
Structure Characterization of Zinc Finger Motif 1 and 2 of GLI1 DNA Binding Region

 and
and
Abstract

1. Introduction
2. Results
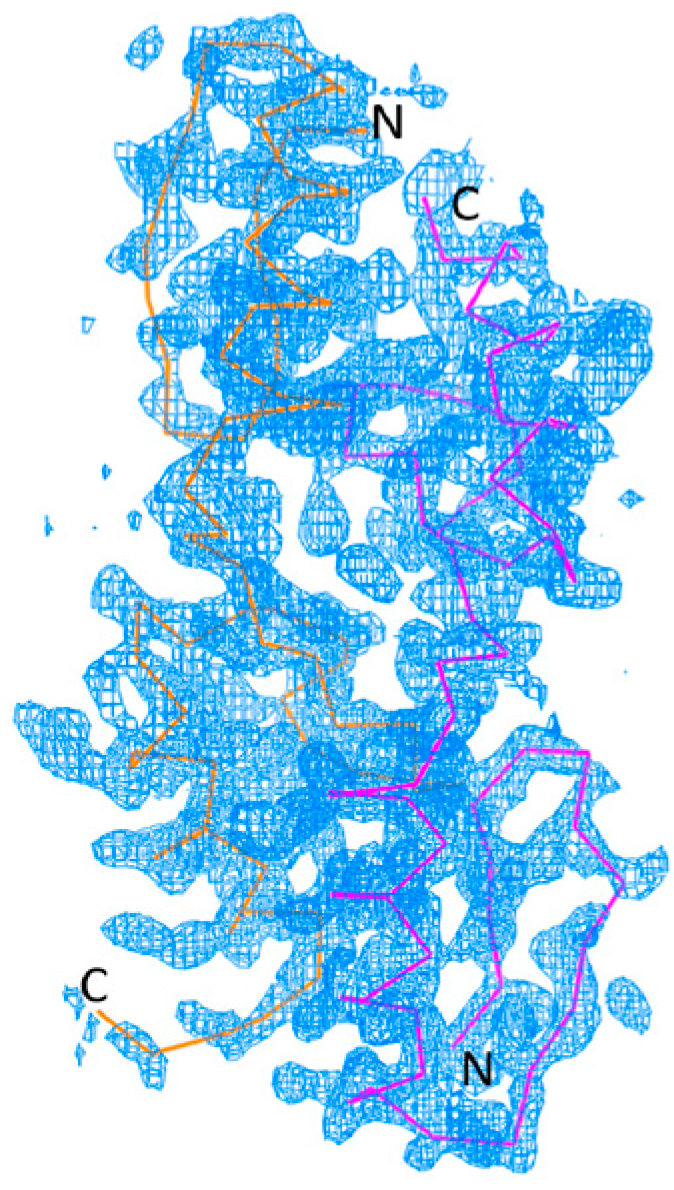
2.1. Structure Determination
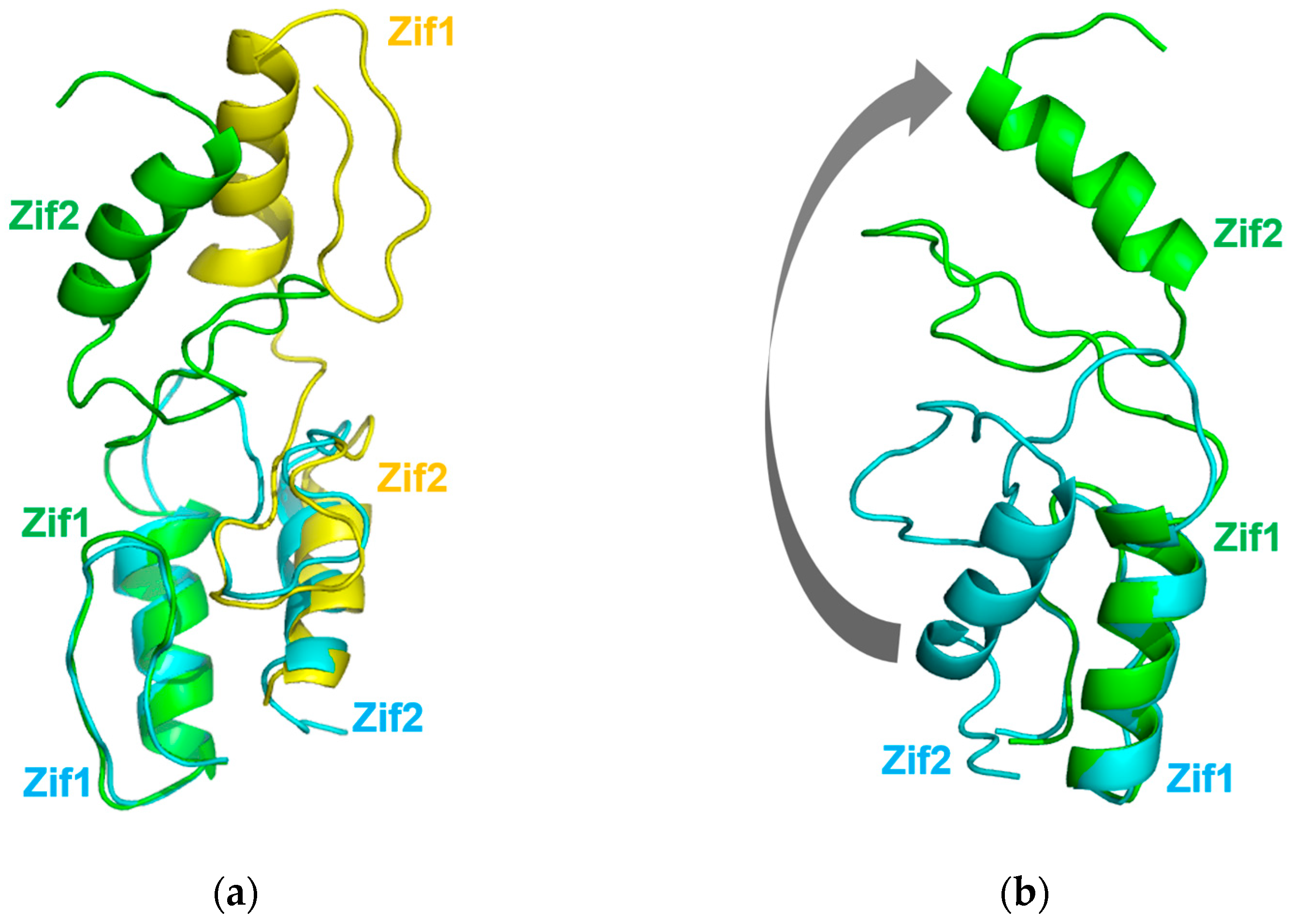
2.2. Overall Structure of Zif1-2
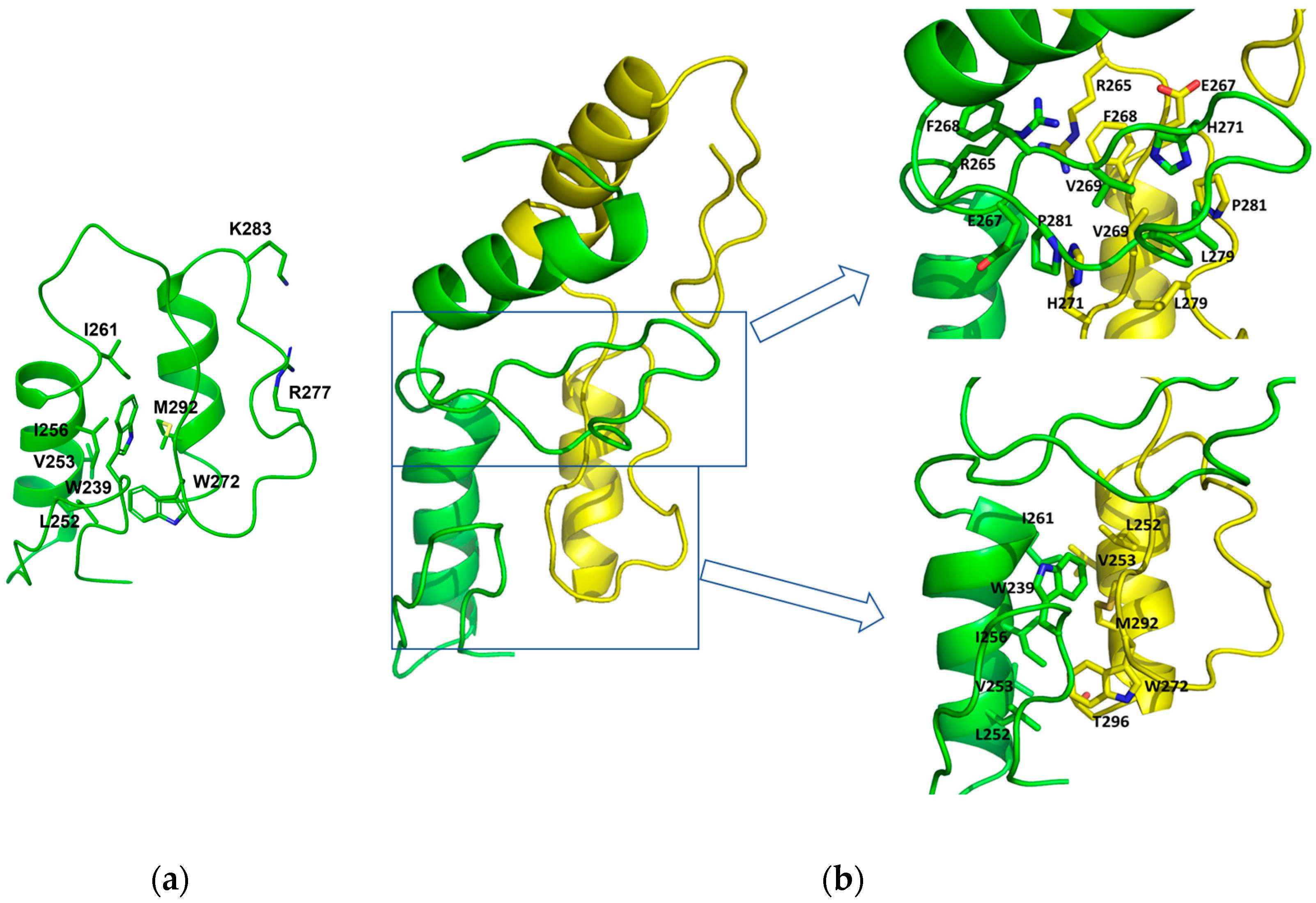
2.3. Dimerization of Zif1-2 and Zif1-5
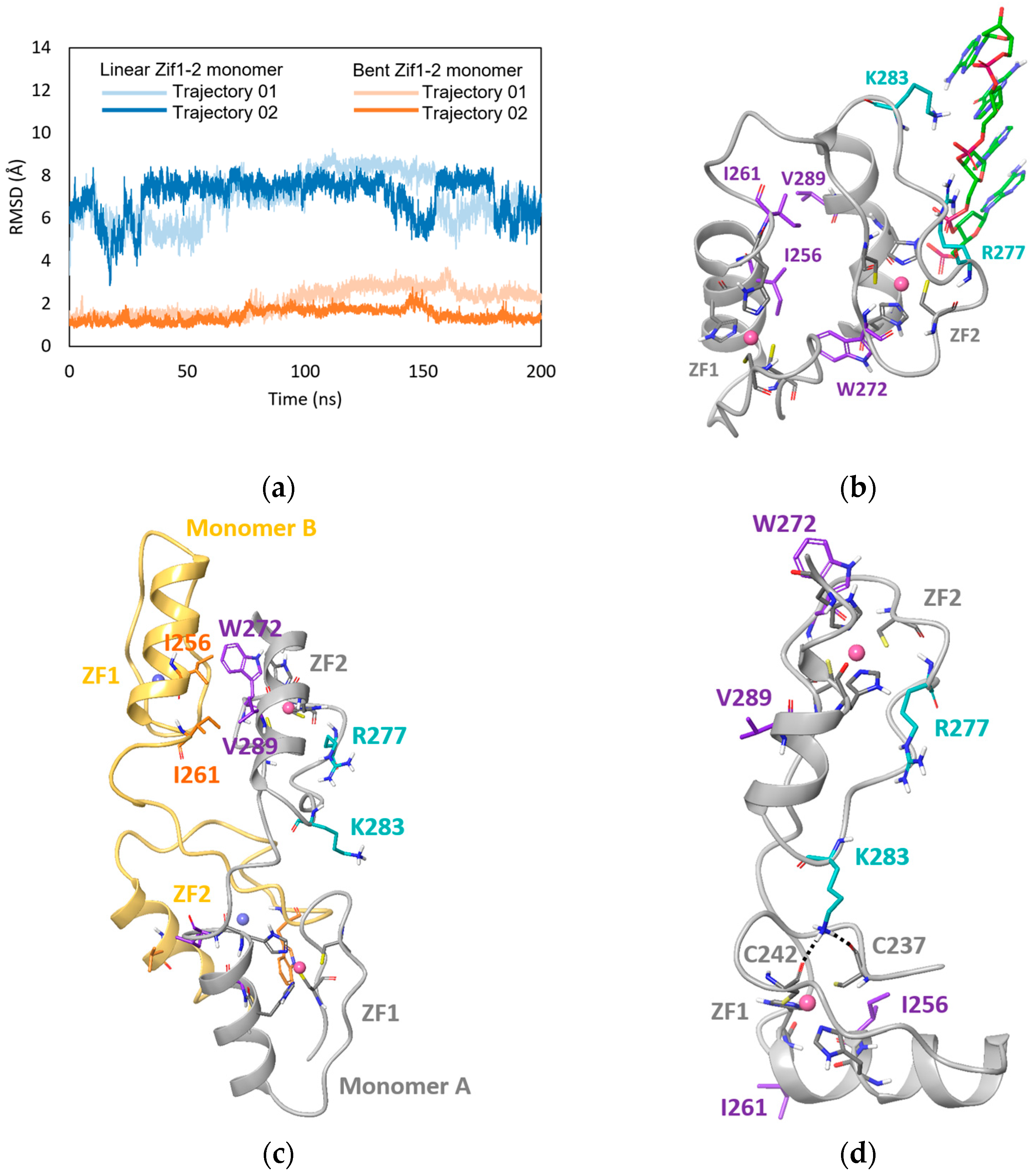
2.4. MD Simulation Study of GLI1 Zif1-2
2.5. In Silico Mutagenesis
3. Discussion
4. Materials and Methods
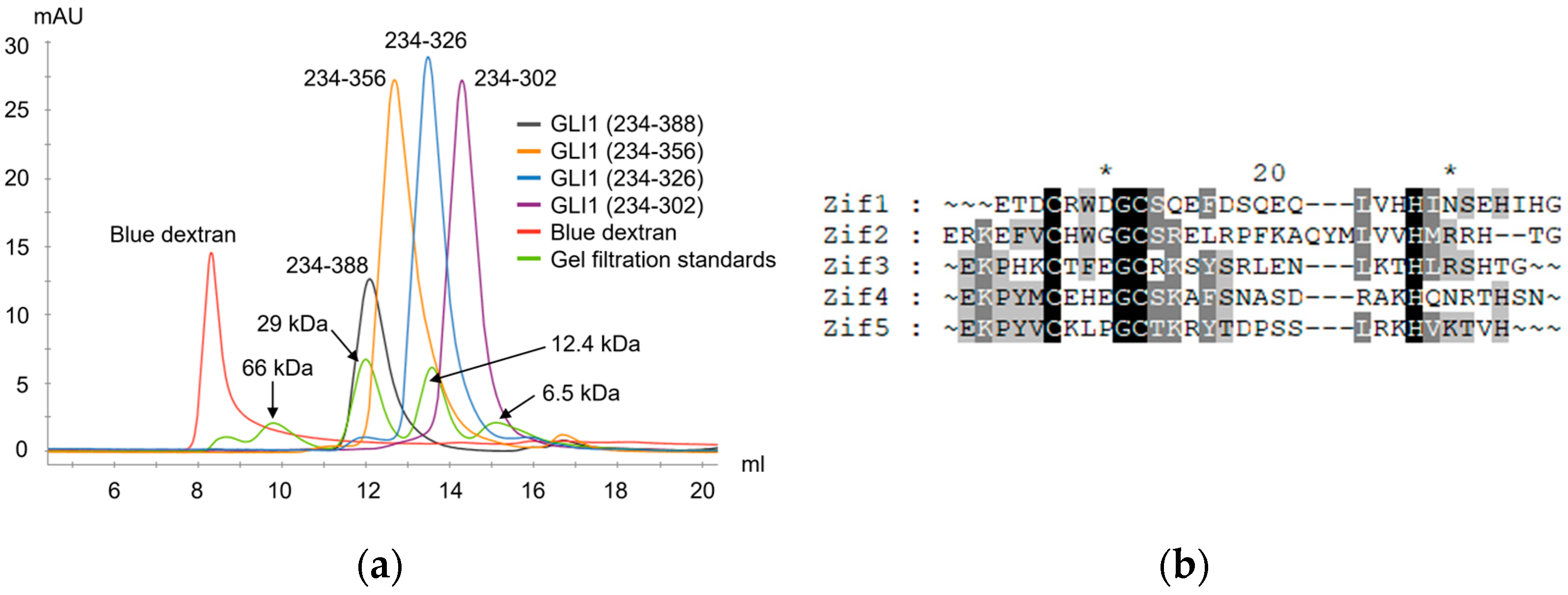
4.1. Molecular Cloning, Protein Expression, and Purification
4.2. Crystallization
4.3. Data Collection and Processing
4.4. Structure Solution and Refinement
4.5. MD Simulation
4.6. In Silico Mutagenesis and Free Energy Calculation
4.7. Large Language AI Model to Predict Protein Stability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef]
- Avery, J.T.; Zhang, R.; Boohaker, R.J. GLI1: A therapeutic target for cancer. Front. Oncol. 2021, 11, 673154. [Google Scholar] [CrossRef]
- Zhang, R.; Ma, J.; Avery, J.T.; Sambandam, V.; Nguyen, T.H.; Xu, B.; Suto, M.J.; Boohaker, R.J. GLI1 inhibitor SRI-38832 attenuates chemotherapeutic resistance by downregulating NBS1 transcription in BRAF(V600E) colorectal cancer. Front. Oncol. 2020, 10, 241. [Google Scholar] [CrossRef]
