
Red Blood Cell Contribution to Thrombosis in Polycythemia Vera and Essential Thrombocythemia


Abstract
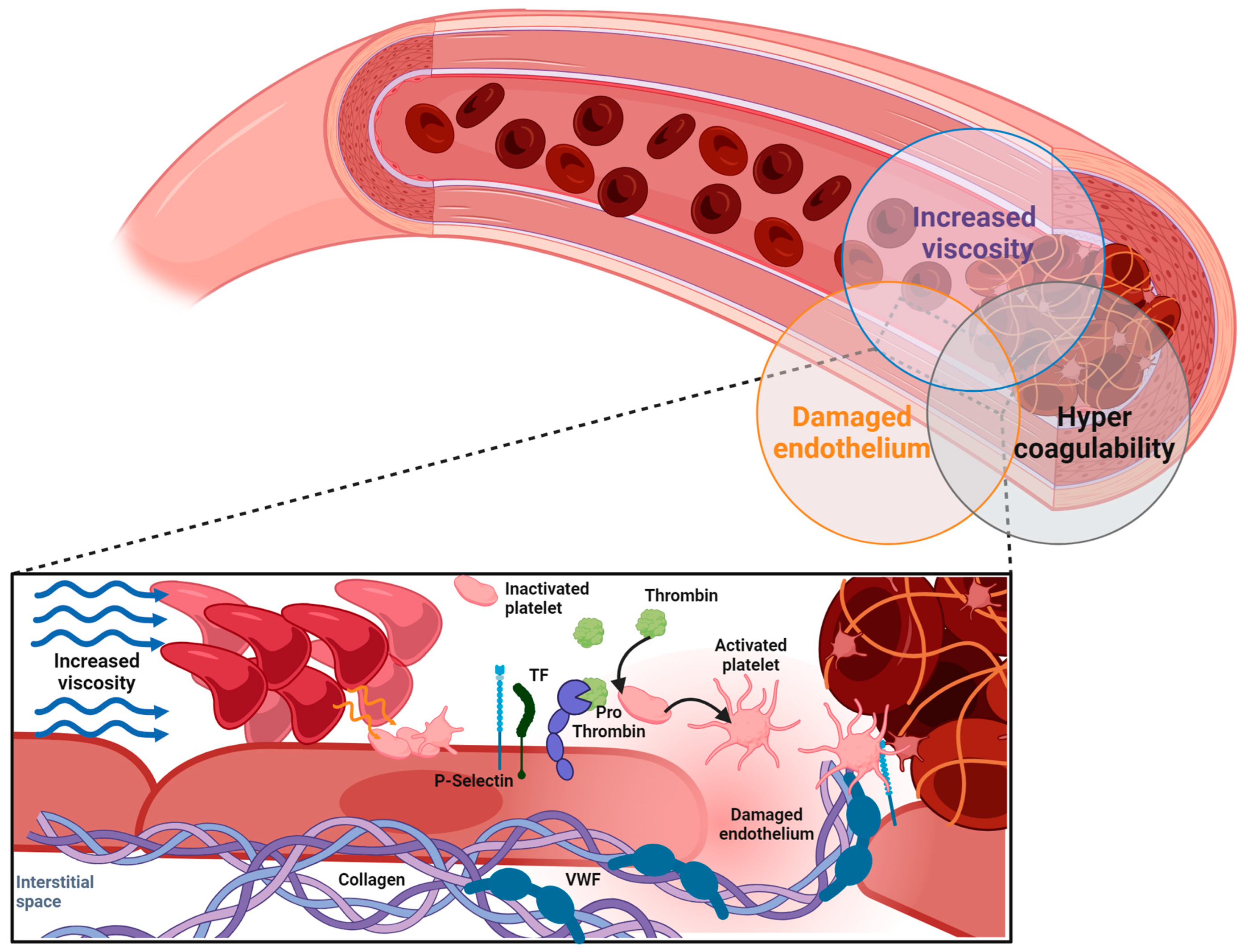
1. Introduction
2. Quantitative and Qualitative Features of PV and ET RBCs in Relation to Thrombosis
2.1. Elevated Hematocrit and Rheological Parameters
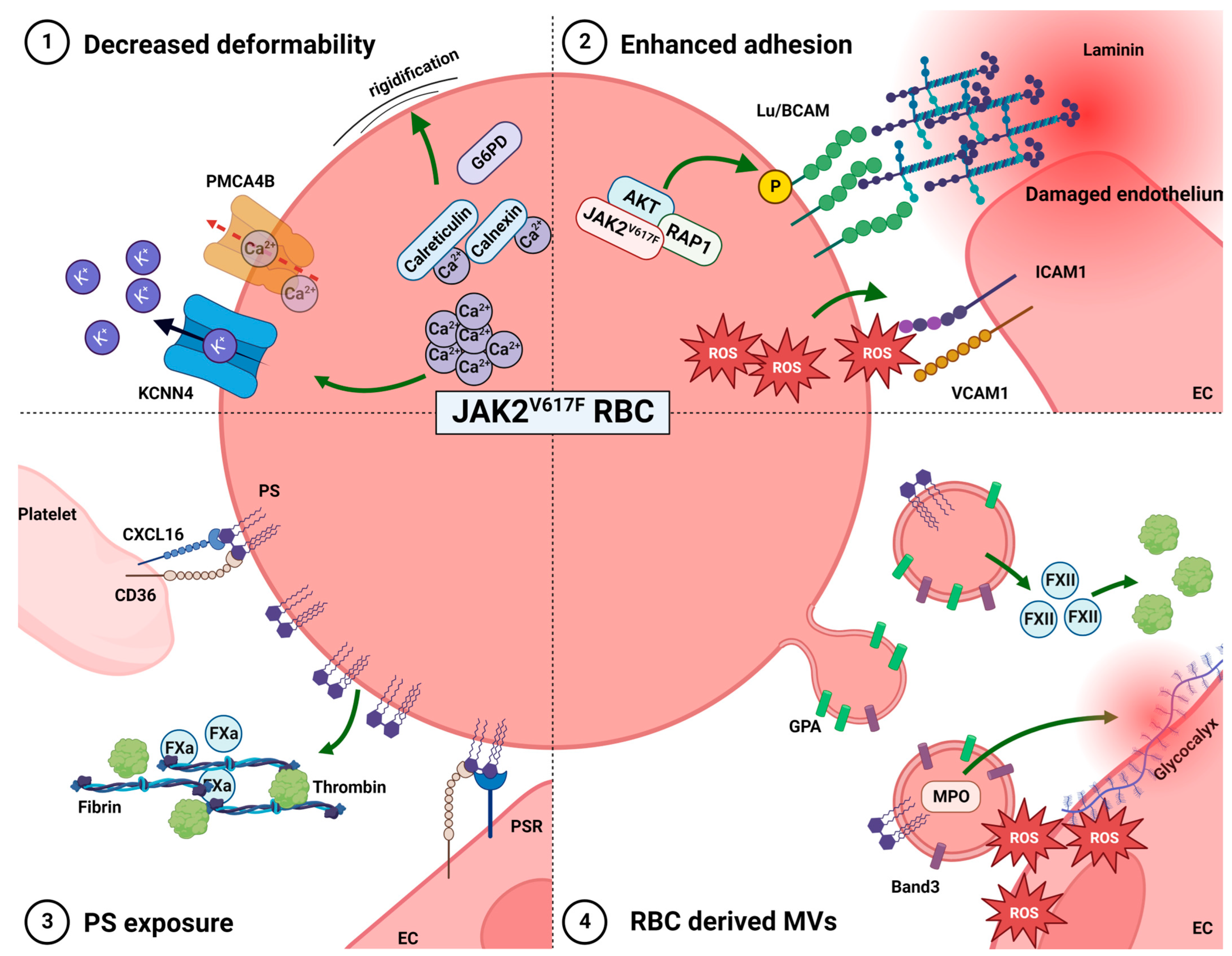
2.2. Altered Deformability
2.3. Enhanced RBC Adhesion to the Vascular Endothelium
2.4. Phosphatidylserine Exposure
2.5. RBC-Derived Microvesicles
2.6. Clinical Relevance of RBC Properties in Thrombotic Risk
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef]
- Elliott, M.A. Chronic neutrophilic leukemia: A contemporary review. Curr. Hematol. Rep. 2004, 3, 210–217. [Google Scholar]
- Marchioli, R.; Finazzi, G.; Landolfi, R.; Kutti, J.; Gisslinger, H.; Patrono, C.; Marilus, R.; Villegas, A.; Tognoni, G.; Barbui, T. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J. Clin. Oncol. 2005, 23, 2224–2232. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. New and treatment-relevant risk stratification for thrombosis in essential thrombocythemia and polycythemia vera. Am. J. Hematol. 2015, 90, 683–685. [Google Scholar] [CrossRef]
- Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; Vannucchi, A.M.; Antonioli, E.; et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: An international study of 891 patients. Blood 2011, 117, 5857–5859. [Google Scholar] [CrossRef] [PubMed]
- Griesshammer, M.; Bangerter, M.; Sauer, T.; Wennauer, R.; Bergmann, L.; Heimpel, H. Aetiology and clinical significance of thrombocytosis: Analysis of 732 patients with an elevated platelet count. J. Intern. Med. 1999, 245, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; de Nully Brown, P.; Lund, B.V.; Nielsen, O.J.; Hasselbalch, H.C. Increased platelet activation and abnormal membrane glycoprotein content and redistribution in myeloproliferative disorders. Br. J. Haematol. 2000, 110, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Royer, Y.; Staerk, J.; Costuleanu, M.; Courtoy, P.J.; Constantinescu, S.N. Janus kinases affect thrombopoietin receptor cell surface localization and stability. J. Biol. Chem. 2005, 280, 27251–27261. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Tanaka, T.; Ohnishi, H.; Kitanaka, A.; Okutani, Y.; Taminato, T.; Ishida, T.; Kamano, H. Constitutively activated phosphatidylinositol 3-kinase primes platelets from patients with chronic myelogenous leukemia for thrombopoietin-induced aggregation. Leukemia 2004, 18, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Landolfi, R.; Di Gennaro, L.; Barbui, T.; De Stefano, V.; Finazzi, G.; Marfisi, R.; Tognoni, G.; Marchioli, R.; European Collaboration on Low-Dose Aspirin in Polycythemia Vera. Leukocytosis as a major thrombotic risk factor in patients with polycythemia vera. Blood 2007, 109, 2446–2452. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Brambilla, M.; Camera, M.; Carbone, A.; Tremoli, E.; Donati, M.B.; de Gaetano, G.; Cerletti, C. Human polymorphonuclear leukocytes produce and express functional tissue factor upon stimulation. J. Thromb. Haemost. 2006, 4, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M.; Evangelista, V.; Vignoli, A.; Licini, M.; Balicco, M.; Manarini, S.; Finazzi, G.; Cerletti, C.; Barbui, T. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood 2000, 96, 4261–4266. [Google Scholar] [CrossRef]
- Sozer, S.; Fiel, M.I.; Schiano, T.; Xu, M.; Mascarenhas, J.; Hoffman, R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 2009, 113, 5246–5249. [Google Scholar] [CrossRef]
- Guadall, A.; Lesteven, E.; Letort, G.; Awan Toor, S.; Delord, M.; Pognant, D.; Brusson, M.; Verger, E.; Maslah, N.; Giraudier, S.; et al. Endothelial Cells Harbouring the JAK2V617F Mutation Display Pro-Adherent and Pro-Thrombotic Features. Thromb. Haemost. 2018, 118, 1586–1599. [Google Scholar] [CrossRef]
- Guy, A.; Gourdou-Latyszenok, V.; Le Lay, N.; Peghaire, C.; Kilani, B.; Dias, J.V.; Duplaa, C.; Renault, M.A.; Denis, C.; Villeval, J.L.; et al. Vascular endothelial cell expression of JAK2(V617F) is sufficient to promote a pro-thrombotic state due to increased P-selectin expression. Haematologica 2019, 104, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, J.R.; Wolberg, A.S. Red blood cells in thrombosis. Blood 2017, 130, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Red blood cells: The forgotten player in hemostasis and thrombosis. J. Thromb. Haemost. 2019, 17, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.E., Jr.; Merrill, E.W. Influence of flow properties of blood upon viscosity-hematocrit relationships. J. Clin. Investig. 1962, 41, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Walton, B.L.; Lehmann, M.; Skorczewski, T.; Holle, L.A.; Beckman, J.D.; Cribb, J.A.; Mooberry, M.J.; Wufsus, A.R.; Cooley, B.C.; Homeister, J.W.; et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood 2017, 129, 2537–2546. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Baker, R.; Lopez, J.A.; Spencer, S. Myeloproliferative disorders and the hyperviscosity syndrome. Hematol. Oncol. Clin. N. Am. 2010, 24, 585–602. