1. Introduction
Protein is an important living organism component, regulating life activities, controlling growth and energy metabolism, etc. Post-translational modifications (PTMs) are important cellular control mechanisms that increase the functional diversity of the proteome through the covalent addition of functional groups or proteins, the hydrolytic cleavage of regulatory subunits, or the degradation of whole proteins [
1]. Protein phosphorylation is the most studied PTM, and the most studied phosphorylation to date is on serine (S), threonine (T), and tyrosine (Y). Phosphorylation site prediction can help researchers understand protein function and interactions, particularly in the areas of signal transduction and metabolic pathways [
2]. For example, it can help researchers to study the relationship between proteins and biological processes such as cell cycle, apoptosis, and cell proliferation. And phosphorylation site prediction can provide important information for protein structure studies, as phosphorylation can change the conformation and charge of proteins and can help researchers predict protein secondary structures, tertiary structures, and interactions [
3]. In addition, some studies have shown that more than 30% of eukaryotic proteins can be phosphorylated, and most of these proteins are closely associated with the development of human diseases, such as cancer [
4]. Therefore, the study of the principles, types, and mechanisms of protein phosphorylation is important for understanding the pathogenesis of human diseases. Research on protein phosphorylation site prediction can also help to identify new drug targets, thus providing new ideas and directions for disease treatment.
Early researchers performed many bioassay identification methods to identify phosphorylation sites, such as low-throughput 32P labeling [
5] and high-throughput mass spectrometry [
6], but such methods are quite time-consuming and laborious. With the rapid development of natural language processing, artificial intelligence, and other disciplines, and the integration of various new technologies with bioinformatics, the difficulties of traditional experimental methods have been effectively solved. Dozens of different methods, including machine learning, statistics, feature extraction, and other techniques [
7,
8,
9], have been used to predict and identify phosphorylation sites. For example, Musite, proposed in 2010, used multiple features and algorithms to improve prediction accuracy by using local amino acid sequence frequencies, k-nearest neighbor features, and protein disorder scores. This was followed by MusiteDeep [
10], the first deep learning framework for predicting general and kinase-specific phosphorylation sites, which was proposed by the team in 2017, using a convolutional neural network with a novel two-dimensional attention mechanism for prediction. In 2019, Fenglin Luo et al. proposed DeepPhos [
11], which utilizes densely connected convolutional neuron network blocks to capture multiple representations of sequences. Final phosphorylation prediction was performed through intra-block crosstalk layers and inter-block crosstalk layers. These deep learning architectures for phosphorylation site prediction are far superior to traditional experimental methods. However, there is still room for improvement in prediction accuracy and prediction efficiency. It is essential to develop a prediction tool that performs well at S, T, and Y sites.
These deep learning architectures for phosphorylation site prediction are far superior to traditional experimental methods. However, there are more negative samples than positive samples for almost all PTM types, including protein phosphorylation [
10], which makes the performance of these methods limited. By training with unbalanced datasets, the model can have better generalization ability in real situations. In addition, there is still a lot of room for improvement in the generalization ability of these models, which are not yet able to achieve a high level of prediction performance on S, T, and Y sites with small sample sizes at the same time. Therefore, there is still room for further improvement in terms of prediction accuracy and efficiency. Therefore, it has become crucial to develop a prediction tool that can perform well on both balanced and unbalanced datasets with higher accuracy and better generalization ability.
