
Altered Protein Kinase A-Dependent Phosphorylation of Cav1.2 in Left Ventricular Myocardium from Cacna1c Haploinsufficient Rat Hearts
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Expression of Cav1.3 and Cavβ2 in Cacna1c+/− Left Ventricular Myocardium
2.2. Phosphorylation of Cav1.2 at S1928
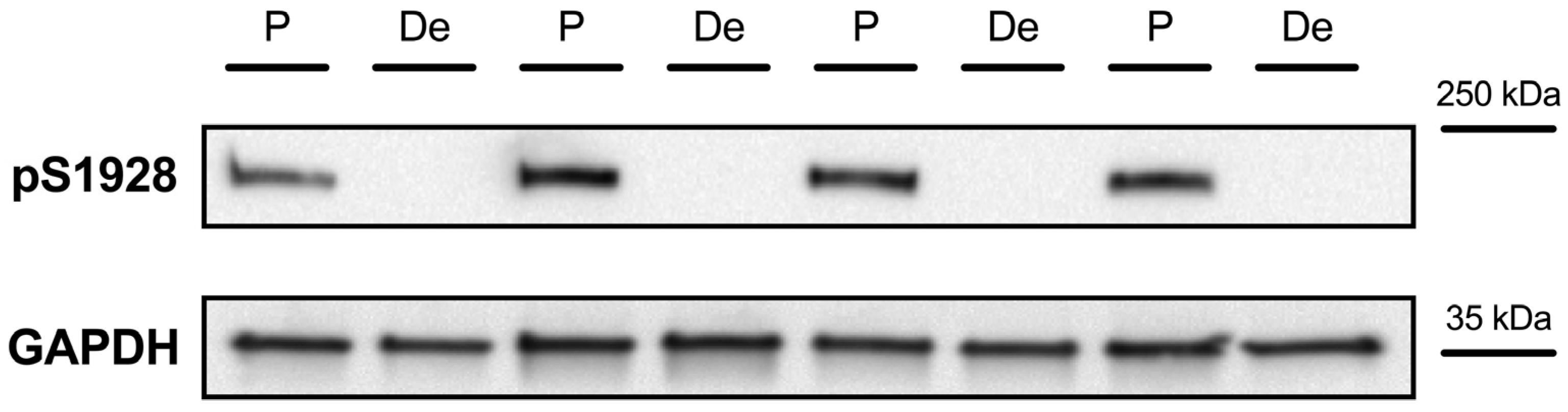
2.2.1. Detection of Cav1.2 Phosphorylation at S1928
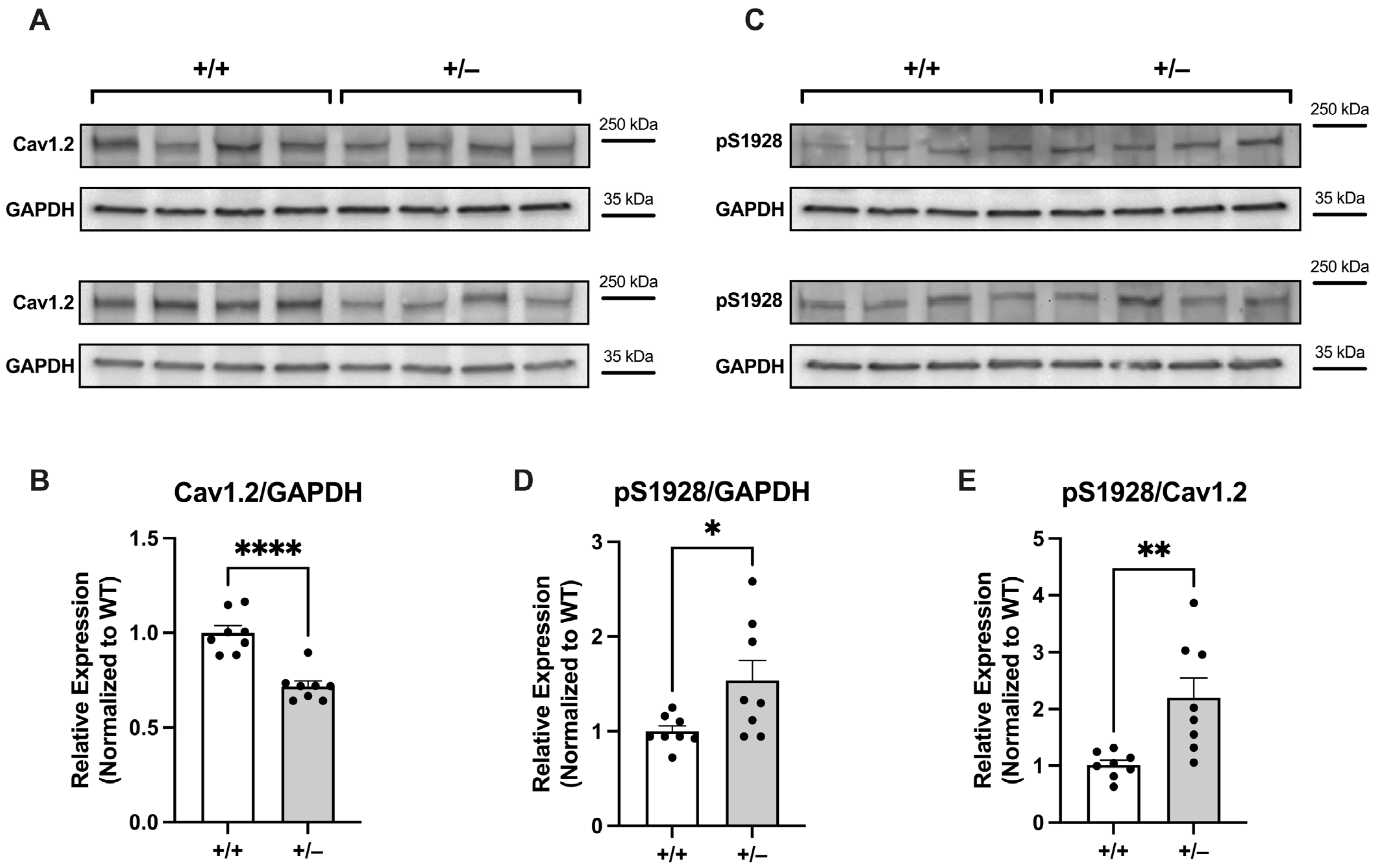
2.2.2. Increased Basal Phosphorylation of Cav1.2 at S1928 in Cacna1c+/− LV Myocardium
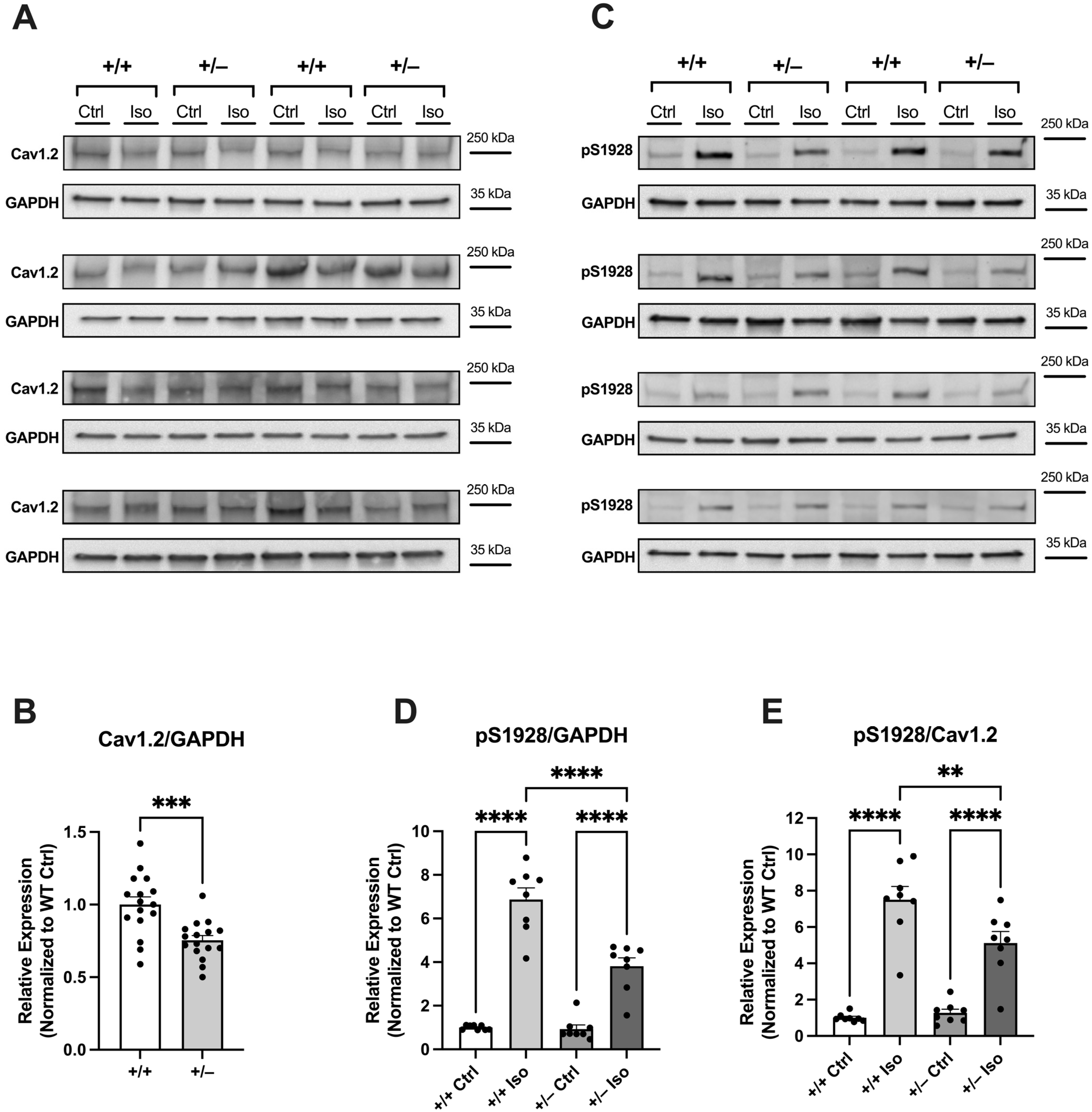
2.2.3. Attenuated Increase in Phosphorylation of Cav1.2 at S1928 in Cacna1c+/− LV Myocardium During Sympathetic Stress
3. Discussion
3.1. Potential Role of Altered Cav1.2 Phosphorylation for Basal Ca2+ Handling in Ventricular Myocardium from Cacna1c+/− Rats
3.2. Potential Role of Altered Cav1.2 Phosphorylation for the Impaired Sympathetic Stress Response in Ventricular Myocardium from Cacna1c+/− Rats
3.3. Altered Phosphorylation of Cav1.2 in Human Cardiac Disease
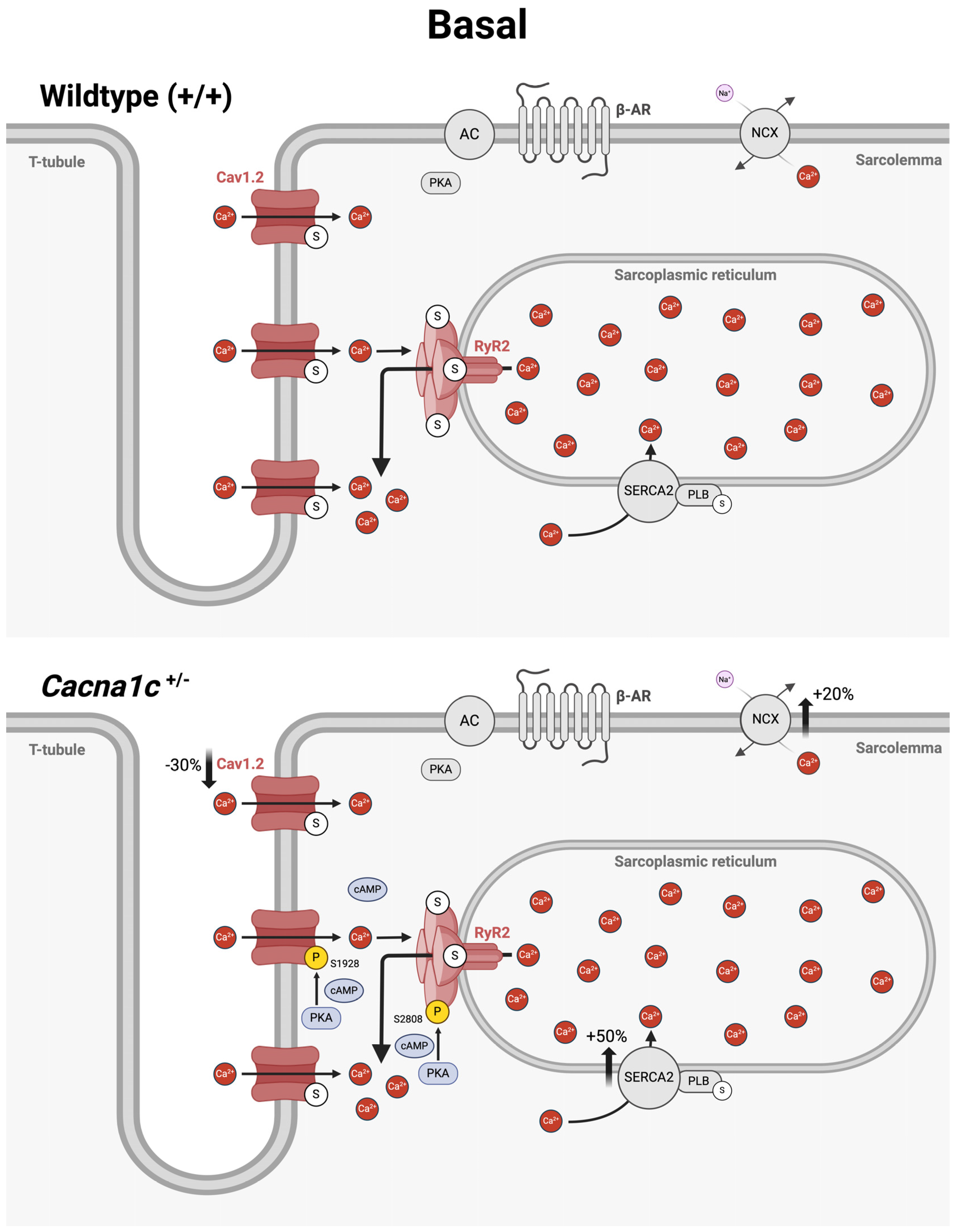
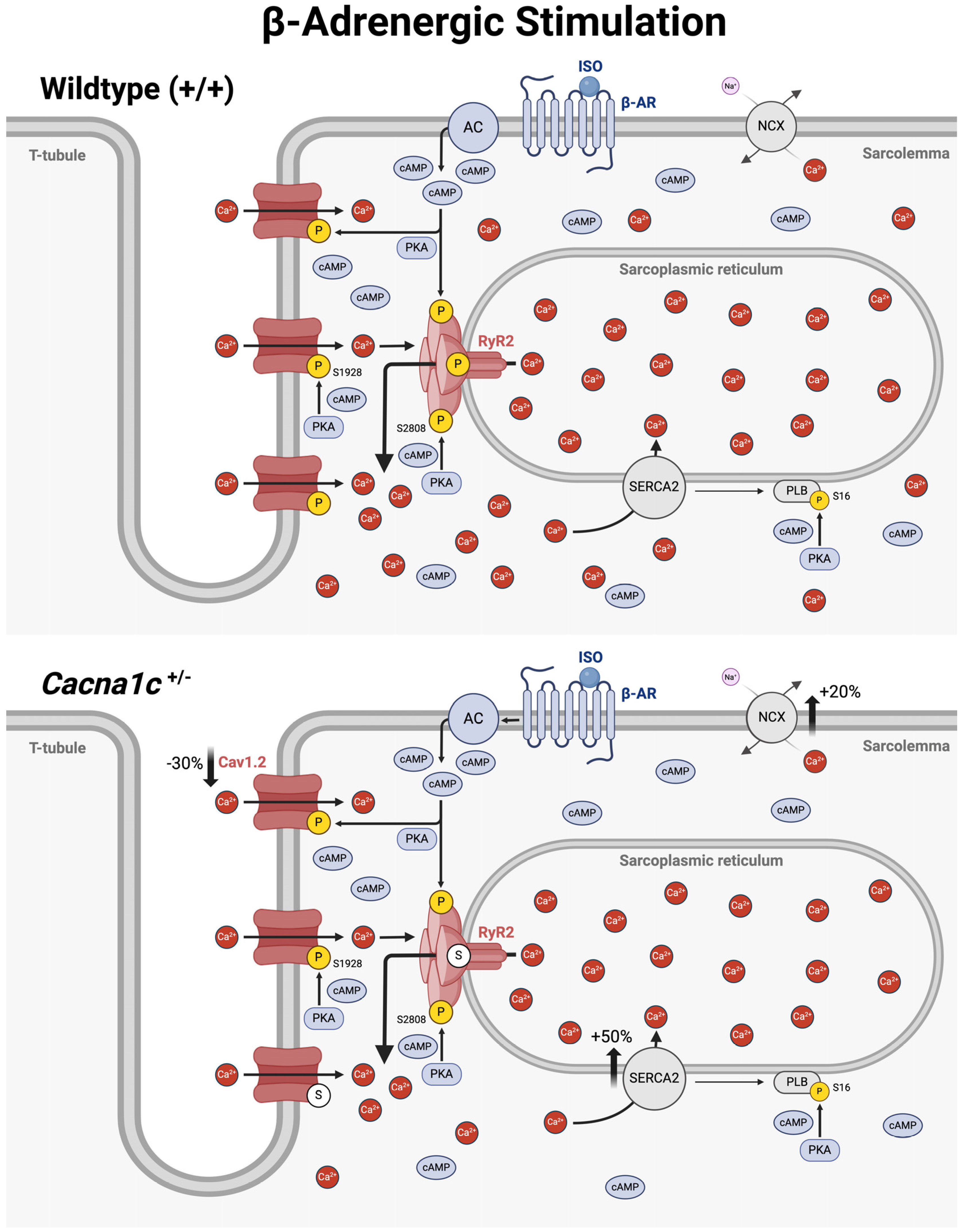
3.4. An Overview on Altered Cellular Ca2+ Handling in Ventricular Myocardium from Cacna1c+/− Rats Under Basal Conditions and During Sympathetic Stress
3.5. Conclusions and Perspectives
4. Materials and Methods
4.1. Animals
4.2. Left Ventricular (LV) Tissue Isolation
4.3. Langendorff Perfusion and Treatment of Whole Hearts