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.C.; Wetherley-Mein, G. Vascular occlusive episodes and venous haematocrit in primary proliferative polycythaemia. Lancet 1978, 2, 1219–1222. [Google Scholar] [CrossRef]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular events and intensity of treatment in polycythemia vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef]
- Santisakultarm, T.P.; Paduano, C.Q.; Stokol, T.; Southard, T.L.; Nishimura, N.; Skoda, R.C.; Olbricht, W.L.; Schafer, A.I.; Silver, R.T.; Schaffer, C.B. Stalled cerebral capillary blood flow in mouse models of essential thrombocythemia and polycythemia vera revealed by in vivo two-photon imaging. J. Thromb. Haemost. 2014, 12, 2120–2130. [Google Scholar] [CrossRef]
- Crodel, C.C.; Jentsch-Ullrich, K.; Reiser, M.; Jacobasch, L.; Sauer, A.; Tesch, H.; Ulshofer, T.; Wunschel, R.; Palandri, F.; Heidel, F.H. Cytoreductive treatment in real life: A chart review analysis on 1440 patients with polycythemia vera. J. Cancer Res. Clin. Oncol. 2022, 148, 2693–2705. [Google Scholar] [CrossRef]
- Lo Presti, R.; Caracciolo, C.; Montana, M.; Barone, R.; Catania, A.; Caimi, G. Erythrocyte deformability evaluated by laser diffractometry in polycythemia vera. Clin. Hemorheol. Microcirc. 2012, 50, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, Z.; Dybowicz, A.J.; Marchewka, A.; Teleglow, A.; Skotnicki, A.; Zdunczyk, A.; Aleksander, P.; Filar-Mierzwa, K. Elongation index of erythrocytes, study of activity of chosen erythrocyte enzymes, and the levels of glutathione, malonyldialdehyde in polycythemia vera (PV). Clin. Hemorheol. Microcirc. 2011, 47, 169–176. [Google Scholar] [CrossRef]
- Kuznetsova, P.I.; Raskurazhev, A.A.; Shabalina, A.A.; Melikhyan, A.L.; Subortseva, I.N.; Tanashyan, M.M. Red Blood Cell Morphodynamics in Patients with Polycythemia Vera and Stroke. Int. J. Mol. Sci. 2022, 23, 2247. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Makhro, A.; Wang, J.; Lipp, P.; Kaestner, L. Calcium in red blood cells-a perilous balance. Int. J. Mol. Sci. 2013, 14, 9848–9872. [Google Scholar] [CrossRef] [PubMed]
- Buks, R.; Dagher, T.; Rotordam, M.G.; Monedero Alonso, D.; Cochet, S.; Gautier, E.F.; Chafey, P.; Cassinat, B.; Kiladjian, J.J.; Becker, N.; et al. Altered Ca2+ Homeostasis in Red Blood Cells of Polycythemia Vera Patients Following Disturbed Organelle Sorting during Terminal Erythropoiesis. Cells 2021, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Brusson, M.; Cochet, S.; Leduc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; Le Van Kim, C.; Cassinat, B.; Kiladjian, J.J.; et al. Enhanced calreticulin expression in red cells of polycythemia vera patients harboring the JAK2(V617F) mutation. Haematologica 2017, 102, e241–e244. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P. Adhesion of sickle red cells to endothelium: Myths and future directions. Transfus. Clin. Biol. 2008, 15, 14–18. [Google Scholar] [CrossRef]
- Embury, S.H.; Matsui, N.M.; Ramanujam, S.; Mayadas, T.N.; Noguchi, C.T.; Diwan, B.A.; Mohandas, N.; Cheung, A.T. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood 2004, 104, 3378–3385. [Google Scholar] [CrossRef]
- Ballas, S.K. Sickle cell anemia with few painful crises is characterized by decreased red cell deformability and increased number of dense cells. Am. J. Hematol. 1991, 36, 122–130. [Google Scholar] [CrossRef]
- Lizarralde-Iragorri, M.A.; Lefevre, S.D.; Cochet, S.; El Hoss, S.; Brousse, V.; Filipe, A.; Dussiot, M.; Azouzi, S.; Le Van Kim, C.; Rodrigues-Lima, F.; et al. Oxidative stress activates red cell adhesion to laminin in sickle cell disease. Haematologica 2021, 106, 2478–2488. [Google Scholar] [CrossRef]
- Udani, M.; Zen, Q.; Cottman, M.; Leonard, N.; Jefferson, S.; Daymont, C.; Truskey, G.; Telen, M.J. Basal cell adhesion molecule/lutheran protein. The receptor critical for sickle cell adhesion to laminin. J. Clin. Investig. 1998, 101, 2550–2558. [Google Scholar] [CrossRef] [PubMed]
- El Nemer, W.; Gane, P.; Colin, Y.; Bony, V.; Rahuel, C.; Galacteros, F.; Cartron, J.P.; Le Van Kim, C. The Lutheran blood group glycoproteins, the erythroid receptors for laminin, are adhesion molecules. J. Biol. Chem. 1998, 273, 16686–16693. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, E.; Rahuel, C.; Wautier, M.P.; El Nemer, W.; Gane, P.; Wautier, J.L.; Cartron, J.P.; Colin, Y.; Le Van Kim, C. Protein kinase A-dependent phosphorylation of Lutheran/basal cell adhesion molecule glycoprotein regulates cell adhesion to laminin α5. J. Biol. Chem. 2005, 280, 30055–30062. [Google Scholar] [CrossRef] [PubMed]
- Wautier, M.P.; El Nemer, W.; Gane, P.; Rain, J.D.; Cartron, J.P.; Colin, Y.; Le Van Kim, C.; Wautier, J.L. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin α5 chain and Lu/BCAM. Blood 2007, 110, 894–901. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, M.; Cambot, M.; Wautier, M.P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-κB activation in endothelial cells. Biochim. Biophys. Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef]
