
Integrated Proteomics and Machine Learning Approach Reveals PYCR1 as a Novel Biomarker to Predict Prognosis of Sinonasal Squamous Cell Carcinoma
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
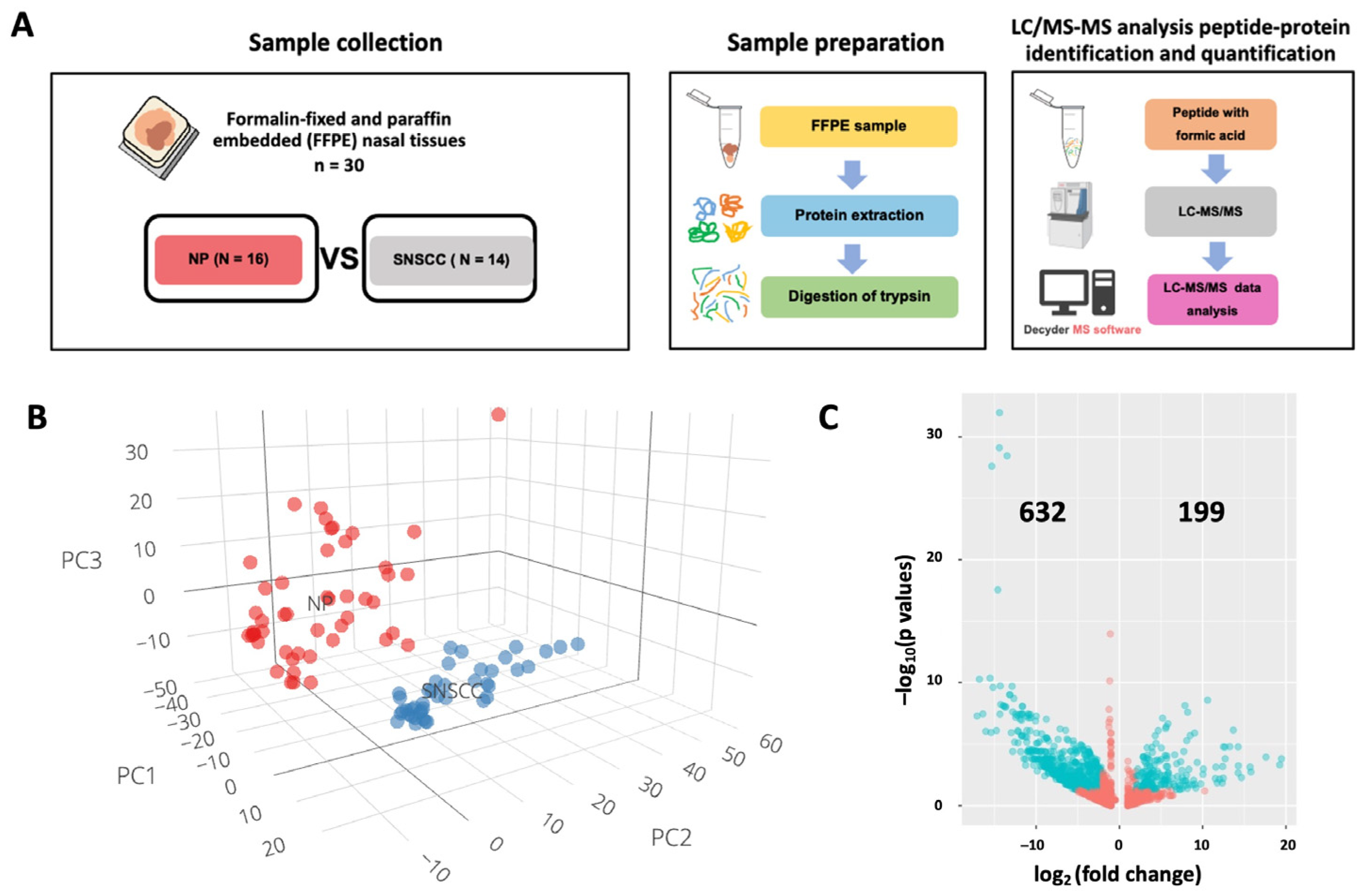
2.1. Label-Free Quantification of Nasal Polyps and Sinonasal Squamous Cell Carcinoma
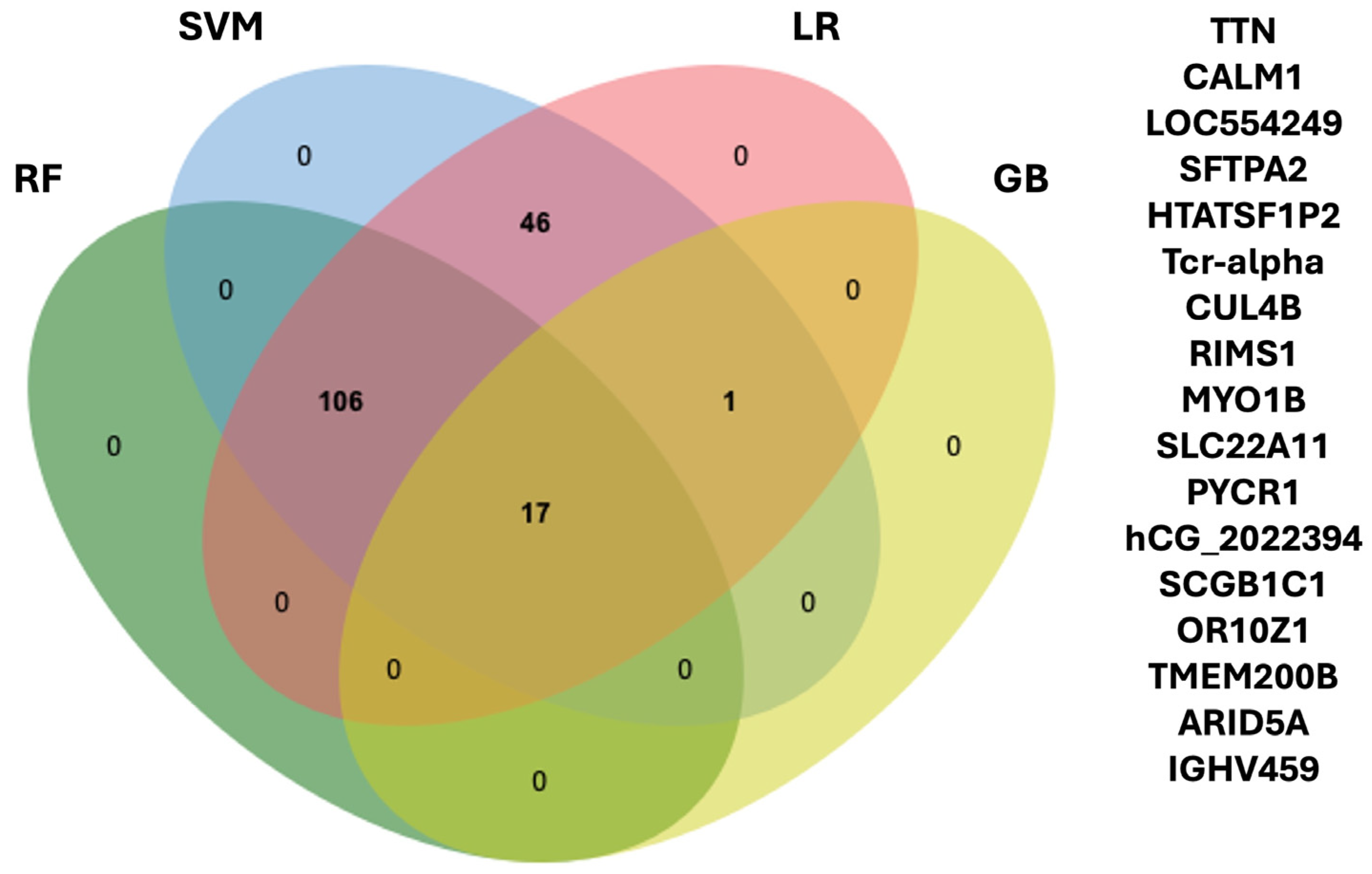
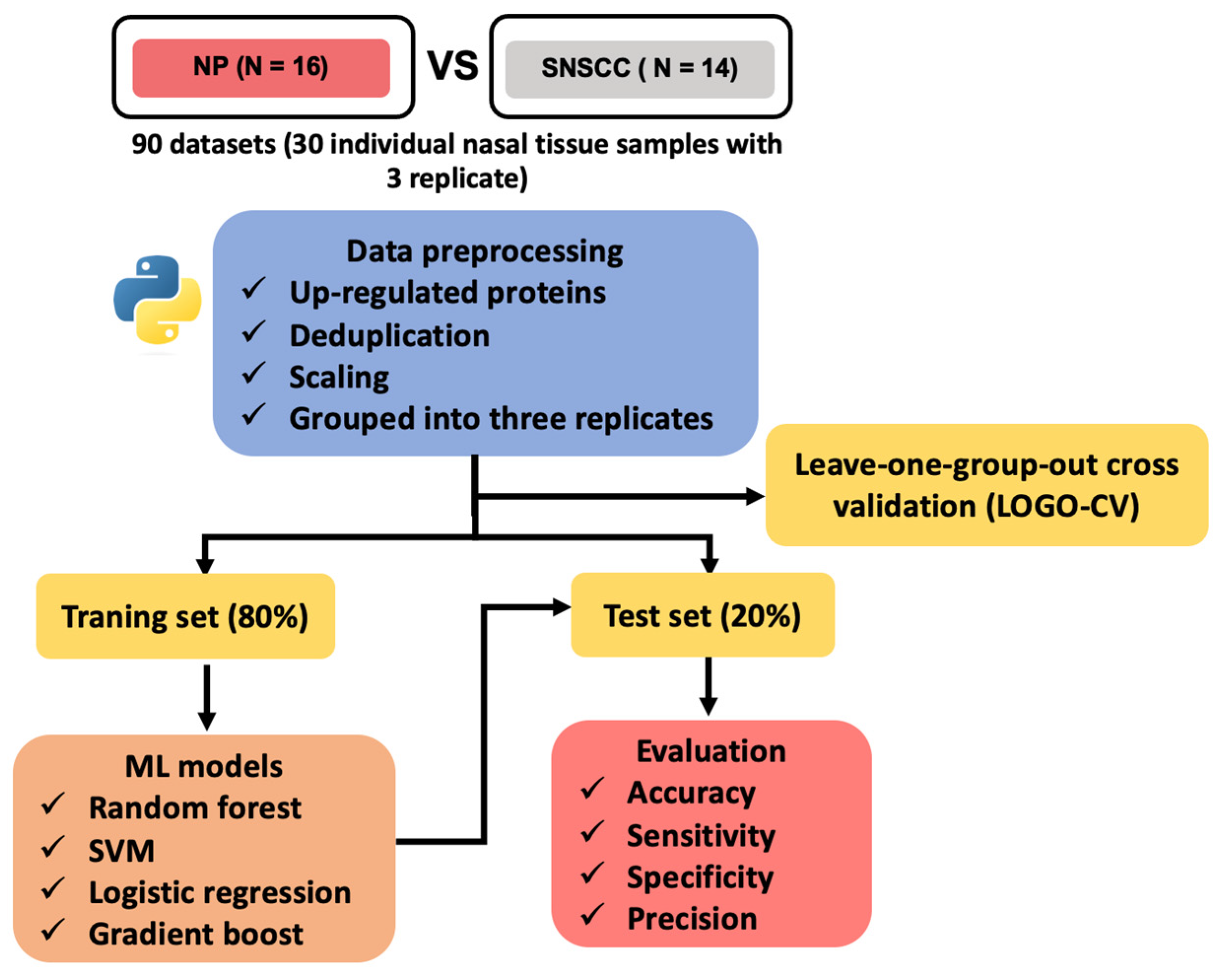
2.2. Machine Learning for Biomarker Discovery
2.3. Selection of Candidate Biomarkers
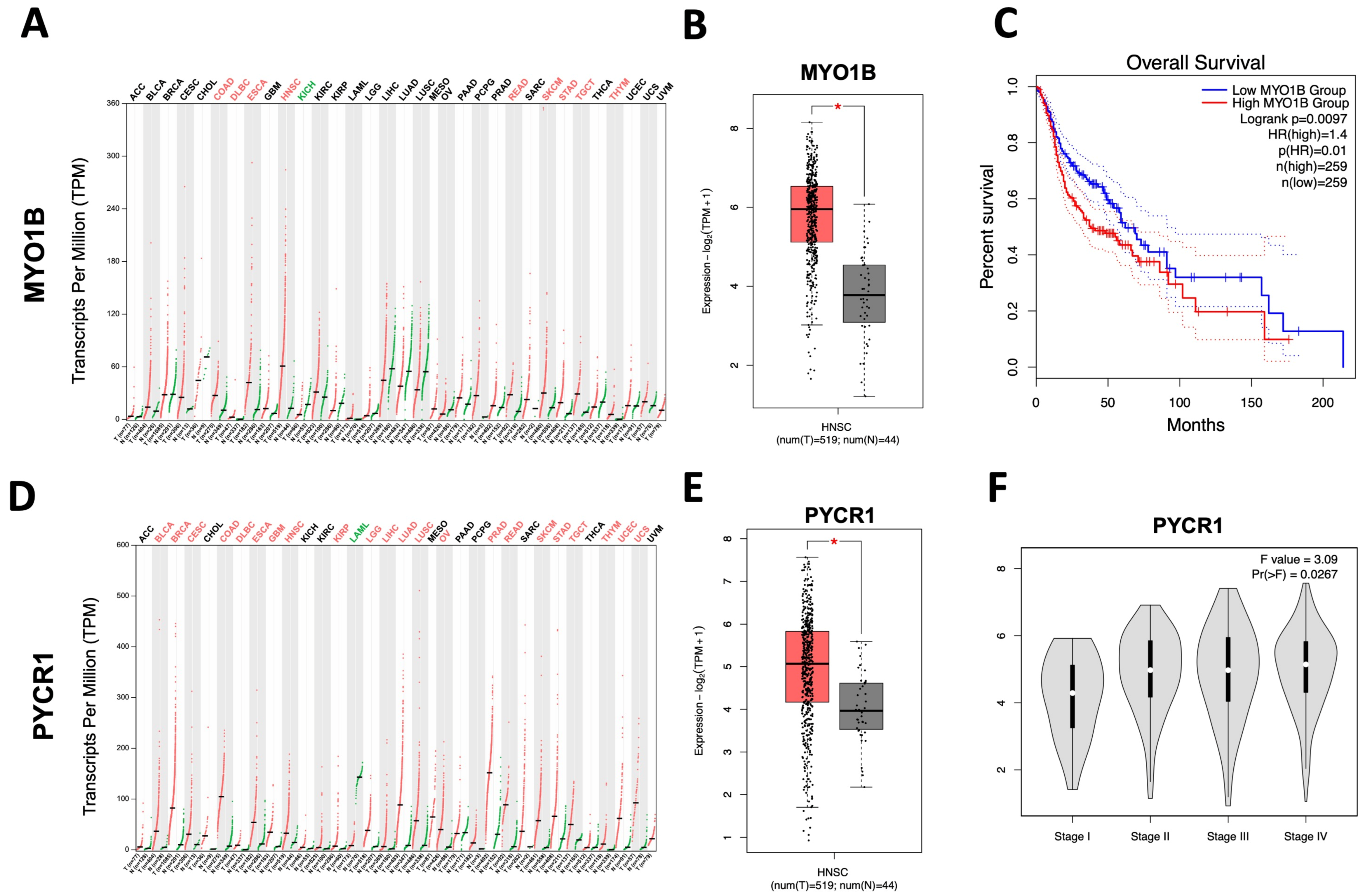
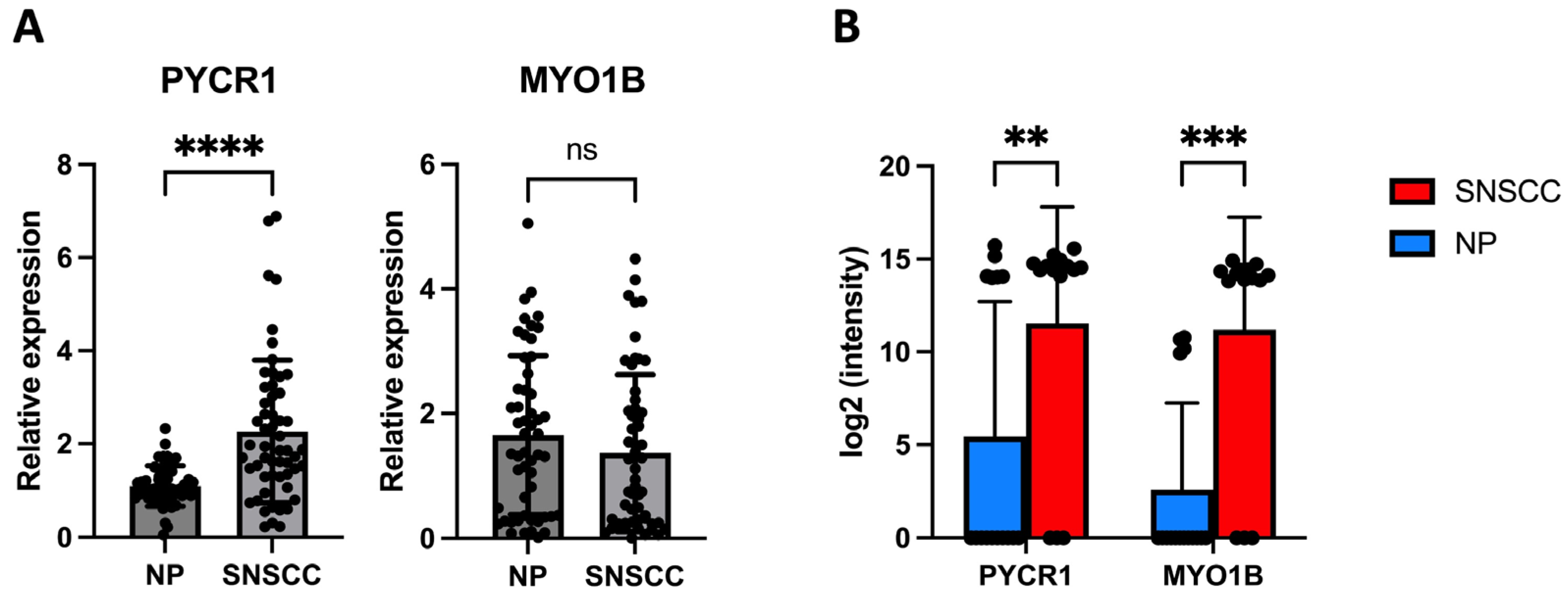
2.4. PYCR1 Might Serve as a Tumor-Associated Biomarker for SNSCC
2.5. Reviewing of PYCR1 Expression in Different Tumor Tissues and Its Association with Clinicopathological Characteristics in SNSCC Patients
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Trypsin-Digested Peptides, LC–MS/MS, and Data Analysis
4.3. Principal Component Analysis and Identification of Differentially Expressed Genes
4.4. Machine Learning Models
4.5. In Silico Analysis of PYCR1 and MYO1B Gene Expression Based on Pan-Cancer Database
4.6. Relative Gene Expression by qRT-PCR
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turner, J.H.; Reh, D.D. Incidence and Survival in Patients with Sinonasal Cancer: A Historical Analysis of Population-Based Data. Head Neck 2012, 34, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Llorente, J.L.; López, F.; Suárez, C.; Hermsen, M.A. Sinonasal Carcinoma: Clinical, Pathological, Genetic and Therapeutic Advances. Nat. Rev. Clin. Oncol. 2014, 11, 460–472. [Google Scholar] [CrossRef]
- Binazzi, A.; Ferrante, P.; Marinaccio, A. Occupational Exposure and Sinonasal Cancer: A Systematic Review and Meta-Analysis. BMC Cancer 2015, 15, 49. [Google Scholar] [CrossRef]
