
An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
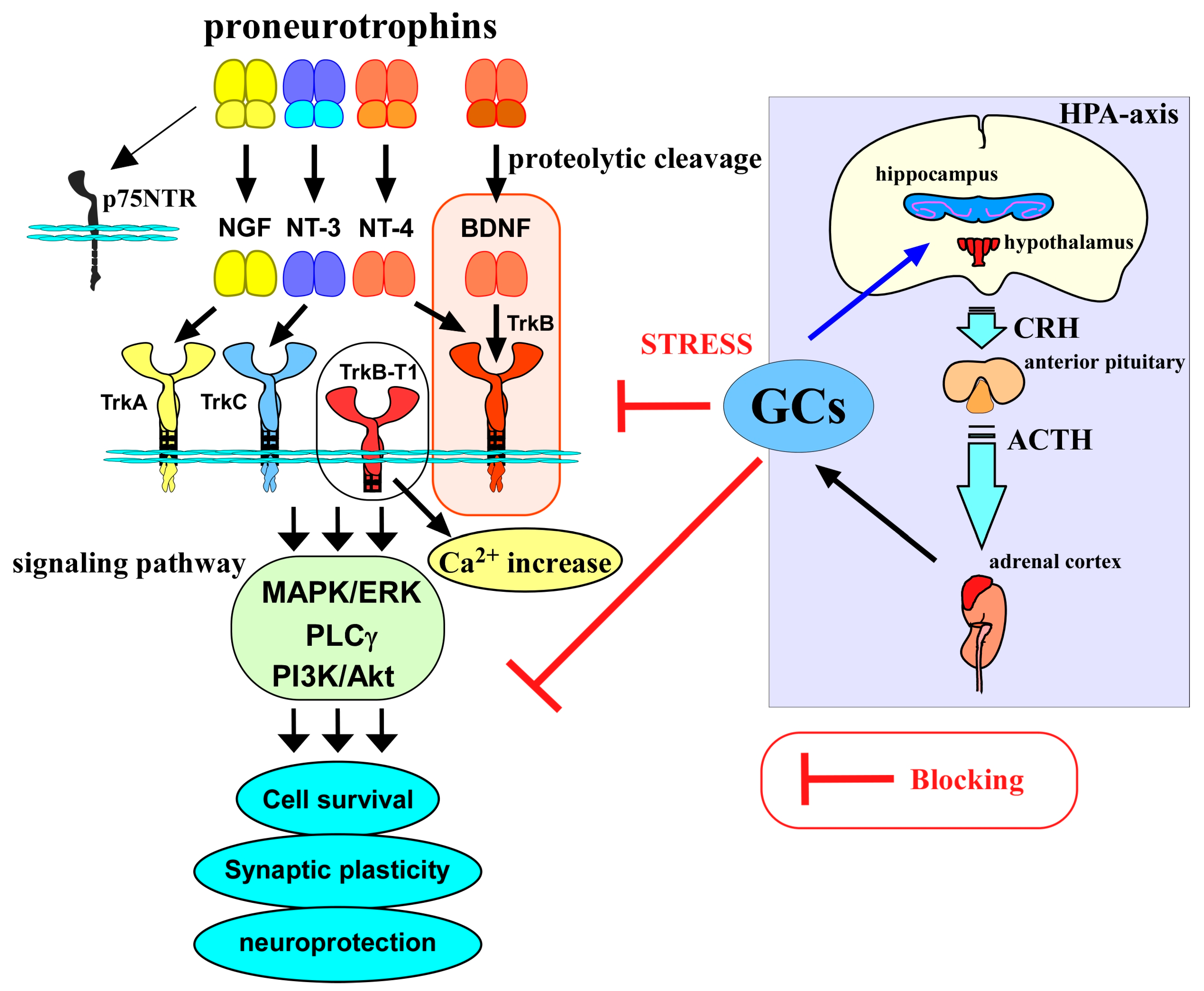
2. BDNF/TrkB System in Neuronal Function
3. Glucocorticoids and Neuronal Functions
4. Glucocorticoid Stress, BDNF, and Neuronal Functions
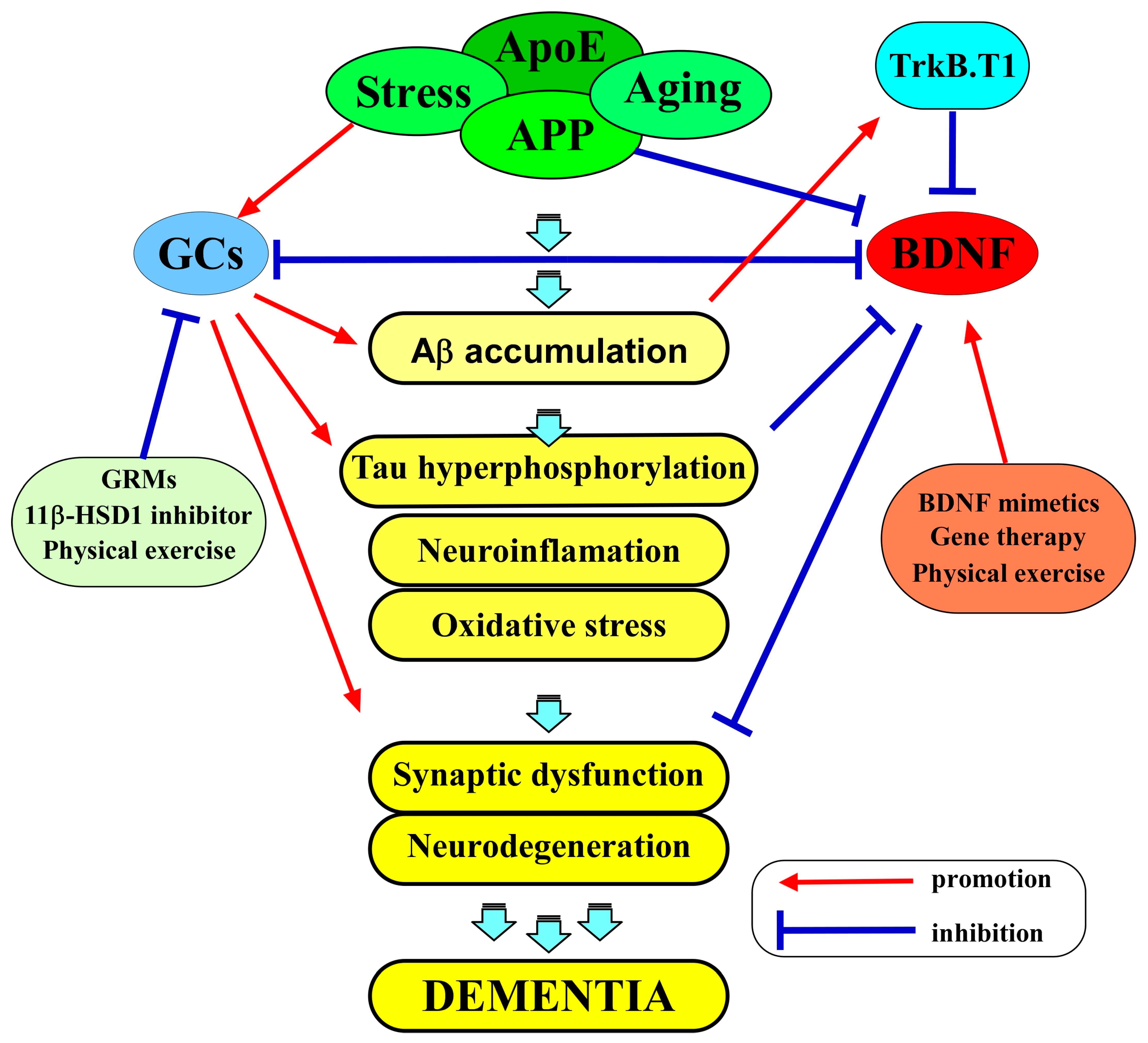
5. The Interplay of BDNF and Glucocorticoids in AD
5.1. The Role of BDNF in AD
5.2. The Role of Glucocorticoids in AD
5.3. BDNF and GCs as Therapeutic Targets in AD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen. Res. 2015, 10, 721–725. [Google Scholar] [CrossRef]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Pisani, A.; Paciello, F.; Del Vecchio, V.; Malesci, R.; De Corso, E.; Cantone, E.; Fetoni, A.R. The Role of BDNF as a Biomarker in Cognitive and Sensory Neurodegeneration. J. Pers. Med. 2023, 13, 652. [Google Scholar] [CrossRef]
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer’s disease: Insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957. [Google Scholar] [CrossRef]
- Autry, A.E. Function of brain-derived neurotrophic factor in the hypothalamus: Implications for depression pathology. Front. Mol. Neurosci. 2022, 15, 1028223. [Google Scholar] [CrossRef]
- Kim, E.J.; Pellman, B.; Kim, J.J. Stress effects on the hippocampus: A critical review. Learn. Mem. 2015, 22, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front. Mol. Neurosci. 2023, 16, 1247422. [Google Scholar] [CrossRef] [PubMed]
- Bassil, K.; Krontira, A.C.; Leroy, T.; Escoto, A.I.H.; Snijders, C.; Pernia, C.D.; Pasterkamp, R.J.; de Nijs, L.; van den Hove, D.; Kenis, G.; et al. In vitro modeling of the neurobiological effects of glucocorticoids: A review. Neurobiol. Stress 2023, 23, 100530. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H. Brain-derived neurotrophic factor and neurogenesis. In Factors Affecting Neurodevelopment; Elsevier: Amsterdam, The Netherlands, 2021; pp. 121–131. [Google Scholar]
- Cao, T.; Matyas, J.J.; Renn, C.L.; Faden, A.I.; Dorsey, S.G.; Wu, J. Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells 2020, 9, 1194. [Google Scholar] [CrossRef]
- Fan, W.L.; Liu, P.; Wang, G.; Pu, J.G.; Xue, X.; Zhao, J.H. Transplantation of hypoxic preconditioned neural stem cells benefits functional recovery via enhancing neurotrophic secretion after spinal cord injury in rats. J. Cell. Biochem. 2018, 119, 4339–4351. [Google Scholar] [CrossRef]
- Wang, J.; Cai, Y.; Sun, J.; Feng, H.; Zhu, X.; Chen, Q.; Gao, F.; Ni, Q.; Mao, L.; Yang, M.; et al. Administration of intramuscular AAV-BDNF and intranasal AAV-TrkB promotes neurological recovery via enhancing corticospinal synaptic connections in stroke rats. Exp. Neurol. 2023, 359, 114236. [Google Scholar] [CrossRef]
- Buch, L.; Lipi, B.; Langhnoja, J.; Jaldeep, L.; Pillai, P.P.; Prakash, P. Role of astrocytic MeCP2 in regulation of CNS myelination by affecting oligodendrocyte and neuronal physiology and axo-glial interactions. Exp. Brain Res. 2018, 236, 3015–3027. [Google Scholar] [CrossRef]
- Datta, I.; Ganapathy, K.; Razdan, R.; Bhonde, R. Location and Number of Astrocytes Determine Dopaminergic Neuron Survival and Function Under 6-OHDA Stress Mediated Through Differential BDNF Release. Mol. Neurobiol. 2018, 55, 5505–5525. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Turati, J.; Ramírez, D.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Astrocyte truncated tropomyosin receptor kinase B mediates brain-derived neurotrophic factor anti-apoptotic effect leading to neuroprotection. J. Neurochem. 2018, 146, 686–702. [Google Scholar] [CrossRef]
- Harley, S.B.R.; Willis, E.F.; Shaikh, S.N.; Blackmore, D.G.; Sah, P.; Ruitenberg, M.J.; Bartlett, P.F.; Vukovic, J. Selective Ablation of BDNF from Microglia Reveals Novel Roles in Self-Renewal and Hippocampal Neurogenesis. J. Neurosci. 2021, 41, 4172–4186. [Google Scholar] [CrossRef] [PubMed]
- Bayas, A.; Hummel, V.; Kallmann, B.A.; Karch, C.; Toyka, K.V.; Rieckmann, P. Human cerebral endothelial cells are a potential source for bioactive BDNF. Cytokine 2002, 19, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Marie, C.; Pedard, M.; Quirié, A.; Tessier, A.; Garnier, P.; Totoson, P.; Demougeot, C. Brain-derived neurotrophic factor secreted by the cerebral endothelium: A new actor of brain function? J. Cereb. Blood Flow Metab. 2018, 38, 935–949. [Google Scholar] [CrossRef]
