

Fixation of Expression Divergences by Natural Selection in Arabidopsis Coding Genes
 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
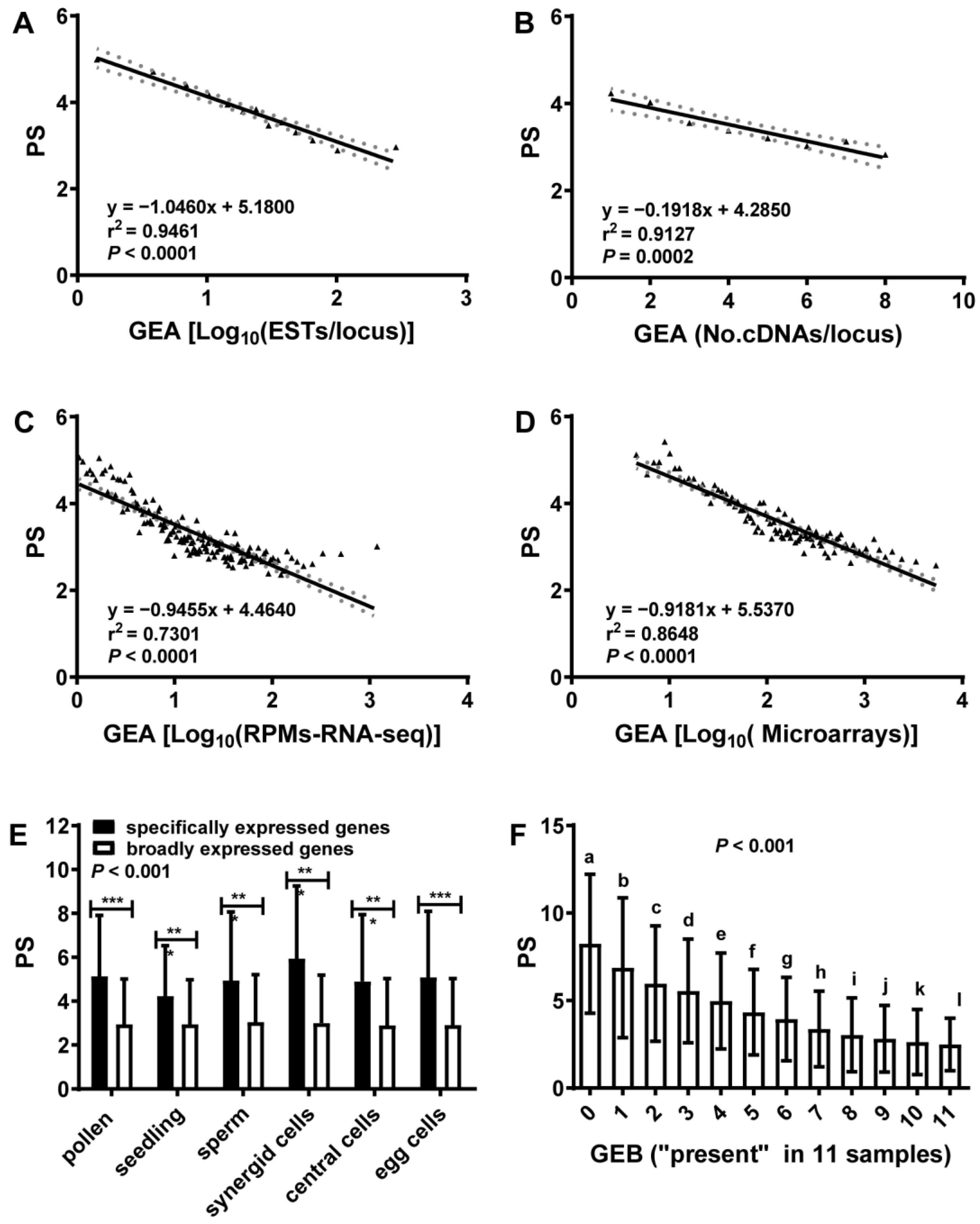
2.1. Gene Expression Abundance (GEA) Was Anti-Correlated with Phylostratum (PS)
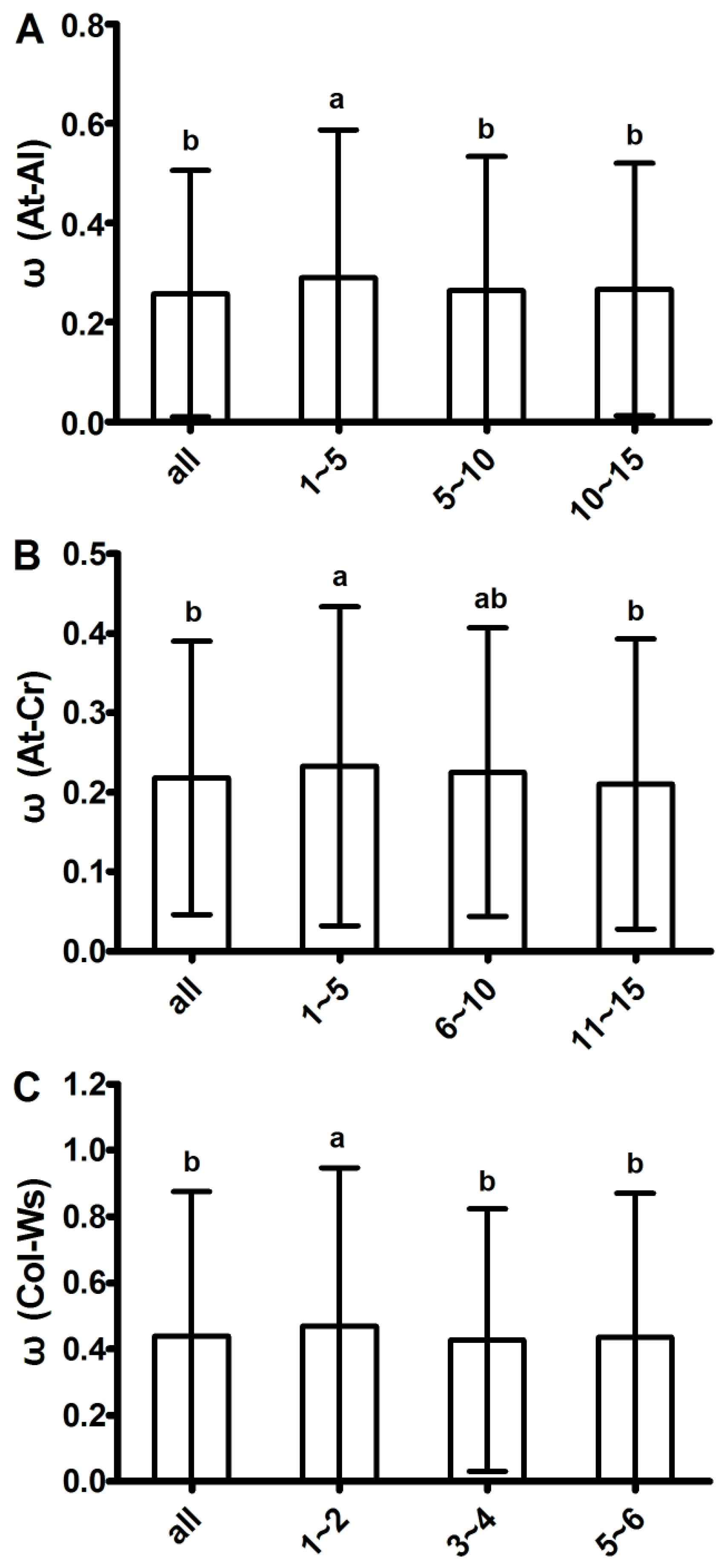
2.2. Evolutionary Analysis and Identification of Loci with Positive Selection (ω > 1) in Arabidopsis
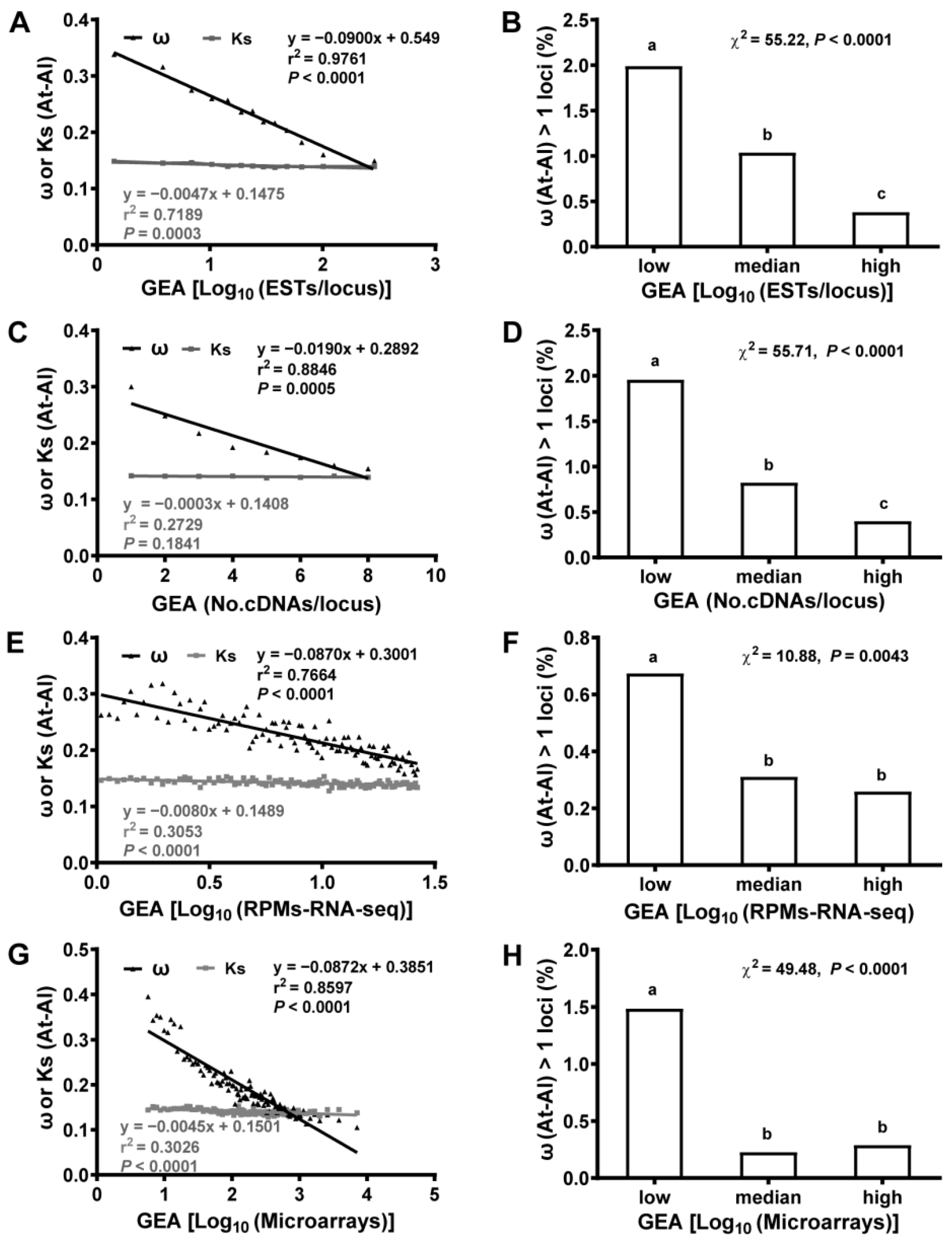
2.3. Gene Expression Abundance (GEA) Was Anti-Correlated with Selective Constraint (ω)
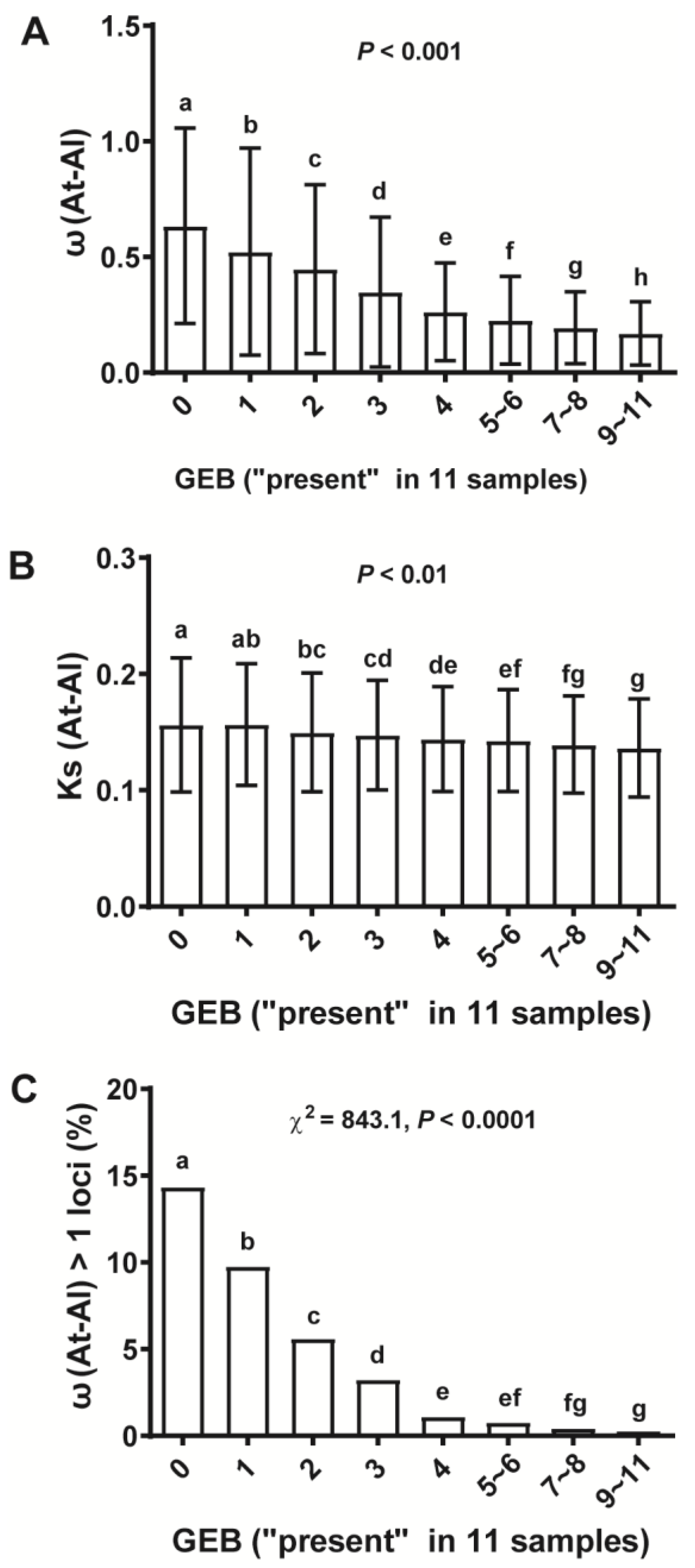
2.4. Gene Expression Breadth (GEB) Was Strongly Anti-Correlated with Selective Constraint (ω)
2.5. Functional Differences Between Gene Age and Gene Expression Patterns Supported by Mutant Analysis in Arabidopsis
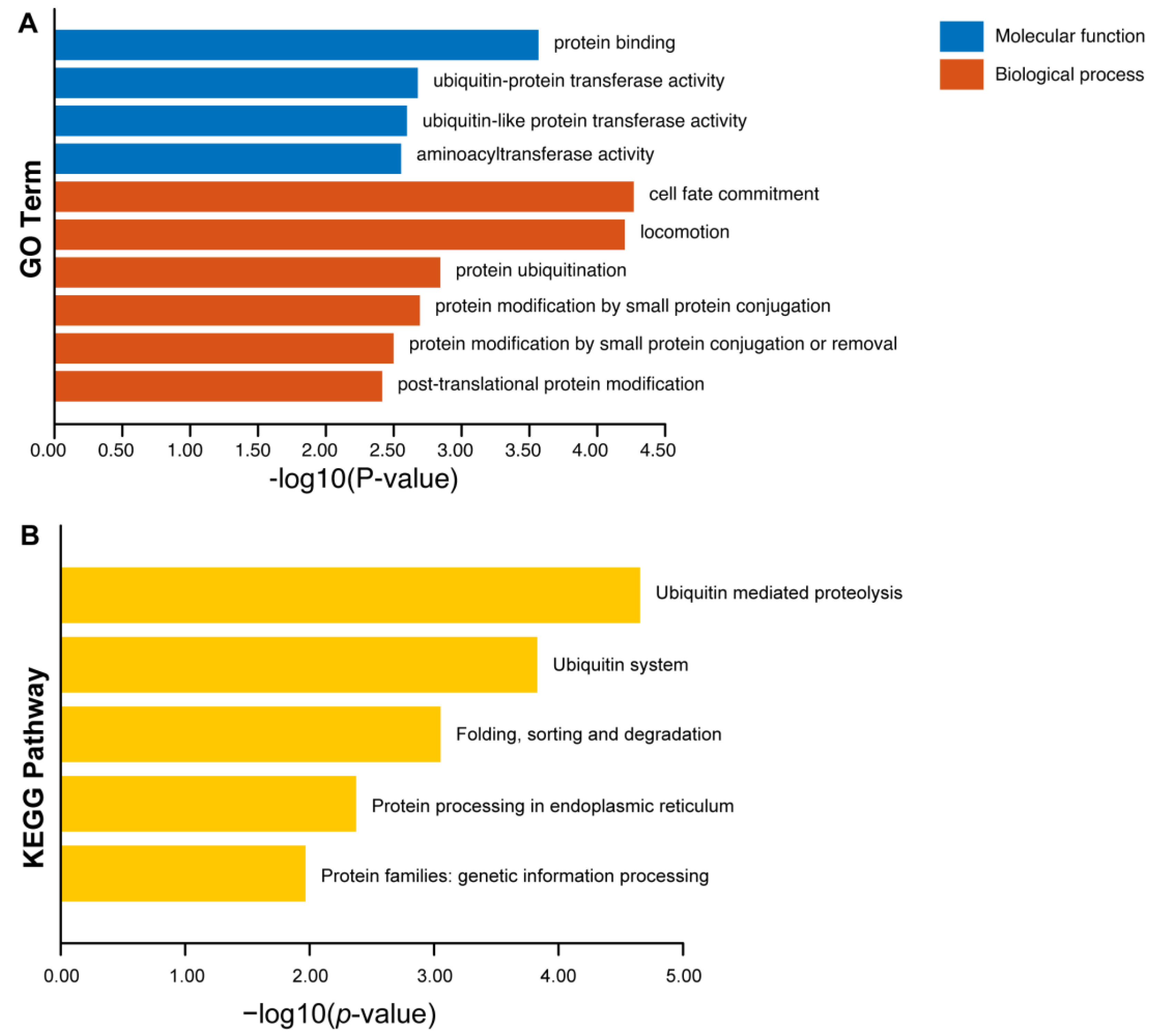
2.6. Functional Enrichments of Putative ω >1 Loci in Arabidopsis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Data Mining
5.2. Calculation of Ka/Ks (ω) Values
5.3. Data Grouping and Statistical Analysis for Gene Expression Abundance and Breadth
5.4. Normal Approximations of Ks
- Scripts for normal.test.r:# Takes a numeric vector x and number of partitions k.# Tests to see if x is reasonably approximately normal.# Generates a vector of same length as x using normal distribution with mean(x) and sd(x)# Bins both vectors k partitions.# Uses chi-sq test to see if counts are similar enough.norm.test = function(x, k){x = na.omit(x)mean.x = mean(x)sd.x = sd(x)norms = rnorm(length(x), mean.x, sd.x)breaks = seq(min(norms), max(norms), l=k+1)breaks [1] = −Infbreaks [2] = Infnormcounts = hist(norms, breaks, plot=FALSE)$countsxcounts = hist(x, breaks, plot=FALSE)$countsreturn(chisq.test(xcounts, normcounts))}atal = read.csv(“./S8.LociwithAt-Al.csv”, header=TRUE, skip=3)katal = atal$Kswatal = atal$X.norm.test(katal, 45) # Figure S1A (should be “reasonably well-approximated”)atcr = read.csv(“./S8.LociwithAt-Cr.csv”, header=TRUE, skip=2)katcr = atcr$Kswatcr = atcr$X.norm.test(katcr, 45) # Figure S1C (should NOT be “reasonably well-approximated”)Scripts for approximated normal distribution (Table S8):>norm.test(katal, 40) # Figure S1A (should be “reasonably well-approximated”)Pearson’s Chi-squared testdata: xcounts and normcountsX-squared = 1257.143, df = 1184, p-value = 0.06853>norm.test(katcr, 40) # Figure S1C (should NOT be “reasonably well-approximated”)Pearson’s Chi-squared testdata: xcounts and normcountsX-squared = 1240, df = 1116, p-value = 0.005428>norm.test(katal, 30) # Figure S1A (should be “reasonably well-approximated”)Pearson’s Chi-squared testdata: xcounts and normcountsX-squared = 720, df = 696, p-value = 0.2566>norm.test(katcr, 30) # Figure S1C (should be “reasonably well-approximated”)Pearson’s Chi-squared testdata: xcounts and normcountsX-squared = 720, df = 696, p-value = 0.2566