- Taverna, C.; Agaimy, A.; Franchi, A. Towards a Molecular Classification of Sinonasal Carcinomas: Clinical Implications and Opportunities. Cancers 2022, 14, 1463. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Zhang, Z.; Zhou, L.; Han, C.; Huang, C.; Nice, E.C. Proteomics, Personalized Medicine and Cancer. Cancers 2021, 13, 2512. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.-H.; Wang, C.-C.; Wu, Y.-L.; Hsu, K.-C.; Lee, T.-H. Machine Learning Approaches for Biomarker Discovery to Predict Large-Artery Atherosclerosis. Sci. Rep. 2023, 13, 15139. [Google Scholar] [CrossRef] [PubMed]
- Aziz, S.; Ahmed, S.; Alouini, M.-S. ECG-Based Machine-Learning Algorithms for Heartbeat Classification. Sci. Rep. 2021, 11, 18738. [Google Scholar] [CrossRef]
- Xu, H.; Usuyama, N.; Bagga, J.; Zhang, S.; Rao, R.; Naumann, T.; Wong, C.; Gero, Z.; González, J.; Gu, Y.; et al. A Whole-Slide Foundation Model for Digital Pathology from Real-World Data. Nature 2024, 630, 181–188. [Google Scholar] [CrossRef]
- Torun, F.M.; Virreira Winter, S.; Doll, S.; Riese, F.M.; Vorobyev, A.; Mueller-Reif, J.B.; Geyer, P.E.; Strauss, M.T. Transparent Exploration of Machine Learning for Biomarker Discovery from Proteomics and Omics Data. J. Proteome Res. 2023, 22, 359–367. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, Y.; Liu, W.; Li, Q.; Hou, W.; Huang, Y.; Lv, P.; Meng, L.; Li, Y.; Jia, Y.; et al. Increased PYCR1 mRNA Predicts Poor Prognosis in Kidney Adenocarcinoma: A Study Based on TCGA Database. Medicine 2021, 100, e27145. [Google Scholar] [CrossRef]
- Xiao, S.; Li, S.; Yuan, Z.; Zhou, L. Pyrroline-5-Carboxylate Reductase 1 (PYCR1) Upregulation Contributes to Gastric Cancer Progression and Indicates Poor Survival Outcome. Ann. Transl. Med. 2020, 8, 937. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Miao, Y.; Liu, C.; Wu, T.; Shen, S.; Su, X.; Shi, Y. Pyrroline-5-carboxylate Reductase 1 Promotes Proliferation and Inhibits Apoptosis in Non-small Cell Lung Cancer. Oncol. Lett. 2018, 15, 731–740. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, X.; Wang, E.; Yang, Y.; Hu, L.; Xu, H.; Zhang, B. PYCR1 Promotes the Malignant Progression of Lung Cancer through the JAK-STAT3 Signaling Pathway via PRODH-Dependent Glutamine Synthesize. Transl. Oncol. 2023, 32, 101667. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mao, W.; Lou, W.; Jin, D.; Wu, W.; Wang, D.; Kuang, T.; Rong, Y.; Xu, X.; Zhang, L. PYCR1: A Potential Prognostic Biomarker in Pancreatic Ductal Adenocarcinoma. J. Cancer 2022, 13, 1501–1511. [Google Scholar] [CrossRef]
- Xiao, H.; Huang, J.; Wu, H.; Li, Y.; Wang, Y. Pro-Tumorigenic Activity of PYCR1 in Gastric Cancer through Regulating the PI3K/AKT Signaling. Heliyon 2024, 10, e26883. [Google Scholar] [CrossRef]
- Fu, W.; Xie, Z.; Zheng, S.; Yang, X.; Ding, Q.; Liu, J.; Liang, Q.; Chen, Y.; Mi, H.; Liu, D.; et al. The Clinical Significance of PYCR1 Expression in Renal Cell Carcinoma. Medicine 2019, 98, e16384. [Google Scholar] [CrossRef]
- Ding, J.; Kuo, M.-L.; Su, L.; Xue, L.; Luh, F.; Zhang, H.; Wang, J.; Lin, T.G.; Zhang, K.; Chu, P.; et al. Human Mitochondrial Pyrroline-5-Carboxylate Reductase 1 Promotes Invasiveness and Impacts Survival in Breast Cancers. Carcinogenesis 2017, 38, 519–531. [Google Scholar] [CrossRef]
- Xu, Y.; Zuo, W.; Wang, X.; Zhang, Q.; Gan, X.; Tan, N.; Jia, W.; Liu, J.; Li, Z.; Zhou, B.; et al. Deciphering the Effects of PYCR1 on Cell Function and Its Associated Mechanism in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2021, 17, 2223–2239. [Google Scholar] [CrossRef]
- Jurmeister, P.; Glöß, S.; Roller, R.; Leitheiser, M.; Schmid, S.; Mochmann, L.H.; Payá Capilla, E.; Fritz, R.; Dittmayer, C.; Friedrich, C.; et al. DNA Methylation-Based Classification of Sinonasal Tumors. Nat. Commun. 2022, 13, 7148. [Google Scholar] [CrossRef]