- Stanton, L.M.; Price, A.J.; Manning, E.E. Hypothalamic corticotrophin releasing hormone neurons in stress-induced psychopathology: Revaluation of synaptic contributions. J. Neuroendocrinol. 2023, 35, e13268. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Kumar, V.; Moore, S.; Li, F.; Murphy, G.G.; Watson, S.J.; Akil, H. High emotional reactivity is associated with activation of a molecularly distinct hippocampal-amygdala circuit modulated by the glucocorticoid receptor. Neurobiol. Stress 2023, 27, 100581. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.K.R.; Amelinez-Robles, N.; Polsfuss, I.; Herbert, K.; Kim, C.; Varghese, N.; Parry, T.J.; Buller, B.; Verdoorn, T.A.; Billing, C.B., Jr.; et al. NTS-105 decreased cell death and preserved long-term potentiation in an in vitro model of moderate traumatic brain injury. Exp. Neurol. 2023, 371, 114608. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Numakawa, T.; Murata, T.; Kawaminami, M.; Himi, T. Enhanced social reward response and anxiety-like behavior with downregulation of nucleus accumbens glucocorticoid receptor in BALB/c mice. J. Vet. Med. Sci. 2023, 85, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kumamaru, E.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Kunugi, H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc. Natl. Acad. Sci. USA 2009, 106, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Odaka, H.; Adachi, N.; Chiba, S.; Ooshima, Y.; Matsuno, H.; Nakajima, S.; Yoshimura, A.; Fumimoto, K.; Hirai, Y.; et al. Basic fibroblast growth factor increased glucocorticoid receptors in cortical neurons through MAP kinase pathway. Neurochem. Int. 2018, 118, 217–224. [Google Scholar] [CrossRef]
- McCann, K.E.; Lustberg, D.J.; Shaughnessy, E.K.; Carstens, K.E.; Farris, S.; Alexander, G.M.; Radzicki, D.; Zhao, M.; Dudek, S.M. Novel role for mineralocorticoid receptors in control of a neuronal phenotype. Mol. Psychiatry 2021, 26, 350–364. [Google Scholar] [CrossRef]
- Montgomery, K.R.; Bridi, M.S.; Folts, L.M.; Marx-Rattner, R.; Zierden, H.C.; Wulff, A.B.; Kodjo, E.A.; Thompson, S.M.; Bale, T.L. Chemogenetic activation of CRF neurons as a model of chronic stress produces sex-specific physiological and behavioral effects. Neuropsychopharmacology 2024, 49, 443–454. [Google Scholar] [CrossRef]
- You, I.J.; Bae, Y.; Beck, A.R.; Shin, S. Lateral hypothalamic proenkephalin neurons drive threat-induced overeating associated with a negative emotional state. Nat. Commun. 2023, 14, 6875. [Google Scholar] [CrossRef]
- Dos-Santos, R.C.; Sweeten, B.L.W.; Stelly, C.E.; Tasker, J.G. The Neuroendocrine Impact of Acute Stress on Synaptic Plasticity. Endocrinology 2023, 164, bqad149. [Google Scholar] [CrossRef]
- Ciubuc-Batcu, M.T.; Stapelberg, N.J.C.; Headrick, J.P.; Renshaw, G.M.C. A mitochondrial nexus in major depressive disorder: Integration with the psycho-immune-neuroendocrine network. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1870, 166920. [Google Scholar] [CrossRef]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef]
- Daviu, N.; Bruchas, M.R.; Moghaddam, B.; Sandi, C.; Beyeler, A. Neurobiological links between stress and anxiety. Neurobiol. Stress 2019, 11, 100191. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Glucocorticoids Orchestrate Adult Hippocampal Plasticity: Growth Points and Translational Aspects. Biochemistry 2023, 88, 565–589. [Google Scholar] [CrossRef]
- Barfield, E.T.; Gourley, S.L. Prefrontal cortical trkB, glucocorticoids, and their interactions in stress and developmental contexts. Neurosci. Biobehav. Rev. 2018, 95, 535–558. [Google Scholar] [CrossRef] [PubMed]
- Arango-Lievano, M.; Borie, A.M.; Dromard, Y.; Murat, M.; Desarmenien, M.G.; Garabedian, M.J.; Jeanneteau, F. Persistence of learning-induced synapses depends on neurotrophic priming of glucocorticoid receptors. Proc. Natl. Acad. Sci. USA 2019, 116, 13097–13106. [Google Scholar] [CrossRef] [PubMed]