- Brusson, M.; De Grandis, M.; Cochet, S.; Bigot, S.; Marin, M.; Leduc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; et al. Impact of hydroxycarbamide and interferon-α on red cell adhesion and membrane protein expression in polycythemia vera. Haematologica 2018, 103, 972–981. [Google Scholar] [CrossRef]
- Nguyen, D.B.; Wagner-Britz, L.; Maia, S.; Steffen, P.; Wagner, C.; Kaestner, L.; Bernhardt, I. Regulation of phosphatidylserine exposure in red blood cells. Cell Physiol. Biochem. 2011, 28, 847–856. [Google Scholar] [CrossRef]
- Peyrou, V.; Lormeau, J.C.; Herault, J.P.; Gaich, C.; Pfliegger, A.M.; Herbert, J.M. Contribution of erythrocytes to thrombin generation in whole blood. Thromb. Haemost. 1999, 81, 400–406. [Google Scholar] [CrossRef]
- Klatt, C.; Kruger, I.; Zey, S.; Krott, K.J.; Spelleken, M.; Gowert, N.S.; Oberhuber, A.; Pfaff, L.; Luckstadt, W.; Jurk, K.; et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J. Clin. Investig. 2018, 128, 3906–3925. [Google Scholar] [CrossRef]
- Tan, X.; Shi, J.; Fu, Y.; Gao, C.; Yang, X.; Li, J.; Wang, W.; Hou, J.; Li, H.; Zhou, J. Role of erythrocytes and platelets in the hypercoagulable status in polycythemia vera through phosphatidylserine exposure and microparticle generation. Thromb. Haemost. 2013, 109, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Whelihan, M.F.; Zachary, V.; Orfeo, T.; Mann, K.G. Prothrombin activation in blood coagulation: The erythrocyte contribution to thrombin generation. Blood 2012, 120, 3837–3845. [Google Scholar] [CrossRef] [PubMed]
- Wali, R.K.; Jaffe, S.; Kumar, D.; Kalra, V.K. Alterations in organization of phospholipids in erythrocytes as factor in adherence to endothelial cells in diabetes mellitus. Diabetes 1988, 37, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, R.A.; McEvoy, L.; Williamson, P. Membrane phospholipid asymmetry and the adherence of loaded red blood cells. Bibl. Haematol. 1985, 51, 150–156. [Google Scholar] [CrossRef]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef]
- Setty, B.N.; Kulkarni, S.; Stuart, M.J. Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood 2002, 99, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Towhid, S.T.; Schmid, E.; Hoffmann, S.M.; Abed, M.; Munzer, P.; Vogel, S.; Neis, F.; Brucker, S.; Gawaz, M.; et al. Dynamic adhesion of eryptotic erythrocytes to immobilized platelets via platelet phosphatidylserine receptors. Am. J. Physiol. Cell Physiol. 2014, 306, C291–C297. [Google Scholar] [CrossRef]