- Jung, M.; Lee, C.; Han, D.; Kim, K.; Yang, S.; Nikas, I.P.; Moon, K.C.; Kim, H.; Song, M.J.; Kim, B.; et al. Proteomic-Based Machine Learning Analysis Reveals PYGB as a Novel Immunohistochemical Biomarker to Distinguish Inverted Urothelial Papilloma from Low-Grade Papillary Urothelial Carcinoma with Inverted Growth. Front. Oncol. 2022, 12, 841398. [Google Scholar] [CrossRef]
- Vizza, P.; Aracri, F.; Guzzi, P.H.; Gaspari, M.; Veltri, P.; Tradigo, G. Machine Learning Pipeline to Analyze Clinical and Proteomics Data: Experiences on a Prostate Cancer Case. BMC Med. Inform. Decis. Mak. 2024, 24, 93. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Masarone, S.; Watson, D.; Barnes, M.R. The Benefits and Pitfalls of Machine Learning for Biomarker Discovery. Cell Tissue Res. 2023, 394, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-R.; Liu, C.; Balgley, B.M.; Lee, C.; Taylor, C.R. Protein Extraction from Formalin-Fixed, Paraffin-Embedded Tissue Sections: Quality Evaluation by Mass Spectrometry. J. Histochem. Cytochem. 2006, 54, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Crockett, D.K.; Lin, Z.; Vaughn, C.P.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Identification of Proteins from Formalin-Fixed Paraffin-Embedded Cells by LC-MS/MS. Lab. Investig. 2005, 85, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Hieggelke, L.; Heydt, C.; Castiglione, R.; Rehker, J.; Merkelbach-Bruse, S.; Riobello, C.; Llorente, J.L.; Hermsen, M.A.; Buettner, R. Mismatch Repair Deficiency and Somatic Mutations in Human Sinonasal Tumors. Cancers 2021, 13, 6081. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Xu, N.-Z.; Hong, C.; Li, W.-Q.; Zhang, Y.-Q.; Yu, X.-Y.; Huang, Y.-L.; Zhou, J.-Y. Myo1b Promotes Tumor Progression and Angiogenesis by Inhibiting Autophagic Degradation of HIF-1α in Colorectal Cancer. Cell Death Dis. 2022, 13, 939. [Google Scholar] [CrossRef]
- Wen, L.-J.; Hu, X.-L.; Li, C.-Y.; Liu, J.; Li, Z.-Y.; Li, Y.-Z.; Zhou, J.-Y. Myosin 1b Promotes Migration, Invasion and Glycolysis in Cervical Cancer via ERK/HIF-1α Pathway. Am. J. Transl. Res. 2021, 13, 12536–12548. [Google Scholar]