- McCarty, K.J.; Pratt, S.L.; Long, N.M. Effects of Exogenous Glucocorticoid Infusion on Appetitic Center Development in Postnatal Dairy Bull Calves. Animals 2023, 13, 1980. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, F.; Zhang, Y.; Liu, Y.; An, L.; Ma, Z.; Zhang, J.; Yu, S. Neonatal DEX exposure leads to hyperanxious and depressive-like behaviors as well as a persistent reduction of BDNF expression in developmental stages. Biochem. Biophys. Res. Commun. 2020, 527, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Razzoli, M.; Domenici, E.; Carboni, L.; Rantamaki, T.; Lindholm, J.; Castrén, E.; Arban, R. A role for BDNF/TrkB signaling in behavioral and physiological consequences of social defeat stress. Genes Brain Behav. 2011, 10, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.F.; Huang, T.Y.; Chang, C.Y.; Hsu, Y.C.; Chen, S.J.; Yu, L.; Kuo, Y.M.; Jen, C.J. Social instability stress differentially affects amygdalar neuron adaptations and memory performance in adolescent and adult rats. Front. Behav. Neurosci. 2014, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Barfield, E.T.; Gerber, K.J.; Zimmermann, K.S.; Ressler, K.J.; Parsons, R.G.; Gourley, S.L. Regulation of actions and habits by ventral hippocampal trkB and adolescent corticosteroid exposure. PLoS Biol. 2017, 15, e2003000. [Google Scholar] [CrossRef]
- Azogu, I.; Liang, J.; Plamondon, H. Sex-specific differences in corticosterone secretion, behavioral phenotypes and expression of TrkB.T1 and TrkB.FL receptor isoforms: Impact of systemic TrkB inhibition and combinatory stress exposure in adolescence. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 10–23. [Google Scholar] [CrossRef]
- Pagliusi, M., Jr.; Franco, D.; Cole, S.; Morais-Silva, G.; Chandra, R.; Fox, M.E.; Iñiguez, S.D.; Sartori, C.R.; Lobo, M.K. The BDNF-TrkB Pathway Acts Through Nucleus Accumbens D2 Expressing Neurons to Mediate Stress Susceptible Outcomes. Front. Psychiatry 2022, 13, 854494. [Google Scholar] [CrossRef] [PubMed]
- Tessarollo, L.; Yanpallewar, S. TrkB Truncated Isoform Receptors as Transducers and Determinants of BDNF Functions. Front. Neurosci. 2022, 16, 847572. [Google Scholar] [CrossRef]
- Rostami, S.; Haghparast, A.; Fayazmilani, R. The downstream effects of forced exercise training and voluntary physical activity in an enriched environment on hippocampal plasticity in preadolescent rats. Brain Res. 2021, 1759, 147373. [Google Scholar] [CrossRef] [PubMed]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Niyaz, M.; Kudo, M.; Kunugi, H. Glucocorticoid prevents brain-derived neurotrophic factor-mediated maturation of synaptic function in developing hippocampal neurons through reduction in the activity of mitogen-activated protein kinase. Mol. Endocrinol. 2008, 22, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Ferrero Restelli, F.; Federicci, F.; Ledda, F.; Paratcha, G. Sprouty4 at the crossroads of Trk neurotrophin receptor signaling suppression by glucocorticoids. Front. Mol. Neurosci. 2023, 16, 1090824. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Li, R.; Cai, L.; Wu, S.D.; Li, C.M. Ro41-5253, a selective antagonist of retinoic acid receptor α, ameliorates chronic unpredictable mild stress-induced depressive-like behaviors in rats: Involvement of regulating HPA axis and improving hippocampal neuronal deficits. Brain Res. Bull. 2019, 146, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.N.; Meng, Q.Y.; Bao, A.M.; Swaab, D.F.; Wang, G.H.; Zhou, J.N. The involvement of retinoic acid receptor-alpha in corticotropin-releasing hormone gene expression and affective disorders. Biol. Psychiatry 2009, 66, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, O.; Li, N.; Sha, Z.; Zhao, Z.; Xu, J. Association between the BDNF Val66Met polymorphism and major depressive disorder: A systematic review and meta-analysis. Front. Psychiatry 2023, 14, 1143833. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Musazzi, L.; Tornese, P.; Sala, N.; Lee, F.S.; Popoli, M.; Ieraci, A. Acute stress induces an aberrant increase of presynaptic release of glutamate and cellular activation in the hippocampus of BDNF(Val/Met) mice. J. Cell. Physiol. 2022, 237, 3834–3844. [Google Scholar] [CrossRef]