5.5. Correlation Grouping of Gene Expression Divergences and Sequence Divergences
- R code (test.12 for Table S12J and test.13 for Table S13J):>test.s12=test_sim(s12$Ks,s12$X.,s12$Log10.RPMs, Ks)$old.corxyzx1.00000000-0.2074137-0.08300841y-0.207413711.0000000-0.23241716z-0.08300841-0.23241721.00000000$new.coravg.rank.xavg.rank.yavg.rank.zavg.rank.x1.0000000-0.25292290.1087751avg.rank.y-0.25292291.0000000-0.3475637avg.rank.z0.1087751-0.34756371.0000000>test.s13=test_sim(s13$X.1,s13$X.2,s13$X.4, Ks)$old.corxyzx1.00000000-0.2115729-0.07317154y-0.211572921.0000000-0.33954236z-0.07317154-0.33954241.00000000$new.coravg.rank.xavg.rank.yavg.rank.zavg.rank.x1.00000000.3940529-0.4705550avg.rank.y0.39405291.0000000-0.9003214avg.rank.z-0.4705550-0.90032141.0000000
5.6. Permutation Tests for Significant Differences in Correlations of ω and GEA Compared to Ks and GEA
- R code (given x= ω, y = Ks and z = GEA) for permutation tests for significant differences in correlations:perm.test = function(x, y, z){if(length(x) != length(y) || length(x) != length(z)){stop(“x, y, and z must be of equal length”)}if (2^(length(x)) <= 100000){allcombs = expand.grid(rep(list(c(TRUE, FALSE)), length(x)))} else {allcombs = t(replicate(100000, sample(c(TRUE, FALSE), length(x), replace=TRUE)))}allcors = apply(allcombs, 1, function(idx){cor(c(x[idx], y[!idx]), z)})alldiffs = allcors-rev(allcors)xcor = cor(x, z)ycor = cor(y, z)mydiff = xcor-ycorhist(alldiffs)abline(v=mydiff)if (mydiff>=0){return(c(mean((xcor-ycor) <= alldiffs))) } else {return(c(mean((xcor-ycor) >= alldiffs)))}wg.kg.perms = mapply(perm.test, x=ω, y=Ks , z=GEA)cor.sig = data.frame(wg.cor, wg.test.p, ifelse(xz.test.p<0.05, “Yes”, “No”),kg.cor, kg.test.p, ifelse(yz.test.p<0.05, “Yes”, “No”),wg.kg.perms, ifelse(xz.yz.perms<0.05, “Yes”, “No”))names(cor.sig) = c(“Cor.ω.GEA”, “p-value”, “Below 0.05?”,“Cor.Ks.GEA”, “p-value”, “Below 0.05?”,“perms.p-value”, “Below 0.05?”)rownames(cor.sig) = 10:21
5.7. GO and KEGG Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Taher, A.; Bohne, A.; Boileau, N.; Ronco, F.; Indermaur, A.; Widmer, L.; Salzburger, W. Gene expression dynamics during rapid organismal diversification in African cichlid fishes. Nat. Ecol. Evol. 2021, 5, 243–250. [Google Scholar] [CrossRef]