- Tong, D.; Yu, M.; Guo, L.; Li, T.; Li, J.; Novakovic, V.A.; Dong, Z.; Tian, Y.; Kou, J.; Bi, Y.; et al. Phosphatidylserine-exposing blood and endothelial cells contribute to the hypercoagulable state in essential thrombocythemia patients. Ann. Hematol. 2018, 97, 605–616. [Google Scholar] [CrossRef]
- Westerman, M.; Porter, J.B. Red blood cell-derived microparticles: An overview. Blood Cells Mol. Dis. 2016, 59, 134–139. [Google Scholar] [CrossRef]
- Koch, C.G.; Li, L.; Sessler, D.I.; Figueroa, P.; Hoeltge, G.A.; Mihaljevic, T.; Blackstone, E.H. Duration of red-cell storage and complications after cardiac surgery. N. Engl. J. Med. 2008, 358, 1229–1239. [Google Scholar] [CrossRef]
- Kim, Y.; Xia, B.T.; Jung, A.D.; Chang, A.L.; Abplanalp, W.A.; Caldwell, C.C.; Goodman, M.D.; Pritts, T.A. Microparticles from stored red blood cells promote a hypercoagulable state in a murine model of transfusion. Surgery 2018, 163, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Noubouossie, D.F.; Henderson, M.W.; Mooberry, M.; Ilich, A.; Ellsworth, P.; Piegore, M.; Skinner, S.C.; Pawlinski, R.; Welsby, I.; Renne, T.; et al. Red blood cell microvesicles activate the contact system, leading to factor IX activation via 2 independent pathways. Blood 2020, 135, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meijden, P.E.; Van Schilfgaarde, M.; Van Oerle, R.; Renne, T.; Ten Cate, H.; Spronk, H.M. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Zecher, D.; Cumpelik, A.; Schifferli, J.A. Erythrocyte-derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 313–320. [Google Scholar] [CrossRef]
- Charpentier, A.; Lebreton, A.; Rauch, A.; Bauters, A.; Trillot, N.; Nibourel, O.; Tintillier, V.; Wemeau, M.; Demory, J.L.; Preudhomme, C.; et al. Microparticle phenotypes are associated with driver mutations and distinct thrombotic risks in essential thrombocythemia. Haematologica 2016, 101, e365–e368. [Google Scholar] [CrossRef]
- Zhang, W.; Qi, J.; Zhao, S.; Shen, W.; Dai, L.; Han, W.; Huang, M.; Wang, Z.; Ruan, C.; Wu, D.; et al. Clinical significance of circulating microparticles in Ph− myeloproliferative neoplasms. Oncol. Lett. 2017, 14, 2531–2536. [Google Scholar] [CrossRef]
- Poisson, J.; Tanguy, M.; Davy, H.; Camara, F.; El Mdawar, M.B.; Kheloufi, M.; Dagher, T.; Devue, C.; Lasselin, J.; Plessier, A.; et al. Erythrocyte-derived microvesicles induce arterial spasms in JAK2V617F myeloproliferative neoplasm. J. Clin. Investig. 2020, 130, 2630–2643. [Google Scholar] [CrossRef]