4.4. Immunoblotting (Western Blotting)
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrison, P.J.; Husain, S.M.; Lee, H.; Los Angeles, A.; Colbourne, L.; Mould, A.; Hall, N.A.L.; Haerty, W.; Tunbridge, E.M. CACNA1C (Ca(V)1.2) and other L-type calcium channels in the pathophysiology and treatment of psychiatric disorders: Advances from functional genomics and pharmacoepidemiology. Neuropharmacology 2022, 220, 109262. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef]
- Wemhoner, K.; Friedrich, C.; Stallmeyer, B.; Coffey, A.J.; Grace, A.; Zumhagen, S.; Seebohm, G.; Ortiz-Bonnin, B.; Rinne, S.; Sachse, F.B.; et al. Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J. Mol. Cell Cardiol. 2015, 80, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Kisko, T.M.; Braun, M.D.; Michels, S.; Witt, S.H.; Rietschel, M.; Culmsee, C.; Schwarting, R.K.W.; Wohr, M. Cacna1c haploinsufficiency leads to pro-social 50-kHz ultrasonic communication deficits in rats. Dis. Model. Mech. 2018, 11, dmm034116. [Google Scholar] [CrossRef] [PubMed]
- Redecker, T.M.; Kisko, T.M.; Schwarting, R.K.W.; Wohr, M. Effects of Cacna1c haploinsufficiency on social interaction behavior and 50-kHz ultrasonic vocalizations in adult female rats. Behav. Brain Res. 2019, 367, 35–52. [Google Scholar] [CrossRef]