- Mantica, F.; Iniguez, L.P.; Marquez, Y.; Permanyer, J.; Torres-Mendez, A.; Cruz, J.; Franch-Marro, X.; Tulenko, F.; Burguera, D.; Bertrand, S.; et al. Evolution of tissue-specific expression of ancestral genes across vertebrates and insects. Nat. Ecol. Evol. 2024, 8, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.S.; Vande Zande, P.; Wittkopp, P.J. Molecular and evolutionary processes generating variation in gene expression. Nat. Rev. Genet. 2021, 22, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Yang, H. The multiple fates of gene duplications: Deletion, hypofunctionalization, subfunctionalization, neofunctionalization, dosage balance constraints, and neutral variation. Plant Cell 2022, 34, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Sandve, S.R.; Rohlfs, R.V.; Hvidsten, T.R. Subfunctionalization versus neofunctionalization after whole-genome duplication. Nat. Genet. 2018, 50, 908–909. [Google Scholar] [CrossRef]
- Braasch, I.; Bobe, J.; Guiguen, Y.; Postlethwait, J.H. Reply to: ‘Subfunctionalization versus neofunctionalization after whole-genome duplication’. Nat. Genet. 2018, 50, 910–911. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csardi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.G.; Ruvinsky, I.; Gilad, Y. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012, 13, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.L.; Alvarez-Ponce, D. Higher Rates of Protein Evolution in the Self-Fertilizing Plant than in the Out-Crossers. Genome Biol. Evol. 2018, 10, 895–900. [Google Scholar] [CrossRef]
- Zhang, D.; Leng, L.; Chen, C.; Huang, J.; Zhang, Y.; Yuan, H.; Ma, C.; Chen, H.; Zhang, Y.E. Dosage sensitivity and exon shuffling shape the landscape of polymorphic duplicates in Drosophila and humans. Nat. Ecol. Evol. 2022, 6, 273–287. [Google Scholar] [CrossRef]
- Shi, T.; Gao, Z.; Chen, J.; Van de Peer, Y. Dosage sensitivity shapes balanced expression and gene longevity of homoeologs after whole-genome duplications in angiosperms. Plant Cell 2024, 36, 4323–4337. [Google Scholar] [CrossRef]
- Liao, B.Y.; Zhang, J. Low rates of expression profile divergence in highly expressed genes and tissue-specific genes during mammalian evolution. Mol. Biol. Evol. 2006, 23, 1119–1128. [Google Scholar] [CrossRef]
- Taylor, D.J.; Chhetri, S.B.; Tassia, M.G.; Biddanda, A.; Yan, S.M.; Wojcik, G.L.; Battle, A.; McCoy, R.C. Sources of gene expression variation in a globally diverse human cohort. Nature 2024, 632, 122–130. [Google Scholar] [CrossRef]
- Barr, K.A.; Rhodes, K.L.; Gilad, Y. The relationship between regulatory changes in cis and trans and the evolution of gene expression in humans and chimpanzees. Genome Biol. 2023, 24, 207. [Google Scholar] [CrossRef]
- Price, P.D.; Palmer Droguett, D.H.; Taylor, J.A.; Kim, D.W.; Place, E.S.; Rogers, T.F.; Mank, J.E.; Cooney, C.R.; Wright, A.E. Detecting signatures of selection on gene expression. Nat. Ecol. Evol. 2022, 6, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Debes, P.V.; Nousiainen, I.; Kahar, S.; Pukk, L.; Gross, R.; Ozerov, M.; Vasemagi, A. The strength and form of natural selection on transcript abundance in the wild. Mol. Ecol. 2021, 30, 2724–2737. [Google Scholar] [CrossRef] [PubMed]