- Xie, L.; Huang, H.; Zheng, Z.; Yang, Q.; Wang, S.; Chen, Y.; Yu, J.; Cui, C. MYO1B Enhances Colorectal Cancer Metastasis by Promoting the F-Actin Rearrangement and Focal Adhesion Assembly via RhoA/ROCK/FAK Signaling. Ann. Transl. Med. 2021, 9, 1543. [Google Scholar] [CrossRef]
- Yamada, Y.; Koshizuka, K.; Hanazawa, T.; Kikkawa, N.; Okato, A.; Idichi, T.; Arai, T.; Sugawara, S.; Katada, K.; Okamoto, Y.; et al. Passenger Strand of miR-145-3p Acts as a Tumor-Suppressor by Targeting MYO1B in Head and Neck Squamous Cell Carcinoma. Int. J. Oncol. 2017, 52, 166–178. [Google Scholar] [CrossRef]
- Chapman, B.V.; Wald, A.I.; Akhtar, P.; Munko, A.C.; Xu, J.; Gibson, S.P.; Grandis, J.R.; Ferris, R.L.; Khan, S.A. MicroRNA-363 Targets Myosin 1B to Reduce Cellular Migration in Head and Neck Cancer. BMC Cancer 2015, 15, 861. [Google Scholar] [CrossRef]
- D’Aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Wu, K.; Guo, X.; Chen, C.; Huang, T.; Li, L.; Liu, B.; Chang, H.; Zhao, J. The Pan-Cancer Landscape of Glutamate and Glutamine Metabolism: A Comprehensive Bioinformatic Analysis across 32 Solid Cancer Types. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2024, 1870, 166982. [Google Scholar] [CrossRef]
- Wang, D.; Wang, L.; Zhang, Y.; Yan, Z.; Liu, L.; Chen, G. PYCR1 Promotes the Progression of Non-Small-Cell Lung Cancer under the Negative Regulation of miR-488. Biomed. Pharmacother. 2019, 111, 588–595. [Google Scholar] [CrossRef]
- Wang, Q.-L.; Liu, L. PYCR1 Is Associated with Papillary Renal Cell Carcinoma Progression. Open Med. 2019, 14, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Lareo, A.C.; Luce, D.; Leclerc, A.; Rodriguez, J.; Brugère, J. History of Previous Nasal Diseases and Sinonasal Cancer: A Case-Control Study. Laryngoscope 1992, 102, 439–442. [Google Scholar] [CrossRef]