- Raju, S.; Notaras, M.; Grech, A.M.; Schroeder, A.; van den Buuse, M.; Hill, R.A. BDNF Val66Met genotype and adolescent glucocorticoid treatment induce sex-specific disruptions to fear extinction and amygdala GABAergic interneuron expression in mice. Horm. Behav. 2022, 144, 105231. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J.S.; Mielke, J.G. The combined effects of corticosterone and brain-derived neurotrophic factor on plasticity-related receptor phosphorylation and expression at the synaptic surface in male Sprague-Dawley rats. Horm. Behav. 2022, 145, 105233. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Yan, X.J.; Lei, F.; Wang, M.L.; He, L.L.; Luo, Y.Y.; Gao, H.W.; Feng, Y.L.; Yang, S.L.; Li, J.; et al. Proteomic profiling of the neurons in mice with depressive-like behavior induced by corticosterone and the regulation of berberine: Pivotal sites of oxidative phosphorylation. Mol. Brain 2019, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.H.; Chao, H.W.; Lin, P.Y.; Lin, S.H.; Liu, T.H.; Chen, H.W.; Huang, Y.S. CPEB3-dowregulated Nr3c1 mRNA translation confers resilience to developing posttraumatic stress disorder-like behavior in fear-conditioned mice. Neuropsychopharmacology 2021, 46, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.; Mathes, S.; Köhler, K.; Achtzehn, S.; Bloch, W.; Mester, J. Acute metabolic, hormonal, and psychological responses to different endurance training protocols. Horm. Metab. Res. 2013, 45, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Jeanneteau, F.; Chao, M.V. Are BDNF and glucocorticoid activities calibrated? Neuroscience 2013, 239, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Hermann, R.; Schaller, A.; Lay, D.; Bloch, W.; Albus, C.; Petrowski, K. Effect of acute psychosocial stress on the brain-derived neurotrophic factor in humans—A randomized cross within trial. Stress 2021, 24, 442–449. [Google Scholar] [CrossRef] [PubMed]
- de Assis, G.G.; Gasanov, E.V. BDNF and Cortisol integrative system–Plasticity vs. degeneration: Implications of the Val66Met polymorphism. Front. Neuroendocrinol. 2019, 55, 100784. [Google Scholar] [CrossRef]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The amyloid cascade hypothesis: An updated critical review. Brain 2023, 146, 3969–3990. [Google Scholar] [CrossRef]
- Numakawa, T.; Kajihara, R. Neurotrophins and Other Growth Factors in the Pathogenesis of Alzheimer’s Disease. Life 2023, 13, 647. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300. [Google Scholar] [CrossRef]
- Garzon, D.; Yu, G.; Fahnestock, M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J. Neurochem. 2002, 82, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef]
- Angelucci, F.; Veverova, K.; Katonová, A.; Vyhnalek, M.; Hort, J. Serum PAI-1/BDNF Ratio Is Increased in Alzheimer’s Disease and Correlates with Disease Severity. ACS Omega 2023, 8, 36025–36031. [Google Scholar] [CrossRef]
- Mori, Y.; Tsuji, M.; Oguchi, T.; Kasuga, K.; Kimura, A.; Futamura, A.; Sugimoto, A.; Kasai, H.; Kuroda, T.; Yano, S.; et al. Serum BDNF as a Potential Biomarker of Alzheimer’s Disease: Verification through Assessment of Serum, Cerebrospinal Fluid, and Medial Temporal Lobe Atrophy. Front. Neurol. 2021, 12, 653267. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Laws, S.M.; Perin, S.; Pietrzak, R.H.; Fowler, C.; Masters, C.L.; Maruff, P. BDNF VAL66MET polymorphism and memory decline across the spectrum of Alzheimer’s disease. Genes Brain Behav. 2021, 20, e12724. [Google Scholar] [CrossRef]
- Bessi, V.; Mazzeo, S.; Bagnoli, S.; Padiglioni, S.; Carraro, M.; Piaceri, I.; Bracco, L.; Sorbi, S.; Nacmias, B. The implication of BDNF Val66Met polymorphism in progression from subjective cognitive decline to mild cognitive impairment and Alzheimer’s disease: A 9-year follow-up study. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 471–482. [Google Scholar] [CrossRef]
- del Toro, D.; Canals, J.M.; Ginés, S.; Kojima, M.; Egea, G.; Alberch, J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J. Neurosci. 2006, 26, 12748–12757. [Google Scholar] [CrossRef]
- Brown, D.T.; Vickers, J.C.; Stuart, K.E.; Cechova, K.; Ward, D.D. The BDNF Val66Met Polymorphism Modulates Resilience of Neurological Functioning to Brain Ageing and Dementia: A Narrative Review. Brain Sci. 2020, 10, 195. [Google Scholar] [CrossRef]