- Reznick, D.N.; Ricklefs, R.E. Darwin’s bridge between microevolution and macroevolution. Nature 2009, 457, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Aboitiz, F. Mechanisms of adaptive evolution. Darwinism and Lamarckism restated. Med. Hypotheses 1992, 38, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Glazko, G.; Mushegian, A. Measuring gene expression divergence: The distance to keep. Biol. Direct 2010, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, S.A.; Kim, J.; White, K.P. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat. Genet. 2003, 33, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Kalinka, A.T.; Varga, K.M.; Gerrard, D.T.; Preibisch, S.; Corcoran, D.L.; Jarrells, J.; Ohler, U.; Bergman, C.M.; Tomancak, P. Gene expression divergence recapitulates the developmental hourglass model. Nature 2010, 468, 811–814. [Google Scholar] [CrossRef]
- Quint, M.; Drost, H.G.; Gabel, A.; Ullrich, K.K.; Bonn, M.; Grosse, I. A transcriptomic hourglass in plant embryogenesis. Nature 2012, 490, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Loso, T.; Tautz, D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 2010, 468, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Bedford, T.; Hartl, D.L. Optimization of gene expression by natural selection. Proc. Natl. Acad. Sci. USA 2009, 106, 1133–1138. [Google Scholar] [CrossRef]
- Subramanian, S.; Kumar, S. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics 2004, 168, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Barkai, N. Evolution of gene sequence and gene expression are not correlated in yeast. Trends Genet. 2008, 24, 109–113. [Google Scholar] [CrossRef]
- Cruickshank, T.; Wade, M.J. Microevolutionary support for a developmental hourglass: Gene expression patterns shape sequence variation and divergence in Drosophila. Evol. Dev. 2008, 10, 583–590. [Google Scholar] [CrossRef]
- Wuest, S.E.; Vijverberg, K.; Schmidt, A.; Weiss, M.; Gheyselinck, J.; Lohr, M.; Wellmer, F.; Rahnenfuhrer, J.; von Mering, C.; Grossniklaus, U. Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr. Biol. 2010, 20, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Ford, M.J.; Cusanovich, D.A.; Mitrano, A.; Pritchard, J.K.; Gilad, Y. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 2013, 342, 1100–1104. [Google Scholar] [CrossRef]