- Kaewseekhao, B.; Roytrakul, S.; Yingchutrakul, Y.; Salao, K.; Reechaipichitkul, W.; Faksri, K. Proteomic Analysis of Infected Primary Human Leucocytes Revealed PSTK as Potential Treatment-Monitoring Marker for Active and Latent Tuberculosis. PLoS ONE 2020, 15, e0231834. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Malat, P.; Ekalaksananan, T.; Heawchaiyaphum, C.; Suebsasana, S.; Roytrakul, S.; Yingchutrakul, Y.; Pientong, C. Andrographolide Inhibits Epstein-Barr Virus Lytic Reactivation in EBV-Positive Cancer Cell Lines through the Modulation of Epigenetic-Related Proteins. Molecules 2022, 27, 4666. [Google Scholar] [CrossRef]
- Oudaert, I.; Satilmis, H.; Vlummens, P.; De Brouwer, W.; Maes, A.; Hose, D.; De Bruyne, E.; Ghesquière, B.; Vanderkerken, K.; De Veirman, K.; et al. Pyrroline-5-Carboxylate Reductase 1: A Novel Target for Sensitizing Multiple Myeloma Cells to Bortezomib by Inhibition of PRAS40-Mediated Protein Synthesis. J. Exp. Clin. Cancer Res. 2022, 41, 45. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | LOGO-CV (95%Cl) | Accuracy (95%Cl) | Sensitivity (95%Cl) | Specificity (95%Cl) | Precision (95%Cl) |
|---|---|---|---|---|---|
| Random forest (RF) | 0.97 (0.91, 0.99) | 0.94 (0.73, 1.00) | 1.00 (0.74, 1.00) | 0.83 (0.36, 1.00) | 0.92 (0.64, 1.00) |
| Support vector machine (SVM) | 0.92 (0.85, 0.97) | 0.89 (0.65, 0.99) | 1.00 (0.74, 1.00) | 0.67 (0.22, 0.96) | 0.86 (0.57, 0.98) |
| Logistic regression (LR) | 0.92 (0.85, 0.97) | 0.89 (0.65, 0.99) | 1.00 (0.74, 1.00) | 0.67 (0.22, 0.96) | 0.86 (0.57, 0.98) |
| Gradient boost (GB) | 0.73 (0.63, 0.82) | 0.78 (0.52, 0.94) | 0.67 (0.35, 0.90) | 1.00 (0.54, 1.00) | 1.00 (0.63, 1.00) |
| Cancer Type | Year | Molecular Mechanism Findings | Association with Clinical Features | Association with Prognosis/Diagnostic | Ref. |
|---|---|---|---|---|---|
| Adenocarcinoma of the kidney | 2021 | - | High PYCR1 expression were associated with high histologic grade, advanced clinical stage, and presence of metastasis. | Increased PYCR1 expression was associated with a worse prognosis. | [10] |