- Eggert, S.; Kins, S.; Endres, K.; Brigadski, T. Brothers in arms: ProBDNF/BDNF and sAPPα/Aβ-signaling and their common interplay with ADAM10, TrkB, p75NTR, sortilin, and sorLA in the progression of Alzheimer’s disease. Biol. Chem. 2022, 403, 43–71. [Google Scholar] [CrossRef]
- Zheng, Z.; Sabirzhanov, B.; Keifer, J. Oligomeric amyloid-{beta} inhibits the proteolytic conversion of brain-derived neurotrophic factor (BDNF), AMPA receptor trafficking, and classical conditioning. J. Biol. Chem. 2010, 285, 34708–34717. [Google Scholar] [CrossRef]
- Yan, P.; Xue, Z.; Li, D.; Ni, S.; Wang, C.; Jin, X.; Zhou, D.; Li, X.; Zhao, X.; Chen, X.; et al. Dysregulated CRTC1-BDNF signaling pathway in the hippocampus contributes to Aβ oligomer-induced long-term synaptic plasticity and memory impairment. Exp. Neurol. 2021, 345, 113812. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, Y.; Lian, Y.; Chen, Y.; Wu, T.; Zheng, Y.; Zong, H.; Sun, L.; Zhang, R.; Wang, Z.; et al. Brain-derived neurotrophic factor ameliorates learning deficits in a rat model of Alzheimer’s disease induced by aβ1-42. PLoS ONE 2015, 10, e0122415. [Google Scholar] [CrossRef]
- Angelucci, F.; Čechová, K.; Průša, R.; Hort, J. Amyloid beta soluble forms and plasminogen activation system in Alzheimer’s disease: Consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci. Ther. 2019, 25, 303–313. [Google Scholar] [CrossRef]
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Jerónimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lérias, S.; Caetano, A.P.; Buée-Scherrer, V.; Castrén, E.; Valente, C.A.; Blum, D.; Sebastião, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-β Peptide is Mediated by Calpain. Cereb. Cortex 2015, 25, 3107–3121. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr. Neuropharmacol. 2016, 14, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, Y.; Dong, Y. Acute and subacute IL-1β administrations differentially modulate neuroimmune and neurotrophic systems: Possible implications for neuroprotection and neurodegeneration. J. Neuroinflamm. 2013, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Balazs, R.; Soiampornkul, R.; Thangnipon, W.; Cotman, C.W. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol. Aging 2008, 29, 1380–1393. [Google Scholar] [CrossRef] [PubMed]
- Barbereau, C.; Yehya, A.; Silhol, M.; Cubedo, N.; Verdier, J.M.; Maurice, T.; Rossel, M. Neuroprotective brain-derived neurotrophic factor signaling in the TAU-P301L tauopathy zebrafish model. Pharmacol. Res. 2020, 158, 104865. [Google Scholar] [CrossRef]
- Oreshko, A.S.; Rodnyy, A.Y.; Bazovkina, D.V.; Naumenko, V.S. Effects of central administration of the human Tau protein on the Bdnf, Trkb, p75, Mapt, Bax and Bcl-2 genes expression in the mouse brain. Vavilovskii Zhurnal Genet. Selektsii 2023, 27, 342–348. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef]
- Laczó, J.; Cechova, K.; Parizkova, M.; Lerch, O.; Andel, R.; Matoska, V.; Kaplan, V.; Matuskova, V.; Nedelska, Z.; Vyhnalek, M.; et al. The Combined Effect of APOE and BDNF Val66Met Polymorphisms on Spatial Navigation in Older Adults. J. Alzheimers Dis. 2020, 78, 1473–1492. [Google Scholar] [CrossRef]
- Pietzuch, M.; Bindoff, A.; Jamadar, S.; Vickers, J.C. Interactive effects of the APOE and BDNF polymorphisms on functional brain connectivity: The Tasmanian Healthy Brain Project. Sci. Rep. 2021, 11, 14514. [Google Scholar] [CrossRef]
- Viho, E.M.G.; Buurstede, J.C.; Mahfouz, A.; Koorneef, L.L.; van Weert, L.; Houtman, R.; Hunt, H.J.; Kroon, J.; Meijer, O.C. Corticosteroid Action in the Brain: The Potential of Selective Receptor Modulation. Neuroendocrinology 2019, 109, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Koning, A.; Buurstede, J.C.; van Weert, L.; Meijer, O.C. Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. J. Endocr. Soc. 2019, 3, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Pedrazzoli, M.; Losurdo, M.; Paolone, G.; Medelin, M.; Jaupaj, L.; Cisterna, B.; Slanzi, A.; Malatesta, M.; Coco, S.; Buffelli, M. Glucocorticoid receptors modulate dendritic spine plasticity and microglia activity in an animal model of Alzheimer’s disease. Neurobiol. Dis. 2019, 132, 104568. [Google Scholar] [CrossRef] [PubMed]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Dioli, C.; Papadimitriou, G.; Megalokonomou, A.; Marques, C.; Sousa, N.; Sotiropoulos, I. Chronic Stress, Depression, and Alzheimer’s Disease: The Triangle of Oblivion. Adv. Exp. Med. Biol. 2023, 1423, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Forget, H.; Lacroix, A.; Somma, M.; Cohen, H. Cognitive decline in patients with Cushing’s syndrome. J. Int. Neuropsychol. Soc. 2000, 6, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Notarianni, E. Hypercortisolemia and glucocorticoid receptor-signaling insufficiency in Alzheimer’s disease initiation and development. Curr. Alzheimer Res. 2013, 10, 714–731. [Google Scholar] [CrossRef]
- Zheng, B.; Tal, R.; Yang, Z.; Middleton, L.; Udeh-Momoh, C. Cortisol hypersecretion and the risk of Alzheimer’s disease: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 64, 101171. [Google Scholar] [CrossRef]
- Klyubin, I.; Ondrejcak, T.; Hu, N.-W.; Rowan, M.J. Glucocorticoids, synaptic plasticity and Alzheimer’s disease. Curr. Opin. Endocr. Metab. Res. 2022, 25, 100365. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Swerdlow, R.H.; Waites, C.L. Glucocorticoid-driven mitochondrial damage stimulates Tau pathology. Brain 2023, 146, 4378–4394. [Google Scholar] [CrossRef]
- Kulstad, J.J.; McMillan, P.J.; Leverenz, J.B.; Cook, D.G.; Green, P.S.; Peskind, E.R.; Wilkinson, C.W.; Farris, W.; Mehta, P.D.; Craft, S. Effects of chronic glucocorticoid administration on insulin-degrading enzyme and amyloid-beta peptide in the aged macaque. J. Neuropathol. Exp. Neurol. 2005, 64, 139–146. [Google Scholar] [CrossRef]
- Ding, S.; Yang, L.; Huang, L.; Kong, L.; Chen, M.; Su, Y.; Li, X.; Dong, X.; Han, Y.; Li, W.; et al. Chronic glucocorticoid exposure accelerates Aβ generation and neurotoxicity by activating calcium-mediated CN-NFAT1 signaling in hippocampal neurons in APP/PS1 mice. Food Chem. Toxicol. 2022, 168, 113407. [Google Scholar] [CrossRef]
- Siegel, G.; Gerber, H.; Koch, P.; Bruestle, O.; Fraering, P.C.; Rajendran, L. The Alzheimer’s Disease γ-Secretase Generates Higher 42:40 Ratios for β-Amyloid Than for p3 Peptides. Cell Rep. 2017, 19, 1967–1976. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Tang, J.; Song, M.; Xu, X.; Xiong, J.; Li, J.; Bai, Y. Glucocorticoids facilitate astrocytic amyloid-β peptide deposition by increasing the expression of APP and BACE1 and decreasing the expression of amyloid-β-degrading proteases. Endocrinology 2011, 152, 2704–2715. [Google Scholar] [CrossRef]
- Sotiropoulos, I.; Catania, C.; Riedemann, T.; Fry, J.P.; Breen, K.C.; Michaelidis, T.M.; Almeida, O.F. Glucocorticoids trigger Alzheimer disease-like pathobiochemistry in rat neuronal cells expressing human tau. J. Neurochem. 2008, 107, 385–397. [Google Scholar] [CrossRef]
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-mediated activation of GSK3β promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 2017, 57, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Brown, C.; Whitehead, G.; Piers, T.; Lee, Y.S.; Perez, C.M.; Regan, P.; Whitcomb, D.J.; Cho, K. Glucocorticoids activate a synapse weakening pathway culminating in tau phosphorylation in the hippocampus. Pharmacol. Res. 2017, 121, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Milligan Armstrong, A.; Porter, T.; Quek, H.; White, A.; Haynes, J.; Jackaman, C.; Villemagne, V.; Munyard, K.; Laws, S.M.; Verdile, G.; et al. Chronic stress and Alzheimer’s disease: The interplay between the hypothalamic-pituitary-adrenal axis, genetics and microglia. Biol. Rev. Camb. Philos. Soc. 2021, 96, 2209–2228. [Google Scholar] [CrossRef]
- Merighi, S.; Nigro, M.; Travagli, A.; Gessi, S. Microglia and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2990. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Harris-White, M.E.; Chu, T.; Miller, S.A.; Simmons, M.; Teter, B.; Nash, D.; Cole, G.M.; Frautschy, S.A. Estrogen (E2) and glucocorticoid (Gc) effects on microglia and A beta clearance in vitro and in vivo. Neurochem. Int. 2001, 39, 435–448. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, T.G. Navigating Alzheimer’s Disease via Chronic Stress: The Role of Glucocorticoids. Curr. Drug Targets 2020, 21, 433–444. [Google Scholar] [CrossRef]