- Khaitovich, P.; Hellmann, I.; Enard, W.; Nowick, K.; Leinweber, M.; Franz, H.; Weiss, G.; Lachmann, M.; Paabo, S. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 2005, 309, 1850–1854. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.J.; Josephs, E.B.; Platts, A.E.; Hazzouri, K.M.; Haudry, A.; Blanchette, M.; Wright, S.I. Evidence for widespread positive and negative selection in coding and conserved noncoding regions of Capsella grandiflora. PLoS Genet. 2014, 10, e1004622. [Google Scholar] [CrossRef]
- Lawrie, D.S.; Messer, P.W.; Hershberg, R.; Petrov, D.A. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 2013, 9, e1003527. [Google Scholar] [CrossRef] [PubMed]
- Meiklejohn, C.D.; Parsch, J.; Ranz, J.M.; Hartl, D.L. Rapid evolution of male-biased gene expression in Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 9894–9899. [Google Scholar] [CrossRef]
- Arendt, D. The evolution of cell types in animals: Emerging principles from molecular studies. Nat. Rev. 2008, 9, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gaut, B.S. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol. Biol. Evol. 2011, 28, 2359–2369. [Google Scholar] [CrossRef]
- Jordan, I.K.; Marino-Ramirez, L.; Koonin, E.V. Evolutionary significance of gene expression divergence. Gene 2005, 345, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.D.; Hoekstra, H.E. Molecular spandrels: Tests of adaptation at the genetic level. Nat. Rev. Genet. 2011, 12, 767–780. [Google Scholar] [CrossRef]
- Nei, M. Selectionism and neutralism in molecular evolution. Mol. Biol. Evol. 2005, 22, 2318–2342. [Google Scholar] [CrossRef]
- Alba, M.M.; Castresana, J. Inverse relationship between evolutionary rate and age of mammalian genes. Mol. Biol. Evol. 2005, 22, 598–606. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, S.; He, F.; Zhu, J.; Hu, S.; Yu, J. How do variable substitution rates influence Ka and Ks calculations? Genom. Proteom. Bioinform. 2009, 7, 116–127. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Park, S.G.; Choi, S.S. Expression breadth and expression abundance behave differently in correlations with evolutionary rates. BMC Evol. Biol. 2010, 10, 241. [Google Scholar] [CrossRef]
- Loraine, A.E.; McCormick, S.; Estrada, A.; Patel, K.; Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol. 2013, 162, 1092–1109. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Uhlenhaut, N.H.; Godard, F.; Demar, M.; Bressan, R.; Weigel, D.; Lohmann, J.U. Dissection of floral induction pathways using global expression analysis. Development 2003, 130, 6001–6012. [Google Scholar] [CrossRef]