- Fender, H.; Walter, K.; Kiper, A.K.; Plackic, J.; Kisko, T.M.; Braun, M.D.; Schwarting, R.K.W.; Rohrbach, S.; Wohr, M.; Decher, N.; et al. Calcium Handling Remodeling Underlies Impaired Sympathetic Stress Response in Ventricular Myocardium from Cacna1c Haploinsufficient Rats. Int. J. Mol. Sci. 2023, 24, 9795. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Kushner, J.; Marx, S.O. Adrenergic Regulation of Calcium Channels in the Heart. Annu. Rev. Physiol. 2022, 84, 285–306. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Reuter, H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature 1983, 301, 569–574. [Google Scholar] [CrossRef]
- McDonald, T.F.; Pelzer, S.; Trautwein, W.; Pelzer, D.J. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol. Rev. 1994, 74, 365–507. [Google Scholar] [CrossRef] [PubMed]
- van der Heyden, M.A.; Wijnhoven, T.J.; Opthof, T. Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc. Res. 2005, 65, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.T.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 16574–16579. [Google Scholar] [CrossRef]
- Filippini, L.; Ortner, N.J.; Kaserer, T.; Striessnig, J. Ca(v) 1.3-selective inhibitors of voltage-gated L-type Ca(2+) channels: Fact or (still) fiction? Br. J. Pharmacol. 2023, 180, 1289–1303. [Google Scholar] [CrossRef]
- Whitcomb, V.; Wauson, E.; Christian, D.; Clayton, S.; Giles, J.; Tran, Q.K. Regulation of beta adrenoceptor-mediated myocardial contraction and calcium dynamics by the G protein-coupled estrogen receptor 1. Biochem. Pharmacol. 2020, 171, 113727. [Google Scholar] [CrossRef]