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoid Generates ROS to Induce Oxidative Injury in the Hippocampus, Leading to Impairment of Cognitive Function of Rats. J. Clin. Biochem. Nutr. 2010, 47, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Yang, F.; Zheng, Q.; Tang, W.; Li, J. The Potential Role of the NLRP3 Inflammasome Activation as a Link between Mitochondria ROS Generation and Neuroinflammation in Postoperative Cognitive Dysfunction. Front. Cell. Neurosci. 2019, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Wurzelmann, M.; Romeika, J.; Sun, D. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen. Res. 2017, 12, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583. [Google Scholar] [CrossRef]
- Liao, J.; Chen, C.; Ahn, E.H.; Liu, X.; Li, H.; Edgington-Mitchell, L.E.; Lu, Z.; Ming, S.; Ye, K. Targeting both BDNF/TrkB pathway and delta-secretase for treating Alzheimer’s disease. Neuropharmacology 2021, 197, 108737. [Google Scholar] [CrossRef] [PubMed]
- Pak, M.E.; Yang, H.J.; Li, W.; Kim, J.K.; Go, Y. Yuk-Gunja-Tang attenuates neuronal death and memory impairment via ERK/CREB/BDNF signaling in the hippocampi of experimental Alzheimer’s disease model. Front. Pharmacol. 2022, 13, 1014840. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Chitranshi, N.; Gupta, V.; You, Y.; Rajput, R.; Paulo, J.A.; Mirzaei, M.; van den Buuse, M.; Graham, S.L. TrkB Receptor Agonist 7,8 Dihydroxyflavone is Protective Against the Inner Retinal Deficits Induced by Experimental Glaucoma. Neuroscience 2022, 490, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Jeong, Y.J.; Kang, E.J.; Kang, B.S.; Lee, S.H.; Kim, Y.J.; Kang, S.S.; Suh, S.W.; Ahn, E.H. GAP-43 closely interacts with BDNF in hippocampal neurons and is associated with Alzheimer’s disease progression. Front. Mol. Neurosci. 2023, 16, 1150399. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 2013, 33, 15596–15602. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF reduce Aβ production by enhancing α-secretase processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef]
- Kim, D.; Cho, J.; Kang, H. Protective effect of exercise training against the progression of Alzheimer’s disease in 3xTg-AD mice. Behav. Brain Res. 2019, 374, 112105. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, L.; Zhu, L.; Chen, D.; Cai, K.; Liu, Z.; Chen, A. Moderate Exercise Combined with Enriched Environment Enhances Learning and Memory through BDNF/TrkB Signaling Pathway in Rats. Int. J. Environ. Res. Public Health 2021, 18, 8283. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, X.; Zhao, Z. A systematic review and meta-analysis on the efficacy outcomes of selective serotonin reuptake inhibitors in depression in Alzheimer’s disease. BMC Neurol. 2023, 23, 210. [Google Scholar] [CrossRef]
- Casarotto, P.; Umemori, J.; Castrén, E. BDNF receptor TrkB as the mediator of the antidepressant drug action. Front. Mol. Neurosci. 2022, 15, 1032224. [Google Scholar] [CrossRef]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e1219. [Google Scholar] [CrossRef]
- Jia, R.X.; Liang, J.H.; Xu, Y.; Wang, Y.Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: A meta-analysis. BMC Geriatr. 2019, 19, 181. [Google Scholar] [CrossRef]
- Håkansson, K.; Ledreux, A.; Daffner, K.; Terjestam, Y.; Bergman, P.; Carlsson, R.; Kivipelto, M.; Winblad, B.; Granholm, A.C.; Mohammed, A.K. BDNF Responses in Healthy Older Persons to 35 Minutes of Physical Exercise, Cognitive Training, and Mindfulness: Associations with Working Memory Function. J. Alzheimers Dis. 2017, 55, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Peppe, A.; Carlesimo, G.A.; Serafini, F.; Zabberoni, S.; Barban, F.; Shofany, J.; Caltagirone, C.; Costa, A. A pilot study on the effect of cognitive training on BDNF serum levels in individuals with Parkinson’s disease. Front. Hum. Neurosci. 2015, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Gomutbutra, P.; Srikamjak, T.; Sapinun, L.; Kunaphanh, S.; Yingchankul, N.; Apaijai, N.; Shinlapawittayatorn, K.; Phuackchantuck, R.; Chattipakorn, N.; Chattipakorn, S. Effect of intensive weekend mindfulness-based intervention on BDNF, mitochondria function, and anxiety. A randomized, crossover clinical trial. Compr. Psychoneuroendocrinol. 2022, 11, 100137. [Google Scholar] [CrossRef] [PubMed]