- Schmidt, A.; Wuest, S.E.; Vijverberg, K.; Baroux, C.; Kleen, D.; Grossniklaus, U. Transcriptome analysis of the Arabidopsis megaspore mother cell uncovers the importance of RNA helicases for plant germline development. PLoS Biol. 2011, 9, e1001155. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.T.; Pattyn, P.; Bakker, E.G.; Cao, J.; Cheng, J.F.; Clark, R.M.; Fahlgren, N.; Fawcett, J.A.; Grimwood, J.; Gundlach, H.; et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 2011, 43, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Beilstein, M.A.; Nagalingum, N.S.; Clements, M.D.; Manchester, S.R.; Mathews, S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 18724–18728. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Steffen, J.G.; Osborne, E.J.; Toomajian, C. Plant organ evolution revealed by phylotranscriptomics in Arabidopsis thaliana. Sci. Rep. 2017, 7, 7567. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Lv, Y.; Chen, M.; Nikoloski, Z.; Twell, D.; Zhang, D. Young Genes out of the Male: An Insight from Evolutionary Age Analysis of the Pollen Transcriptome. Mol. Plant 2015, 8, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Davison, T.S.; Henz, S.R.; Pape, U.J.; Demar, M.; Vingron, M.; Scholkopf, B.; Weigel, D.; Lohmann, J.U. A gene expression map of Arabidopsis thaliana development. Nat. Genet. 2005, 37, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Hanikenne, M.; Kroymann, J.; Trampczynska, A.; Bernal, M.; Motte, P.; Clemens, S.; Kramer, U. Hard selective sweep and ectopic gene conversion in a gene cluster affording environmental adaptation. PLoS Genet. 2013, 9, e1003707. [Google Scholar] [CrossRef] [PubMed]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105. [Google Scholar] [CrossRef]
- Paape, T.; Bataillon, T.; Zhou, P.; Kono, T.J.Y.; Briskine, R.; Young, N.D.; Tiffin, P. Selection, genome-wide fitness effects and evolutionary rates in the model legume Medicago truncatula. Mol. Ecol. 2013, 22, 3525–3538. [Google Scholar] [CrossRef] [PubMed]
- Drews, G.N.; Koltunow, A.M. The female gametophyte. Arab. Book 2011, 9, e0155. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Song, S.; Li, C.; Zhang, J. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature 2022, 606, 725–731. [Google Scholar] [CrossRef]
- Francisco, M.; Kliebenstein, D.J.; Rodriguez, V.M.; Soengas, P.; Abilleira, R.; Cartea, M.E. Fine mapping identifies NAD-ME1 as a candidate underlying a major locus controlling temporal variation in primary and specialized metabolism in Arabidopsis. Plant J. 2021, 106, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.W.; Juan, C.T.; Wang, C.M.; Jauh, G.Y. Mitochondrial Heat Shock Protein 60s Interact with What’s This Factor 9 to Regulate RNA Splicing of ccmFC and rpl2. Plant Cell Physiol. 2019, 60, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sun, R.; Hicks, G.R.; Raikhel, N.V. Arabidopsis ribosomal proteins control vacuole trafficking and developmental programs through the regulation of lipid metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Guzman, P.; Ecker, J.R. Exploiting the triple response of Arabidopsis to identify ethylene-related mutants. Plant Cell 1990, 2, 513–523. [Google Scholar]