- Bauer, N.C.; Doetsch, P.W.; Corbett, A.H. Mechanisms regulating protein localization. Traffic 2015, 16, 1039–1061. [Google Scholar] [CrossRef]
- Han, Y.; Shi, Q.; Jiang, J. Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 6383–6388. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, E.; Milani, M. Role and inhibition of GLI1 protein in cancer. Lung Cancer 2018, 9, 35–43. [Google Scholar] [CrossRef]
- Ruppert, J.M.; Kinzler, K.W.; Wong, A.J.; Bigner, S.H.; Kao, F.T.; Law, M.L.; Seuanez, H.N.; O’Brien, S.J.; Vogelstein, B. The GLI-Kruppel family of human genes. Mol. Cell. Biol. 1988, 8, 3104–3113. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Pabo, C.O. Crystal structure of a five-finger GLI-DNA complex: New perspectives on zinc fingers. Science 1993, 261, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent advances in the clinical targeting of Hedgehog/GLI signaling in cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, J.; Ferrandon, S.; Glowacki, K.J.; Houghton, J.A. Targeting GLI by GANT61 involves mechanisms dependent on inhibition of both transcription and DNA licensing. Oncotarget 2016, 7, 80190–80207. [Google Scholar] [CrossRef] [PubMed]
- Maresca, L.; Crivaro, E.; Migliorini, F.; Anichini, G.; Giammona, A.; Pepe, S.; Poggialini, F.; Vagaggini, C.; Giannini, G.; Sestini, S.; et al. Targeting GLI1 and GLI2 with small molecule inhibitors to suppress GLI-dependent transcription and tumor growth. Pharmacol. Res. 2023, 195, 106858. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI job dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M., Jr. Rational design of particle mesh Ewald compatible Lennard-Jones parameters for +2 metal cations in explicit solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.B.; Yang, Y.; Wang, B.; Fusti-Molnar, L.; Weaver, M.N.; Merz, K.M., Jr. Structural survey of zinc containing proteins and the development of the zinc AMBER force field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Predicted Interaction Free Energy (kcal/mol) |
|---|---|
| Wild Type | −149.92 |
| W239G | −101.03 |
| W239N | −123.23 |
| L252G | −140.63 |
| L252S | −142.30 |
| V253G | −143.68 |
| V253S | −137.70 |
| I256G | −145.48 |
| I256S | −142.37 |
| I261G | −140.80 |
| I261N | −146.78 |
| I261S | −142.44 |
| W272G | −114.26 |
| W272N | −118.65 |
| W272Y | −142.84 |
| V289G | −151.93 |