- Rosati, B.; Yan, Q.; Lee, M.S.; Liou, S.R.; Ingalls, B.; Foell, J.; Kamp, T.J.; McKinnon, D. Robust L-type calcium current expression following heterozygous knockout of the Cav1.2 gene in adult mouse heart. J. Physiol. 2011, 589, 3275–3288. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Hammer, K.; Auger-Messier, M.; Bodi, I.; Chen, X.; Zhang, H.; Reiken, S.; Elrod, J.W.; Correll, R.N.; York, A.J.; et al. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J. Clin. Investig. 2012, 122, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J.; Nabauer, M.; Erdmann, E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992, 85, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Mewes, T.; Ravens, U. L-type calcium currents of human myocytes from ventricle of non-failing and failing hearts and from atrium. J. Mol. Cell Cardiol. 1994, 26, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J. Contributions of Ca(2+)-influx via the L-type Ca(2+)-current and Ca(2+)-release from the sarcoplasmic reticulum to [Ca2+]i-transients in human myocytes. Basic. Res. Cardiol. 1997, 92 (Suppl. 1), 105–110. [Google Scholar] [CrossRef] [PubMed]
- Piacentino, V., 3rd; Weber, C.R.; Chen, X.; Weisser-Thomas, J.; Margulies, K.B.; Bers, D.M.; Houser, S.R. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 2003, 92, 651–658. [Google Scholar] [CrossRef]