- Canet, G.; Hernandez, C.; Zussy, C.; Chevallier, N.; Desrumaux, C.; Givalois, L. Is AD a Stress-Related Disorder? Focus on the HPA Axis and Its Promising Therapeutic Targets. Front. Aging Neurosci. 2019, 11, 269. [Google Scholar] [CrossRef]
- Watermeyer, T.; Robb, C.; Gregory, S.; Udeh-Momoh, C. Therapeutic implications of hypothalamic-pituitaryadrenal-axis modulation in Alzheimer’s disease: A narrative review of pharmacological and lifestyle interventions. Front. Neuroendocrinol. 2021, 60, 100877. [Google Scholar] [CrossRef]
- Sooy, K.; Webster, S.P.; Noble, J.; Binnie, M.; Walker, B.R.; Seckl, J.R.; Yau, J.L. Partial deficiency or short-term inhibition of 11beta-hydroxysteroid dehydrogenase type 1 improves cognitive function in aging mice. J. Neurosci. 2010, 30, 13867–13872. [Google Scholar] [CrossRef]
- Sooy, K.; Noble, J.; McBride, A.; Binnie, M.; Yau, J.L.; Seckl, J.R.; Walker, B.R.; Webster, S.P. Cognitive and Disease-Modifying Effects of 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibition in Male Tg2576 Mice, a Model of Alzheimer’s Disease. Endocrinology 2015, 156, 4592–4603. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Medeiros, R.; Martinez-Coria, H.; LaFerla, F.M.; Green, K.N. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol. Psychiatry 2013, 74, 357–366. [Google Scholar] [CrossRef]
- Pineau, F.; Canet, G.; Desrumaux, C.; Hunt, H.; Chevallier, N.; Ollivier, M.; Belanoff, J.K.; Givalois, L. New selective glucocorticoid receptor modulators reverse amyloid-β peptide-induced hippocampus toxicity. Neurobiol. Aging 2016, 45, 109–122. [Google Scholar] [CrossRef]
- da Costa Daniele, T.M.; de Bruin, P.F.C.; de Matos, R.S.; de Bruin, G.S.; Maia Chaves, C.J.; de Bruin, V.M.S. Exercise effects on brain and behavior in healthy mice, Alzheimer’s disease and Parkinson’s disease model-A systematic review and meta-analysis. Behav. Brain Res. 2020, 383, 112488. [Google Scholar] [CrossRef] [PubMed]
- Bashiri, H.; Enayati, M.; Bashiri, A.; Salari, A.A. Swimming exercise improves cognitive and behavioral disorders in male NMRI mice with sporadic Alzheimer-like disease. Physiol. Behav. 2020, 223, 113003. [Google Scholar] [CrossRef] [PubMed]
- Campos, H.C.; Ribeiro, D.E.; Hashiguchi, D.; Glaser, T.; Milanis, M.D.S.; Gimenes, C.; Suchecki, D.; Arida, R.M.; Ulrich, H.; Monteiro Longo, B. Neuroprotective effects of resistance physical exercise on the APP/PS1 mouse model of Alzheimer’s disease. Front. Neurosci. 2023, 17, 1132825. [Google Scholar] [CrossRef] [PubMed]
- Irazoki, E.; Contreras-Somoza, L.M.; Toribio-Guzmán, J.M.; Jenaro-Río, C.; van der Roest, H.; Franco-Martín, M.A. Technologies for Cognitive Training and Cognitive Rehabilitation for People With Mild Cognitive Impairment and Dementia. A Systematic Review. Front. Psychol. 2020, 11, 648. [Google Scholar] [CrossRef]
- Cutuli, D.; Decandia, D.; Giacovazzo, G.; Coccurello, R. Physical Exercise as Disease-Modifying Alternative against Alzheimer’s Disease: A Gut-Muscle-Brain Partnership. Int. J. Mol. Sci. 2023, 24, 4686. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numakawa, T.; Kajihara, R. An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 1596. https://doi.org/10.3390/ijms25031596
Numakawa T, Kajihara R. An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer’s Disease. International Journal of Molecular Sciences. 2024; 25(3):1596. https://doi.org/10.3390/ijms25031596
Chicago/Turabian StyleNumakawa, Tadahiro, and Ryutaro Kajihara. 2024. "An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer’s Disease" International Journal of Molecular Sciences 25, no. 3: 1596. https://doi.org/10.3390/ijms25031596
APA StyleNumakawa, T., & Kajihara, R. (2024). An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer’s Disease. International Journal of Molecular Sciences, 25(3), 1596. https://doi.org/10.3390/ijms25031596