- Hartman, C.L.; Ford, D.A. MPO (Myeloperoxidase) Caused Endothelial Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1676–1677. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Y.; Xu, S.; Yin, Y.; Ageno, W.; De Stefano, V.; Zhao, Q.; Qi, X. Clinical significance of neutrophil extracellular traps biomarkers in thrombosis. Thromb. J. 2022, 20, 63. [Google Scholar] [CrossRef]
- Sharma, S.; Hofbauer, T.M.; Ondracek, A.S.; Chausheva, S.; Alimohammadi, A.; Artner, T.; Panzenboeck, A.; Rinderer, J.; Shafran, I.; Mangold, A.; et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood 2021, 137, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.A.; Fuchs, T.A.; Jackson, T.O.; Kremer Hovinga, J.A.; Lammle, B.; Henke, P.K.; Myers, D.D., Jr.; Wagner, D.D.; Wakefield, T.W.; Michigan Research Venous Group. Plasma DNA is Elevated in Patients with Deep Vein Thrombosis. J. Vasc. Surg. Venous Lymphat. Disord. 2013, 1, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cavanaugh, L.; Leung, H.; Yan, F.; Ahmadi, Z.; Chong, B.H.; Passam, F. Quantification of NETs-associated markers by flow cytometry and serum assays in patients with thrombosis and sepsis. Int. J. Lab. Hematol. 2018, 40, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Martos, L.; Oto, J.; Fernandez-Pardo, A.; Plana, E.; Solmoirago, M.J.; Cana, F.; Hervas, D.; Bonanad, S.; Ferrando, F.; Espana, F.; et al. Increase of Neutrophil Activation Markers in Venous Thrombosis-Contribution of Circulating Activated Protein C. Int. J. Mol. Sci. 2020, 21, 5651. [Google Scholar] [CrossRef] [PubMed]
- Gkaliagkousi, E.; Nikolaidou, B.; Gavriilaki, E.; Lazaridis, A.; Yiannaki, E.; Anyfanti, P.; Zografou, I.; Markala, D.; Douma, S. Increased erythrocyte- and platelet-derived microvesicles in newly diagnosed type 2 diabetes mellitus. Diabetes Vasc. Dis. Res. 2019, 16, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, B.; Xu, Z.; Zhang, P.; Qin, T.; Qu, S.; Pan, L.; Sun, X.; Shi, Z.; Huang, H.; et al. RBC distribution width predicts thrombosis risk in polycythemia vera. Leukemia 2022, 36, 566–568. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, M.; Cassinat, B.; Kiladjian, J.J.; Chomienne, C.; El Nemer, W. Lu/BCAM-mediated cell adhesion as biological marker of JAK2V617F activity in erythrocytes of polycythemia vera patients. Am. J. Hematol. 2015, 90, E137–E138. [Google Scholar] [CrossRef] [PubMed]
- Krecak, I.; Krecak, F.; Gveric-Krecak, V. High red blood cell distribution width might predict thrombosis in essential thrombocythemia and polycythemia vera. Blood Cells Mol. Dis. 2020, 80, 102368. [Google Scholar] [CrossRef]