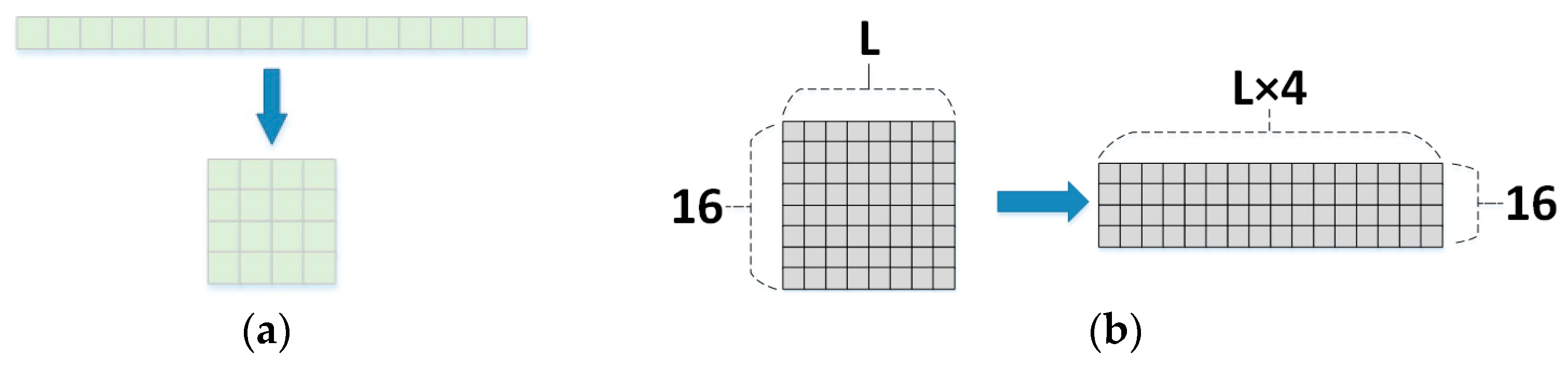
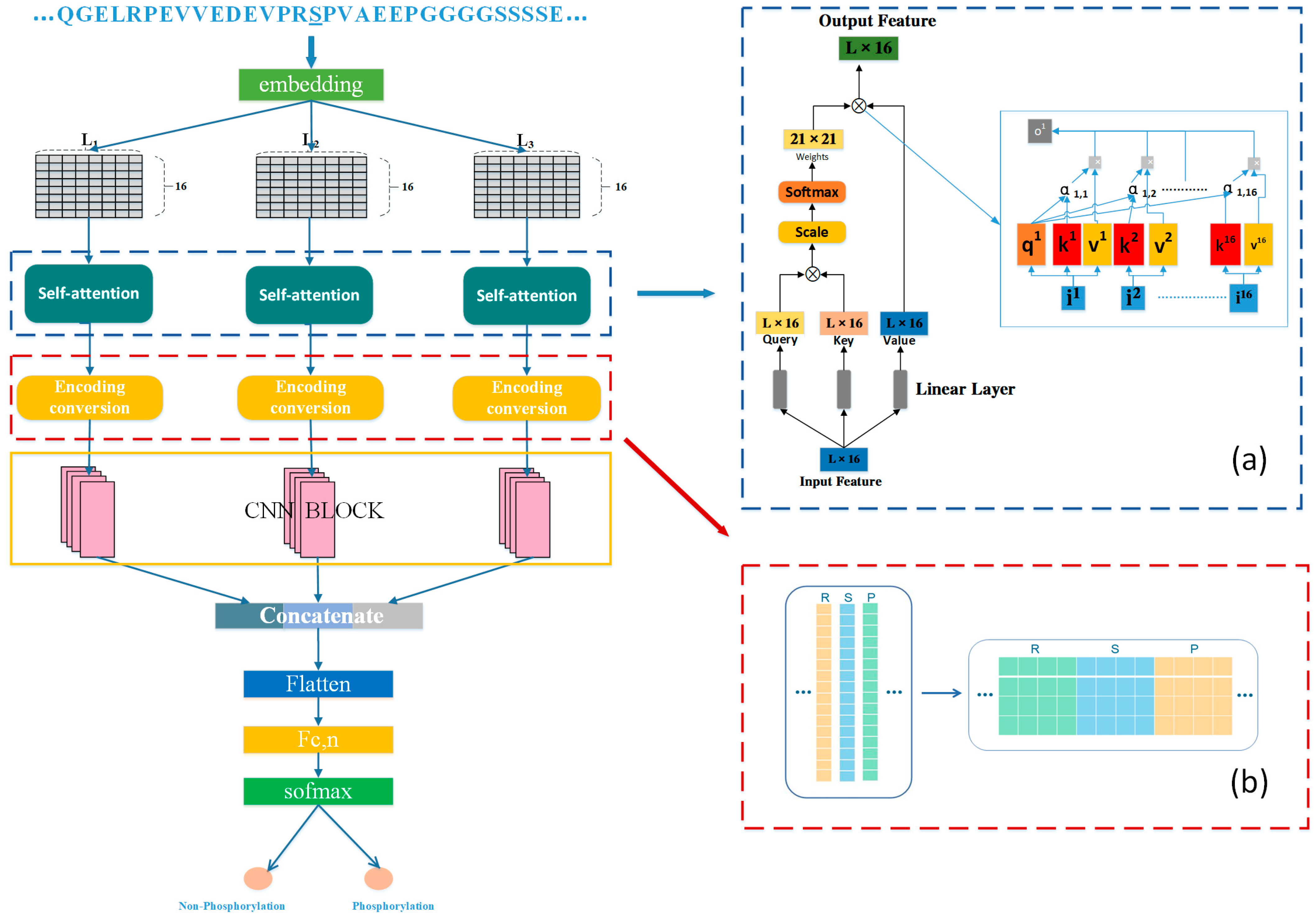
In response to the above challenges, this study proposes a new deep learning framework to accurately predict general phosphorylation sites using protein sequence information with good robustness and generalization ability. Unlike previous deep learning prediction methods, Attenphos consists of self-attention and CNN blocks that use three structurally identical modules with different window sizes to capture significant sequence representations of protein phosphorylation sites. The feature vector output of the attention module is converted using the Encoding conversion module, increasing the representation capability of the neural network. The transformed coded feature matrix has more structural information and learns features more fully than if the output features were processed directly. The final three outputs are further integrated to make the final prediction. To evaluate the performance of Attenphos, we collected a large number of validated phosphorylation examples for model training and evaluation, and we conducted experimental comparisons with some other prediction tools and deep learning methods. The experimental results show that our proposed method outperforms some existing prediction tools on both balanced and unbalanced datasets. Attenphos achieves the best overall prediction performance on T, S, and Y sites with small sample sizes.