- Li, Z.; Wang, M.; Zhong, Z.; Gallego-Bartolomé, J.; Feng, S.; Jami-Alahmadi, Y.; Wang, X.; Wohlschlegel, J.; Bischof, S.; Long, J.A.; et al. The MOM1 complex recruits the RdDM machinery via MORC6 to establish de novo DNA methylation. Nat. Commun. 2023, 14, 4135. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Choi, J.K.; Kim, Y.J. Epigenetic regulation and the variability of gene expression. Nat. Genet. 2008, 40, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Gianoulis, T.A.; Karczewski, K.J.; Zhao, H.; Snyder, M. Regulatory variation within and between species. Annu. Rev. Genom. Hum. Genet. 2011, 12, 327–346. [Google Scholar] [CrossRef]
- Dai, Z.; Dai, X. Gene expression divergence is coupled to evolution of DNA structure in coding regions. PLoS Comput. Biol. 2011, 7, e1002275. [Google Scholar] [CrossRef]
- Fraser, H.B.; Moses, A.M.; Schadt, E.E. Evidence for widespread adaptive evolution of gene expression in budding yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 2977–2982. [Google Scholar] [CrossRef]
- Fraser, H.B.; Babak, T.; Tsang, J.; Zhou, Y.; Zhang, B.; Mehrabian, M.; Schadt, E.E. Systematic detection of polygenic cis-regulatory evolution. PLoS Genet. 2011, 7, e1002023. [Google Scholar] [CrossRef] [PubMed]
- Frankel, N.; Erezyilmaz, D.F.; McGregor, A.P.; Wang, S.; Payre, F.; Stern, D.L. Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature 2011, 474, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, S.A.; Houle, D.; Kim, J.; White, K.P. A mutation accumulation assay reveals a broad capacity for rapid evolution of gene expression. Nature 2005, 438, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Xu, K.; Park, T.; Yi, S.V. What are the determinants of gene expression levels and breadths in the human genome? Hum. Mol. Genet. 2012, 21, 46–56. [Google Scholar] [CrossRef]
- Das, S.; Roymondal, U.; Sahoo, S. Analyzing gene expression from relative codon usage bias in Yeast genome: A statistical significance and biological relevance. Gene 2009, 443, 121–131. [Google Scholar] [CrossRef]
- Tang, C.; Toomajian, C.; Sherman-Broyles, S.; Plagnol, V.; Guo, Y.L.; Hu, T.T.; Clark, R.M.; Nasrallah, J.B.; Weigel, D.; Nordborg, M. The evolution of selfing in Arabidopsis thaliana. Science 2007, 317, 1070–1072. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002, 419, 832–837. [Google Scholar] [CrossRef]