- Verstovsek, S.; Krecak, I.; Heidel, F.H.; De Stefano, V.; Bryan, K.; Zuurman, M.W.; Zaiac, M.; Morelli, M.; Smyth, A.; Redondo, S.; et al. Identifying Patients with Polycythemia Vera at Risk of Thrombosis after Hydroxyurea Initiation: The Polycythemia Vera-Advanced Integrated Models (PV-AIM) Project. Biomedicines 2023, 11, 1925. [Google Scholar] [CrossRef]
- Patel, K.V.; Mohanty, J.G.; Kanapuru, B.; Hesdorffer, C.; Ershler, W.B.; Rifkind, J.M. Association of the red cell distribution width with red blood cell deformability. Adv. Exp. Med. Biol. 2013, 765, 211–216. [Google Scholar] [CrossRef]
- Aarts, P.A.; Banga, J.D.; van Houwelingen, H.C.; Heethaar, R.M.; Sixma, J.J. Increased red blood cell deformability due to isoxsuprine administration decreases platelet adherence in a perfusion chamber: A double-blind cross-over study in patients with intermittent claudication. Blood 1986, 67, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, R.I.; Weisel, J.W. Role of red blood cells in haemostasis and thrombosis. ISBT Sci. Ser. 2017, 12, 176–183. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Parameter | Read Out | Clinical Routine Compatible |
|---|---|---|
| Hematocrit | Blood count | yes |
| RBCs deformability | LoRRca | yes |
| Ca2+ concentration | Fluorescence/Ca2+ chelator/ atomic absorption microscopy | difficult |
| Intracellular proteins | Western Blot/FACS | no |
| Adhesion molecules | FACS | difficult |
| PS exposure | FACS | yes |
| RBC derived MVs | FACS | yes |
| Serum MPO | ELISA | yes |
| Oxidative stress | qPCR/fluorescence | difficult |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grenier, J.M.P.; El Nemer, W.; De Grandis, M. Red Blood Cell Contribution to Thrombosis in Polycythemia Vera and Essential Thrombocythemia. Int. J. Mol. Sci. 2024, 25, 1417. https://doi.org/10.3390/ijms25031417
Grenier JMP, El Nemer W, De Grandis M. Red Blood Cell Contribution to Thrombosis in Polycythemia Vera and Essential Thrombocythemia. International Journal of Molecular Sciences. 2024; 25(3):1417. https://doi.org/10.3390/ijms25031417
Chicago/Turabian StyleGrenier, Julien M. P., Wassim El Nemer, and Maria De Grandis. 2024. "Red Blood Cell Contribution to Thrombosis in Polycythemia Vera and Essential Thrombocythemia" International Journal of Molecular Sciences 25, no. 3: 1417. https://doi.org/10.3390/ijms25031417
APA StyleGrenier, J. M. P., El Nemer, W., & De Grandis, M. (2024). Red Blood Cell Contribution to Thrombosis in Polycythemia Vera and Essential Thrombocythemia. International Journal of Molecular Sciences, 25(3), 1417. https://doi.org/10.3390/ijms25031417