| V289T | −160.63 |
| M292G | −133.47 |
| M292N | −135.87 |
| T296S | −147.12 |
| Parameter | Value |
|---|---|
| Source organism | Homo Sapiens |
| DNA source | Laboratory plasmid |
| Forward primer | CGCGGATCCGAAACTGACTGCCGTTGGGATGGC |
| Reverse primer | CGCGTCGACTTACTTGTGTGGCTTCTCGCCAGTGTG |
| Cloning vector | pET28a (+)-SUMO-TEV |
| Expression vector | pET28a (+)-SUMO-TEV |
| Expression host | E. coli Rosetta pLysS |
| Sequence of full construct | MGSSHHHHHHSSGLVPRGSHMSDSEVNQEAKPEVKPEVKPETHINLKVSDGSSEIFFKIKKTTPLRRLMEAFAKRQGKEMDSLRFLYDGIRIQADQTPEDLDMEDNDIIEAHREQIGGIEENLYFQSGSETDCRWDGCSQEFDSQEQLVHHINSEHIHGERKEFVCHWGGCSRELRPFKAQYMLVVHMRRHTGEKPHK |
| Parameter | Value |
|---|---|
| Method | Sitting-drop |
| Plate type | 24-well plate |
| Temperature (K) | 291 |
| Protein concentration (mg/mL) | 5.4 |
| Buffer for protein solution | 20 mM HEPES, pH = 7.6, 500 mM NaCl, 0.1 mM TCEP |
| Reservoir solution | 29–32% PEG3350, 0.1 M Bis-Tris pH = 6.5 |
| Volume (µL) and ratio of drop | 1:1 |
| Volume of reservoir (µL) | 500 |
| Section | Parameter | Value * |
|---|---|---|
| Data collection | Beamline | 22-ID, APS |
| Processing software | XDS version 2019 | |
| Wavelength (Å) | 1.0 | |