| Gastric cancer | 2020 | Silencing of PYCR1 inhibited cell proliferation and induced apoptosis; PI3K/Akt pathway can affect proline metabolism via PYCR1. | High PYCR1 expression was associated advanced stage, histologic type, and high Ki-67. | Increased PYCR1 expression was associated with a worse prognosis. | [11] |
| Non-small cell lung cancer | 2018 | Silencing of PYCR1 inhibited cell proliferation, induced cell cycle arrest, and increased apoptosis. | - | Increased PYCR1 expression was associated with poor prognosis. | [12] |
| Lung cancer | 2023 | PYCR1 induced cell proliferative, migration, and invasion through the JAK–STAT3 signaling pathway via PRODH-dependent glutamine synthesize. | High PYCR1 expression was associated with advanced stage. | Increased PYCR1 expression was associated with poor prognosis; PYCR1 secretion in serum may use as a diagnostic marker. | [13] |
| Pancreatic ductal adenocarcinoma | 2022 | Silencing of PYCR1 inhibited cell proliferation and induces apoptosis. | - | Increased PYCR1 expression was associated with a worse prognosis. | [14] |
| Gastric cancer | 2024 | PYCR1 induced cell proliferation and metastasis and suppressed the apoptosis via the PI3K/AKT signaling. | - | Increased PYCR1 expression was associated with unfavorable prognosis. | [15] |
| Renal cell carcinoma | 2019 | - | High PYCR1 expression was associated with metastasis. | Increased PYCR1 expression was associated with poor prognosis. | [16] |
| Breast cancer | 2017 | Silencing of PYCR1 inhibited cell proliferation, invasion and enhanced the chemosensitivity to doxorubicin. | High PYCR1 expression were associated with larger tumor size, higher tumor grade, and more invasive molecular subtypes. | Increased PYCR1 expression was associated with poor prognosis. | [17] |