- Benitah, J.-P.; Alvarez, J.L.; Gómez, A.M. L-type Ca2+ current in ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 48, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Allen, P.D.; Lacro, R.V.; Marks, A.R.; Dennis, A.R.; Schoen, F.J.; Grossman, W.; Marsh, J.D.; Izumo, S. Expression of dihydropyridine receptor (Ca2+ channel) and calsequestrin genes in the myocardium of patients with end-stage heart failure. J. Clin. Investig. 1992, 90, 927–935. [Google Scholar] [CrossRef]
- Ouadid, H.; Albat, B.; Nargeot, J. Calcium currents in diseased human cardiac cells. J. Cardiovasc. Pharmacol. 1995, 25, 282–291. [Google Scholar] [CrossRef]
- Schroder, F.; Handrock, R.; Beuckelmann, D.J.; Hirt, S.; Hullin, R.; Priebe, L.; Schwinger, R.H.; Weil, J.; Herzig, S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 1998, 98, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Piacentino, V., 3rd; Furukawa, S.; Goldman, B.; Margulies, K.B.; Houser, S.R. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 2002, 91, 517–524. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Harris, D.M.; Piacentino, V., 3rd; Berretta, R.M.; Margulies, K.B.; Houser, S.R. Reduced effects of BAY K 8644 on L-type Ca2+ current in failing human cardiac myocytes are related to abnormal adrenergic regulation. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2257–H2267. [Google Scholar] [CrossRef]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Grammatika Pavlidou, N.; Dobrev, S.; Beneke, K.; Reinhardt, F.; Pecha, S.; Jacquet, E.; Abu-Taha, I.H.; Schmidt, C.; Voigt, N.; Kamler, M.; et