| Resolution range (Å) | 33.36–2.05 (2.123–2.05) | |
| Space group | P65 | |
| Unit cell a, b, c (Å) | 66.728, 66.728, 65.909 | |
| Unit cell α, β, γ (°) | 90, 90, 120 | |
| Total reflections | 21,071 (2088) | |
| Unique reflections | 10,536 (1042) | |
| Multiplicity | 2.0 (2.0) | |
| Completeness (%) | 99.88 (100.00) | |
| Mean I/sigma (I) | 17.64 (4.41) | |
| Wilson B factor (Å2) | 40.82 | |
| R-merge | 0.03403 (0.1799) | |
| R-means | 0.04813 (0.2544) | |
| R-pim | 0.03403 (0.1799) | |
| CC1/2 | 0.998 (0.899) | |
| Refinement | Reflections used in refinement | 10,527 (1044) |
| Reflections used for Rfree | 509 (56) | |
| Rwork | 0.1985 (0.2403) | |
| Rfree | 0.2258 (0.2982) | |
| CCwork | 0.948 (0.785) | |
| CCfree | 0.934 (0.700) | |
| Number of nonhydrogen atoms | 1160 | |
| Macromolecules | 1084 | |
| Ligands | 4 | |
| Solvent | 72 | |
| Protein residues | 129 | |
| Bond length (Å) | 0.013 | |
| Bond angle (°) | 1.60 | |
| Ramachandran plot | Ramachandran favored (%) | 99.20 |
| Ramachandran allowed (%) | 0.80 | |
| Ramachandran outliers (%) | 0.00 | |
| Rotamer outliers (%) | 1.69 | |
| Clash score | 3.85 | |
| Average B factor (Å2) | 48.57 | |
| Macromolecules | 48.50 | |
| Ligands | 39.66 | |
| Solvent | 50.03 | |