- Wang, E.T.; Kodama, G.; Baldi, P.; Moyzis, R.K. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc. Natl. Acad. Sci. USA 2006, 103, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.A.; Garfield, D.A.; Manier, M.K.; Haygood, R.; Wray, G.A.; Palumbi, S.R. Whole-genome positive selection and habitat-driven evolution in a shallow and a deep-sea urchin. Genome Biol. Evol. 2010, 2, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Suzuki, Y.; Nozawa, M. The neutral theory of molecular evolution in the genomic era. Annu. Rev. Genom. Hum. Genet. 2010, 11, 265–289. [Google Scholar] [CrossRef]
- King, J.L.; Jukes, T.H. Non-Darwinian evolution. Science 1969, 164, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar] [CrossRef]
- Kimura, M. The neutral theory of molecular evolution. Sci. Am. 1979, 241, 98–100, 102, 108 passim. [Google Scholar] [CrossRef]
- Brown, J.C. Role of Gene Length in Control of Human Gene Expression: Chromosome-Specific and Tissue-Specific Effects. Int. J. Genom. 2021, 2021, 8902428. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.Y.; Simons, C.; Firth, A.E.; Brown, C.M.; Hellens, R.P. Effect of 5′UTR introns on gene expression in Arabidopsis thaliana. BMC Genom. 2006, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Ka | Ks | ω | PS | EST | cDNA | RNA-Seq | Microarrays | Mutant |
|---|---|---|---|---|---|---|---|---|---|
| AT2G13560 | 0.0007 | 0.0641 | 0.0108 | 1 | 93 | 3 | 161.28 | 972.10 | nad-me1 [58] |
| AT2G33210 | 0.0008 | 0.0581 | 0.0132 | 1 | 34 | 3 | 51.22 | 1153.88 | hsp60-2-1 [59] |
| AT5G02870 | 0.0021 | 0.1137 | 0.0181 | 1 | 843 | 10 | 315.57 | 4365.06 | rpl4d [60] |
| AT3G18980 | 0.0021 | 0.0097 | 0.2203 | 11 | 44 | 7 | 18.88 | n.a. | ein2 [61] |
| AT2G28240 | 0.0043 | 0.0035 | 1.2273 | 11 | 10 | 2 | 16.14 | n.a. | mom2-2 [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, C.; Wei, Q.; Ye, Y.; Liu, J.; Li, G.; Liang, J.W.; Huang, H.; Wu, G. Fixation of Expression Divergences by Natural Selection in Arabidopsis Coding Genes. Int. J. Mol. Sci. 2024, 25, 13710. https://doi.org/10.3390/ijms252413710
Qi C, Wei Q, Ye Y, Liu J, Li G, Liang JW, Huang H, Wu G. Fixation of Expression Divergences by Natural Selection in Arabidopsis Coding Genes. International Journal of Molecular Sciences. 2024; 25(24):13710. https://doi.org/10.3390/ijms252413710
Chicago/Turabian StyleQi, Cheng, Qiang Wei, Yuting Ye, Jing Liu, Guishuang Li, Jane W. Liang, Haiyan Huang, and Guang Wu. 2024. "Fixation of Expression Divergences by Natural Selection in Arabidopsis Coding Genes" International Journal of Molecular Sciences 25, no. 24: 13710. https://doi.org/10.3390/ijms252413710
APA StyleQi, C., Wei, Q., Ye, Y., Liu, J., Li, G., Liang, J. W., Huang, H., & Wu, G. (2024). Fixation of Expression Divergences by Natural Selection in Arabidopsis Coding Genes. International Journal of Molecular Sciences, 25(24), 13710. https://doi.org/10.3390/ijms252413710