2. Results
2.1. Comparison Experiments on Balanced Datasets
In this section, we provide a discussion of the comparative performance of Attenphos for general phosphorylation site prediction using a balanced dataset from human protein phosphorylation. We compared Attenphos with several existing general phosphorylation site prediction tools, including GPS 2.1 [
12], NetPhos 3.0 [
13], PPRED [
14], Musite [
15], PhosphoSVM [
16], SKIPHOS [
17], Deepphos [
11], and Transphos [
18]. We performed 10-fold cross-validation to train the model using the training datasets of the S, T, and Y sites. We tested it using an independent PPA test set. As shown in
Table 1, the results indicate that our method obtains the best overall performance in S and T bit prediction. And on the Y dataset with a smaller sample size, our method still outperforms existing state-of-the-art deep learning architectures to obtain the best overall performance.
Specifically, at the S site, our model obtains the highest Mcc value of 0.443 and the highest Auc value of 0.788, while other metrics such as Sn, Sp, and F1 values are also in the lead. In the case of the Mcc metric, we achieve an improvement of 11% compared to the state-of-the-art prediction tool Transphos. At the T site, our method shows the best performance, with Attenphos obtaining the highest Mcc and Auc values compared to other methods. Compared to GPS 2.1, NetPhos 3.0, PPRED, Musite, PhosphoSVM, SKIPHOS, Deepphos, and Transphos, our architecture improves the Auc values by 2.1%, 5.1%, 11.3%, 6.9%, 2.6%, 4.8%, 1.7%, and 1.9%, respectively, and obtains the highest Mcc value (0.254). At the Y site, our method obtains the Auc value of 0.625, second only to the Auc value of SKIPHOS (0.634), and the highest Mcc value (0.201). Compared to state-of-the-art deep learning prediction tools such as Deepphos and Transphos, Attenphos improves a lot on Auc metrics, especially on Mcc metrics. To summarize, our model Attenphos performs well in phosphorylation prediction at the S, T, and Y sites, showing the best overall results, with Attenphos being able to accurately predict phosphorylation sites compared to existing state-of-the-art tools.