| Number of TLS groups | 2 | |
| PDB ID | 7T91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; Jahan, N.; Sharp, A.; Ullah, A.; Augelli-Szafran, C.E.; Zhang, S.; Boohaker, R.J. Structure Characterization of Zinc Finger Motif 1 and 2 of GLI1 DNA Binding Region. Int. J. Mol. Sci. 2024, 25, 13368. https://doi.org/10.3390/ijms252413368
Wu M, Jahan N, Sharp A, Ullah A, Augelli-Szafran CE, Zhang S, Boohaker RJ. Structure Characterization of Zinc Finger Motif 1 and 2 of GLI1 DNA Binding Region. International Journal of Molecular Sciences. 2024; 25(24):13368. https://doi.org/10.3390/ijms252413368
Chicago/Turabian StyleWu, Mousheng, Nusrat Jahan, Amanda Sharp, Anwar Ullah, Corinne E. Augelli-Szafran, Sixue Zhang, and Rebecca J. Boohaker. 2024. "Structure Characterization of Zinc Finger Motif 1 and 2 of GLI1 DNA Binding Region" International Journal of Molecular Sciences 25, no. 24: 13368. https://doi.org/10.3390/ijms252413368
APA StyleWu, M., Jahan, N., Sharp, A., Ullah, A., Augelli-Szafran, C. E., Zhang, S., & Boohaker, R. J. (2024). Structure Characterization of Zinc Finger Motif 1 and 2 of GLI1 DNA Binding Region. International Journal of Molecular Sciences, 25(24), 13368. https://doi.org/10.3390/ijms252413368