al. Phosphodiesterase 8 governs cAMP/PKA-dependent reduction of L-type calcium current in human atrial fibrillation: A novel arrhythmogenic mechanism. Eur. Heart J. 2023, 44, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Brette, F.; Orchard, C.H.; Bers, D.M. Na/Ca exchange and Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys. J. 2003, 85, 3388–3396. [Google Scholar] [CrossRef]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Scriven, D.R.L.; Asghari, P.; Schulson, M.N.; Moore, E.D.W. Analysis of Cav1.2 and Ryanodine Receptor Clusters in Rat Ventricular Myocytes. Biophys. J. 2010, 99, 3923–3929. [Google Scholar] [CrossRef]
- Dixon, R.E. Nanoscale Organization, Regulation, and Dynamic Reorganization of Cardiac Calcium Channels. Front. Physiol. 2021, 12, 810408. [Google Scholar] [CrossRef] [PubMed]
- Man, K.N.M.; Bartels, P.; Horne, M.C.; Hell, J.W. Tissue-specific adrenergic regulation of the L-type Ca(2+) channel Ca(V)1.2. Sci. Signal 2020, 13, eabc6438. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Gaburjakova, M.; Gaburjakova, J.; Yang, Y.M.; Rosemblit, N.; Marks, A.R. Phosphorylation-dependent regulation of ryanodine receptors: A novel role for leucine/isoleucine zippers. J. Cell Biol. 2001, 153, 699–708. [Google Scholar] [CrossRef]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Backx, P.H.; Gao, W.D.; Azan-Backx, M.D.; Marban, E. Mechanism of force inhibition by 2,3-butanedione monoxime in rat cardiac muscle: Roles of [Ca2+]i and cross-bridge kinetics. J. Physiol. 