2.2. Comparison Experiments on Unbalanced Datasets
As shown in
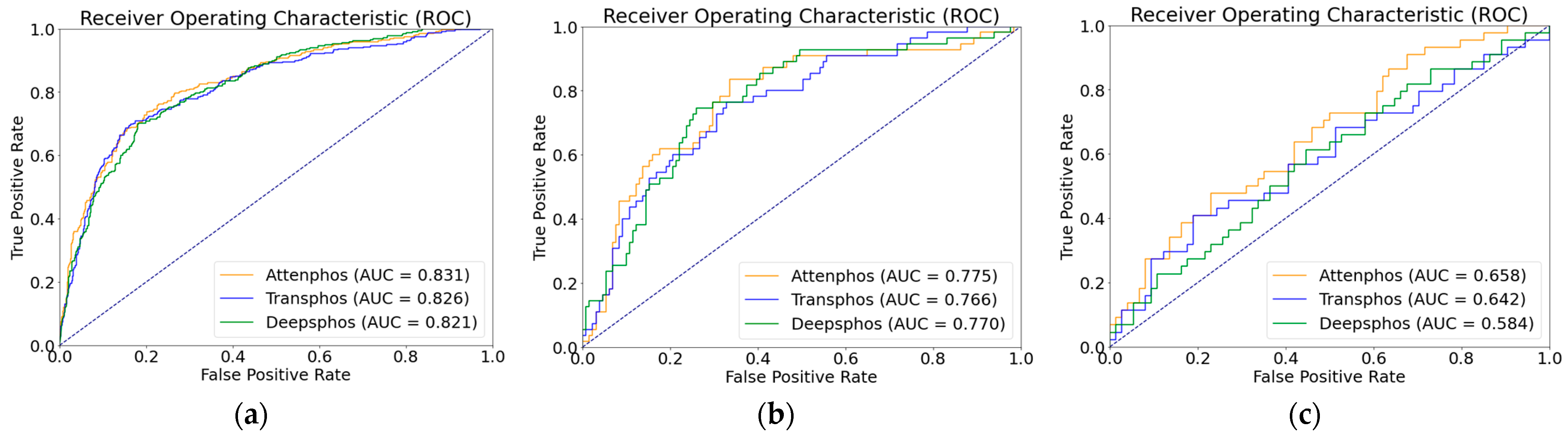
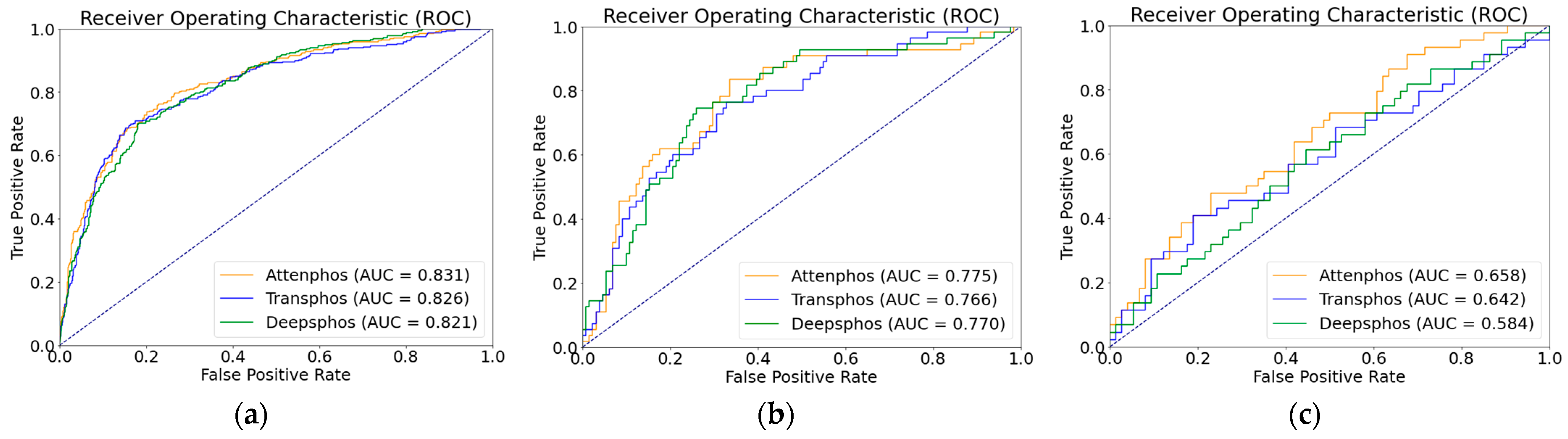
Table 2, we conducted performance comparison experiments on a dataset with positive and negative sample unbalance distribution, which is a phosphorylation dataset from mouse proteins. In order to assess the generalization capability of our model on unbalanced datasets with limited samples, we specifically selected the advanced deep learning models Transphos and Deepphos for a focused comparison. Both Transphos and Deepphos perform well on the balanced dataset. We trained and validated individual models, including Deepphos, Transphos, and Attenphos. Then, we tested them using independent test sets. Considering the unbalance in the number of positive and negative samples, we used evaluation metrics of SN, SP, MCC, AUC, and F1 scores for comparison. The results are shown in the table above. At the T site, Attenphos achieves the highest SP, AUC, MCC, SN, and F1 values of 87.79%, 0.775, 0.416, 50.91%, and 0.566, respectively. Compared with Transphos, its AUC value is increased by 0.9% and its MCC value by 2.2%. At the Y site, Attenphos achieves the highest SP, SN, AUC, F1, and MCC values. Attenphos obtains the highest MCC and F1 values in S-site phosphorylation prediction. Compared to Transphos, Attenphos shows an increase of 0.5% in the AUC value and 0.3% in the F1 score value. In addition, the MCC, SN, and SP values also perform better. Our research findings unveil the superiority of Attenpphos over Deepphos and Transphos, as confirmed by the ROC curve analysis. The ROC curves of Attenpphos consistently exhibit a larger area under the curve (AUC), indicating the better classification performance of our model compared to the other two models. The ROC graph is shown in
Figure 1 below. This indicates that our model can still show excellent performance in the presence of positive and negative sample unbalance.