| Hepatocellular carcinoma | 2021 | Silencing PYCR1 inhibited cell proliferation, invasion, epithelial–mesenchymal transition, and metastasis | High PYCR1 expression were associated with female sex, higher alpha-fetoprotein levels, advanced clinical stage, and younger age (<45 years) | Increased PYCR1 expression was associated with poor prognosis | [18] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panthong, W.; Pientong, C.; Nukpook, T.; Roytrakul, S.; Yingchutrakul, Y.; Teeramatwanich, W.; Aromseree, S.; Ekalaksananan, T. Integrated Proteomics and Machine Learning Approach Reveals PYCR1 as a Novel Biomarker to Predict Prognosis of Sinonasal Squamous Cell Carcinoma. Int. J. Mol. Sci. 2024, 25, 13234. https://doi.org/10.3390/ijms252413234
Panthong W, Pientong C, Nukpook T, Roytrakul S, Yingchutrakul Y, Teeramatwanich W, Aromseree S, Ekalaksananan T. Integrated Proteomics and Machine Learning Approach Reveals PYCR1 as a Novel Biomarker to Predict Prognosis of Sinonasal Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2024; 25(24):13234. https://doi.org/10.3390/ijms252413234
Chicago/Turabian StylePanthong, Watcharapong, Chamsai Pientong, Thawaree Nukpook, Sittiruk Roytrakul, Yodying Yingchutrakul, Watchareporn Teeramatwanich, Sirinart Aromseree, and Tipaya Ekalaksananan. 2024. "Integrated Proteomics and Machine Learning Approach Reveals PYCR1 as a Novel Biomarker to Predict Prognosis of Sinonasal Squamous Cell Carcinoma" International Journal of Molecular Sciences 25, no. 24: 13234. https://doi.org/10.3390/ijms252413234
APA StylePanthong, W., Pientong, C., Nukpook, T., Roytrakul, S., Yingchutrakul, Y., Teeramatwanich, W., Aromseree, S., & Ekalaksananan, T. (2024). Integrated Proteomics and Machine Learning Approach Reveals PYCR1 as a Novel Biomarker to Predict Prognosis of Sinonasal Squamous Cell Carcinoma. International Journal of Molecular Sciences, 25(24), 13234. https://doi.org/10.3390/ijms252413234