1994, 476, 487–500. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Königstein, D.; Fender, H.; Plačkić, J.; Kisko, T.M.; Wöhr, M.; Kockskämper, J. Altered Protein Kinase A-Dependent Phosphorylation of Cav1.2 in Left Ventricular Myocardium from Cacna1c Haploinsufficient Rat Hearts. Int. J. Mol. Sci. 2024, 25, 13713. https://doi.org/10.3390/ijms252413713
Königstein D, Fender H, Plačkić J, Kisko TM, Wöhr M, Kockskämper J. Altered Protein Kinase A-Dependent Phosphorylation of Cav1.2 in Left Ventricular Myocardium from Cacna1c Haploinsufficient Rat Hearts. International Journal of Molecular Sciences. 2024; 25(24):13713. https://doi.org/10.3390/ijms252413713
Chicago/Turabian StyleKönigstein, David, Hauke Fender, Jelena Plačkić, Theresa M. Kisko, Markus Wöhr, and Jens Kockskämper. 2024. "Altered Protein Kinase A-Dependent Phosphorylation of Cav1.2 in Left Ventricular Myocardium from Cacna1c Haploinsufficient Rat Hearts" International Journal of Molecular Sciences 25, no. 24: 13713. https://doi.org/10.3390/ijms252413713
APA StyleKönigstein, D., Fender, H., Plačkić, J., Kisko, T. M., Wöhr, M., & Kockskämper, J. (2024). Altered Protein Kinase A-Dependent Phosphorylation of Cav1.2 in Left Ventricular Myocardium from Cacna1c Haploinsufficient Rat Hearts. International Journal of Molecular Sciences, 25(24), 13713. https://doi.org/10.3390/ijms252413713