In real-world protein phosphorylation problems, where the distributions of positive and negative samples are often unbalanced, our model demonstrates significant performance improvement in phosphorylation prediction at the S, T, and Y sites, achieving higher overall performance. This suggests that Attenphos is not only applicable to datasets with balanced distributions but also to datasets with unbalanced distributions that are close to the real situation.
2.3. Ablation Experiments
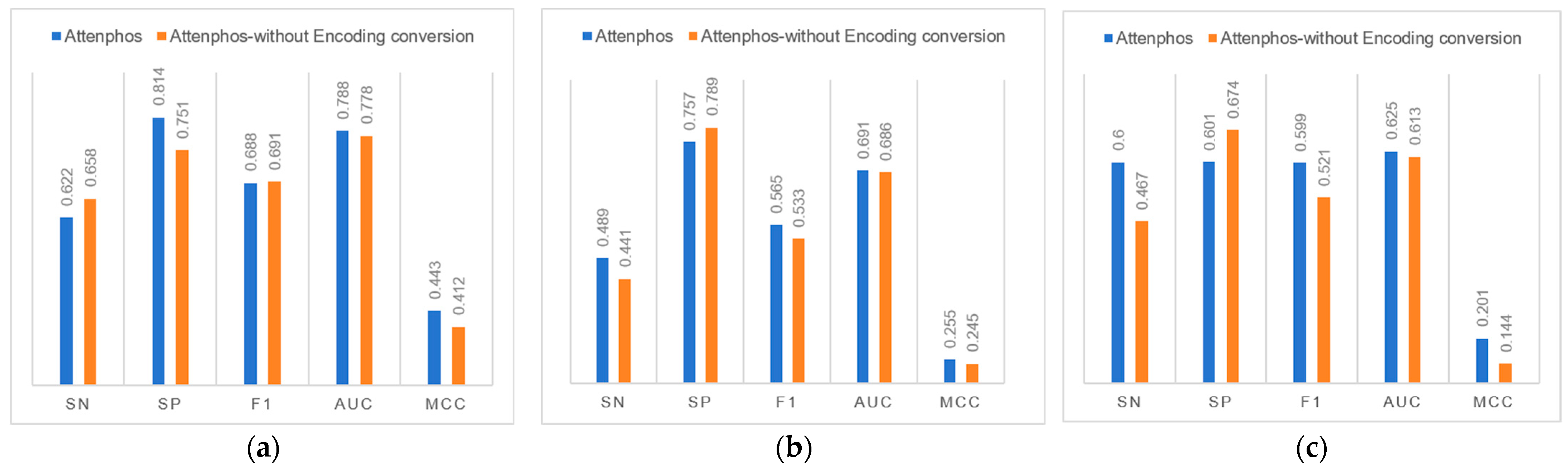
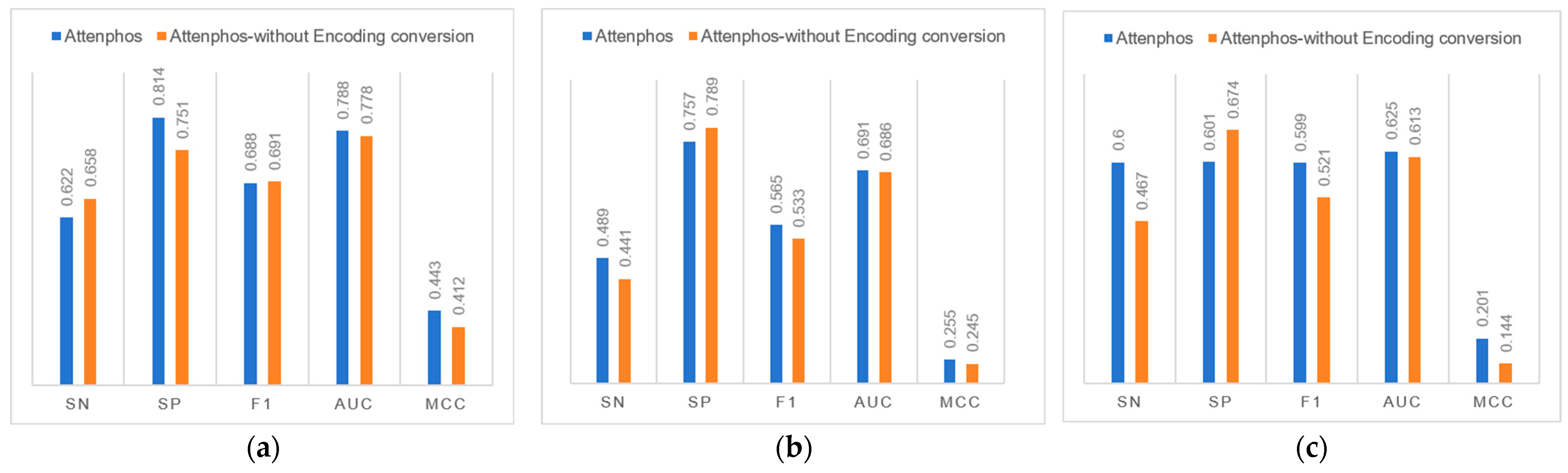
We investigated the contribution of the Encoding conversion block to general phosphorylation site prediction.
