAA)N, (GAT)N | Y | N | Intron | Pathogenic/risk factor |
| rs380390 G>A,C,T | CFH | N | Y | (NG)Y, (GAA)N, (GAT)N | Y | N | Intron | Pathogenic/intron variant |
| rs800292 G>A | CFH | N | Y | (NG)Y, (GAA)Y, (GAT)N | Y | N | Exon | Pathogenic/risk factor, missense variant |
| rs1061147 A>C | CFH | N | Y | (NG)Y, (GAA)Y, (GAT)N | N | Y | Exon | Pathogenic/synonymous variant |
| rs1831282 A>C | CFH | N | N | (NG)Y, (GAA)N, (GAT)Y | N | N | Intron | Pathogenic/intron variant |
| rs1061170 C>T | CFH | Y | N | (NG)Y, (GAA)N, (GAT)Y | Y | N | Exon | Pathogenic/risk factor/missense variant |
| rs1329424 T>G | CFH | N | N | (NG)Y, (GAA)Y, (GAT)N | Y | Y | Intron | Pathogenic/intron variant |
| rs1329428 C>T | CFH | N | N | (NG)Y, (GAA)Y, (GAT)N | N | N | Intron | Pathogenic/intron variant |
| rs10733086 A>C,T | CFH | N | N | (NG)Y, (GAA)N, (GAT)N | N | N | Intron | Pathogenic/intron variant |
| rs10737680 A>C | CFH | N | N | (NG)Y, (GAA)N, (GAT)N | Y | Y | Intron | Pathogenic/intron variant |
| rs10801555 A>G | CFH | N | N | (NG)Y, (GAA)Y, (GAT)N | Y | N | Intron | Pathogenic/intron variant |
| rs10922109 C>A | CFH | N | N | (NG)Y, (GAA)N, (GAT)Y | N | Y | Intron | Pathogenic/intron variant |
| rs35292876 C>T | CFH | N | Y | (NG)Y, (GAA)Y, (GAT)N | Y | Y | Exon | Benign/coding seq variant/Syn variant |
| rs121913059 C>T | CFH | N | N | (NG)Y, (GAA)Y, (GAT)N | N | N | Exon | Benign/missense variant |
| rs641153 G>A/T | CFB | Y | Y | (NG)Y, (GAA)N, (GAT)Y | Y | N | Exon | Protective |
| rs4541862 T>C | CFB | Y | Y | (NG)Y, (GAA)N, (GAT)Y | Y | Y | Exon | Pathogenic/intron variant |
| rs4151667 T>A | CFB | Y | Y | (NG)Y, (GAA)N, (GAT)N | Y | N | Exon | Benign/coding seq variant/missense |
| rs2230199 G>C,T | C3 | N | Y | (NG)Y, (GAA)Y, (GAT)N | Y | N | Exon | Pathogenic/missense variant |
| rs147859257 T>G | C3 | Y | Y | (NG)Y, (GAA)N, (GAT)N | Y | N | Exon | Pathogenic/missense variant |
| rs141853578 C>T | CFI | N | Y | (NG)Y, (GAA)Y, (GAT)N | Y | N | Exon | Pathogenic/missense variant |
| rs10033900 T>C | CFI | N | Y | (NG)Y, (GAA)Y, (GAT)N | Y | N | Intron | Pathogenic/intron variant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salman, A.; McClements, M.E.; MacLaren, R.E. CRISPR Manipulation of Age-Related Macular Degeneration Haplotypes in the Complement System: Potential Future Therapeutic Applications/Avenues. Int. J. Mol. Sci. 2024, 25, 1697. https://doi.org/10.3390/ijms25031697
Salman A, McClements ME, MacLaren RE. CRISPR Manipulation of Age-Related Macular Degeneration Haplotypes in the Complement System: Potential Future Therapeutic Applications/Avenues. International Journal of Molecular Sciences. 2024; 25(3):1697. https://doi.org/10.3390/ijms25031697
Chicago/Turabian StyleSalman, Ahmed, Michelle E. McClements, and Robert E. MacLaren. 2024. "CRISPR Manipulation of Age-Related Macular Degeneration Haplotypes in the Complement System: Potential Future Therapeutic Applications/Avenues" International Journal of Molecular Sciences 25, no. 3: 1697. https://doi.org/10.3390/ijms25031697
APA StyleSalman, A., McClements, M. E., & MacLaren, R. E. (2024). CRISPR Manipulation of Age-Related Macular Degeneration Haplotypes in the Complement System: Potential Future Therapeutic Applications/Avenues. International Journal of Molecular Sciences, 25(3), 1697. https://doi.org/10.3390/ijms25031697