Figure 2 shows that removing the Encoding conversion block in the Attenphos architecture decreases the overall performance of general phosphorylation site prediction. For example, at the S site, removing the Encoding conversion causes the Sp, F1, Auc, and Mcc values to be reduced at the S site. At the T site, the removal of Encoding conversion reduces the values of Sn, F1, Auc, and Mcc. At the Y site, the removal of Encoding conversion reduces the values of Sn, F1, Auc, and Mcc. Therefore, the use of the Encoding conversion module allows the model to better capture the relational features prior to the amino acids, focusing on key information and thus improving the overall predictive performance of the model.
In this section, we performed ablation experiments on the effect of the attention mechanism in the Attenphos method. The experiment results are shown in
Table 3 below. As shown in the table, the performance of Attenphos-noAttention undergoes a large decrease compared to Attenphos at the S, T, and Y sites. Although the F1 score improves by 1% and 0.6% at the S and T sites, other metrics such as Mcc and Auc show a large decrease. At the Y site, Attenphos-noAttention decreases by 6.1% in AUC, 7.2% in mcc, and 5.1% in F1 score. Self-attention mechanisms can focus on the relationship between different amino acids. The self-attention mechanism module enables our model to better and more efficiently capture the long-range dependencies of protein sequences and extract global feature information, thus improving the prediction accuracy.
2.4. Feature Visualization
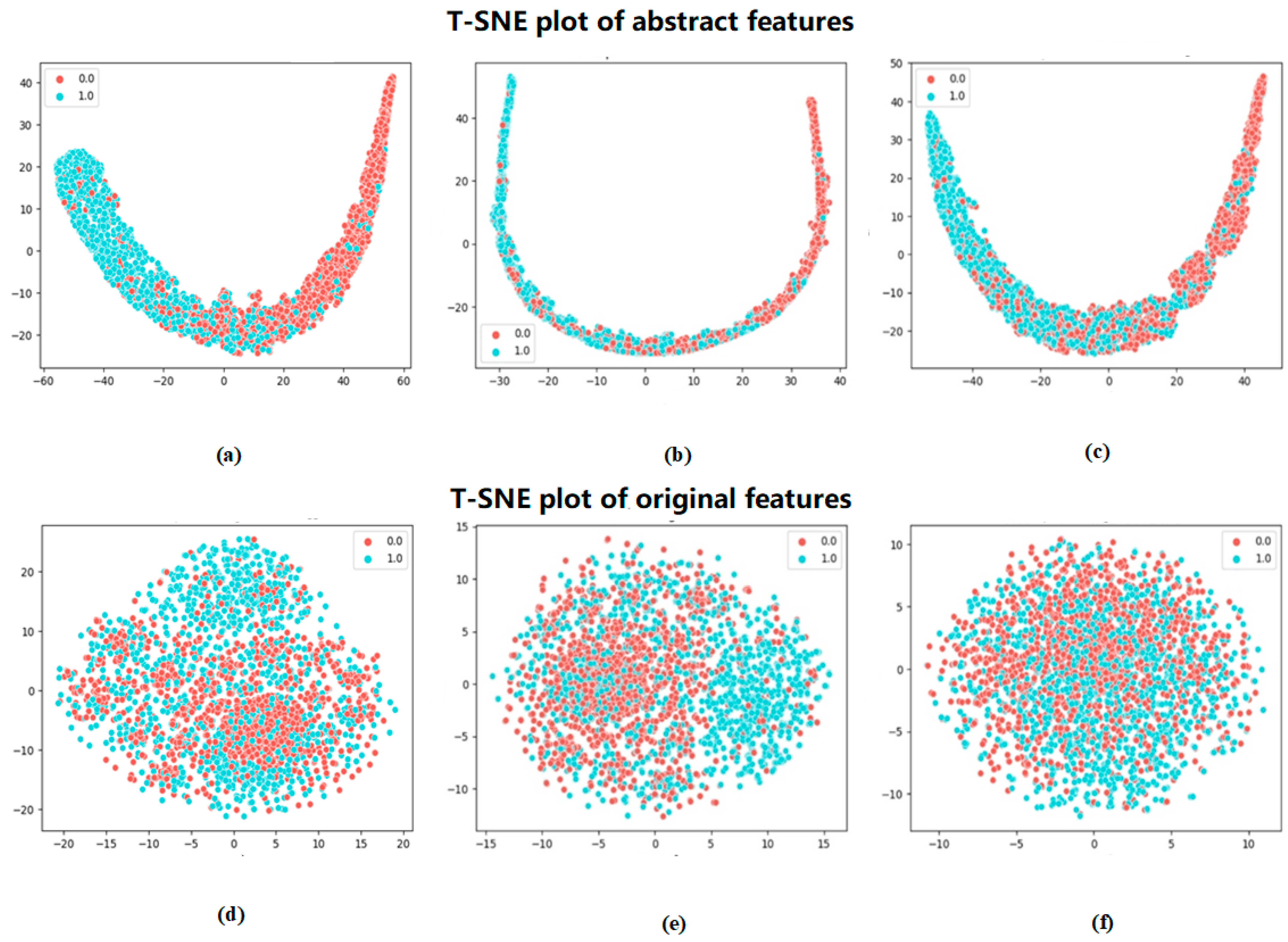
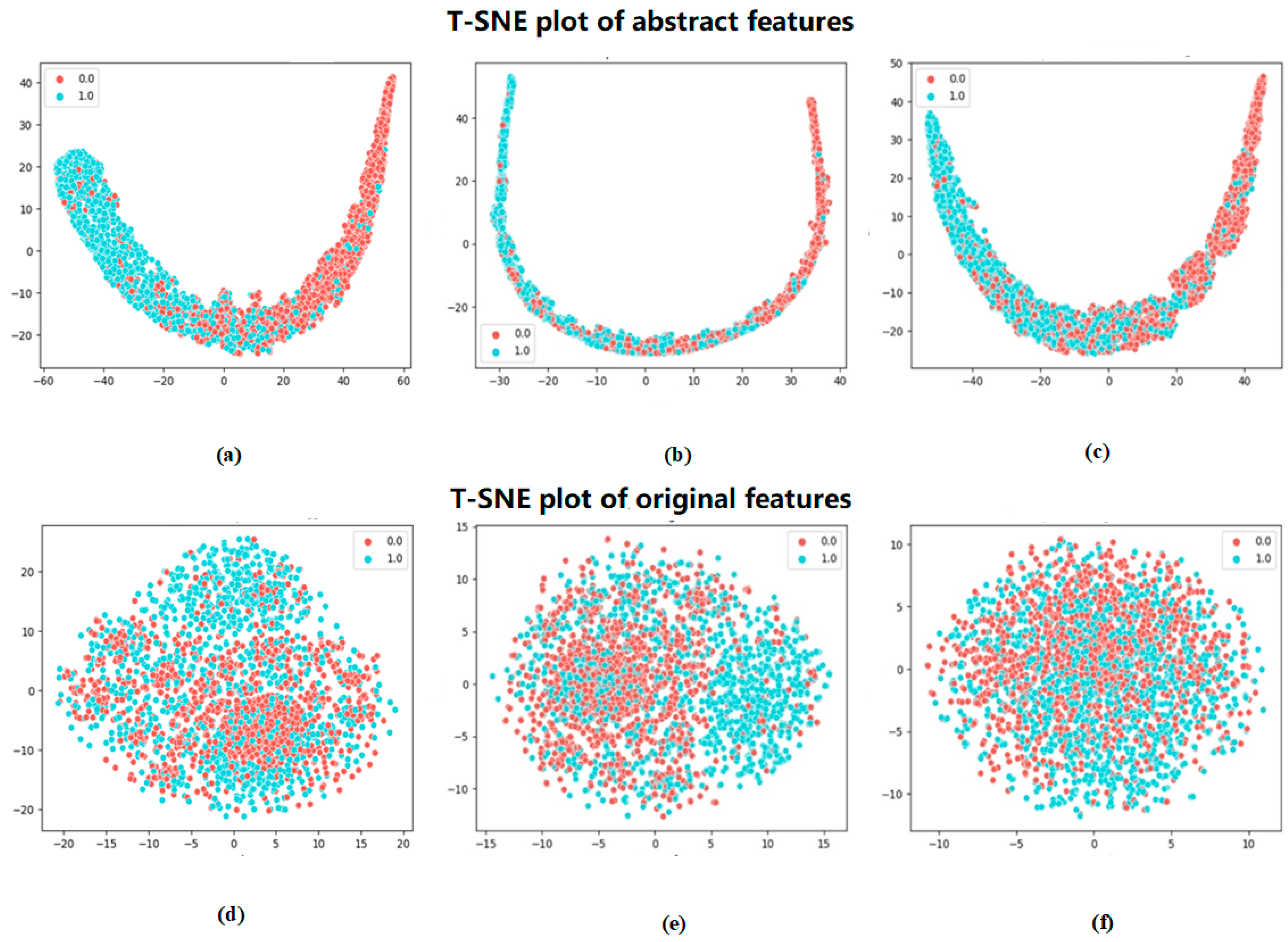
To more visually demonstrate the effectiveness of the method in this paper for the classification of phosphorylated versus non-phosphorylated sites, the features extracted by Attenphos were visualized. We chose a commonly used dimensionality reduction visualization method, t-SNE (t-Distributed Stochastic Neighbor Embedding) [
19], which can be used to map high-dimensional data into a low-dimensional space and preserve the relative distance relationship between the data. It can reduce the dimensionality of the high-dimensional features and visualize the results.
Figure 3 shows the visualization of the original features encoded by embedding and the features extracted by Attenphos. It is clear that the features encoded by embedding are visualized in a cluttered manner. However, the extracted features of Attenphos can easily separate phosphorylated and unphosphorylated samples, indicating that our method can predict the generalized phosphorylated site with high accuracy.
3. Discussion
We propose a new deep learning method named Attenphos. It can predict potential general phosphorylation sites. And experiments comparing our model with existing methods show that our method has better overall performance compared to other existing methods. The protein sequences are centered on the prediction sites, intercepting short sequences with different window sizes. Short sequences are encoded and input into three modules with the same structure, in which the output features of the self-attention block are transformed and encoded. Then, when convolution is used, effective features can be better extracted. Finally, the three parts are combined into a combined feature to fully improve the generalization ability of the enhanced model and the prediction of the model accuracy. And self-attention can effectively solve the problem of long-distance dependence on protein phosphorylation.
Although our method has achieved better performance, there are still some shortcomings for improvement. (1) In future work, we can improve the framework using some deep learning modules that work well for classification, such as BiGRU [
20], which can be explored in the next work. (2) We could attempt to include information other than the protein sequence. For example, protein structure information can provide information about the environment of amino acid residues, such as residue accessibility, local secondary structure, inter-residue distance, etc. PPI [
21] information can provide association information in protein interaction networks, such as the tendency of proteins in the same signaling pathway to share phosphorylation sites. So, we could try to combine protein sequence, structure, and PPI information and use deep learning models to perform prediction, which can be further explored in future work. (3) Due to the small kinase-specific phosphorylation dataset available and the small number of samples, our model does not have good prediction performance on small samples, and more advanced model architecture is needed for kinase phosphorylation prediction, which will be the goal of our next work.
In summary, our deep learning method Attenphos shows superior performance in protein general phosphorylation site prediction with high accuracy and stability. The method is based on an attention mechanism which works by filtering out the most predictive features and optimal model parameters for prediction. Our method can provide a useful tool not only in basic research but also in biomedical and drug discovery applications, helping to accelerate the identification and study of phosphorylation sites. This study provides a strong reference for future bioinformatics and proteomics studies and offers new ideas for understanding the mechanisms of protein phosphorylation regulation.
5. Conclusions
In this study, we propose a new framework, Attenphos, based on deep learning to accurately predict general phosphorylation sites using protein sequence information. Attenphos has good robustness and generalization ability. Unlike previous deep learning prediction methods, Attenphos consists of self-attention and CNN modules that use three modules with the same structure and different window sizes to capture important sequence representations of protein phosphorylation sites. On the balanced dataset, Attenphos achieved AUC values of 0.788, 0.691, and 0.625 at the S, T, and Y sites, respectively. On the unbalanced dataset for mouse species, AUC values of 0.831, 0.775, and 0.658 and MCC values of 0.511, 0.416, and 0.262 were achieved at the S, T, and Y sites, respectively. It was demonstrated that Attenphos achieved better results on both balanced and unbalanced datasets. The experiment proves that our model outperforms other existing methods and deep learning models in all aspects. Also, we experimentally proved the importance of the Encoding conversion block. Adding the Encoding conversion block can significantly improve the performance of the model. We chose a commonly used downscaling visualization method, t-SNE, and the features extracted by our method, Attenphos, can easily separate phosphorylated sites from unphosphorylated sites compared to the features encoded by embedding.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}