
Potent Anti-Cancer Activity of 1-Dehydrodiosgenone from the Product of Microbial Transformation of Steroid Saponins
 ,
,
Abstract
:1. Introduction
2. Results
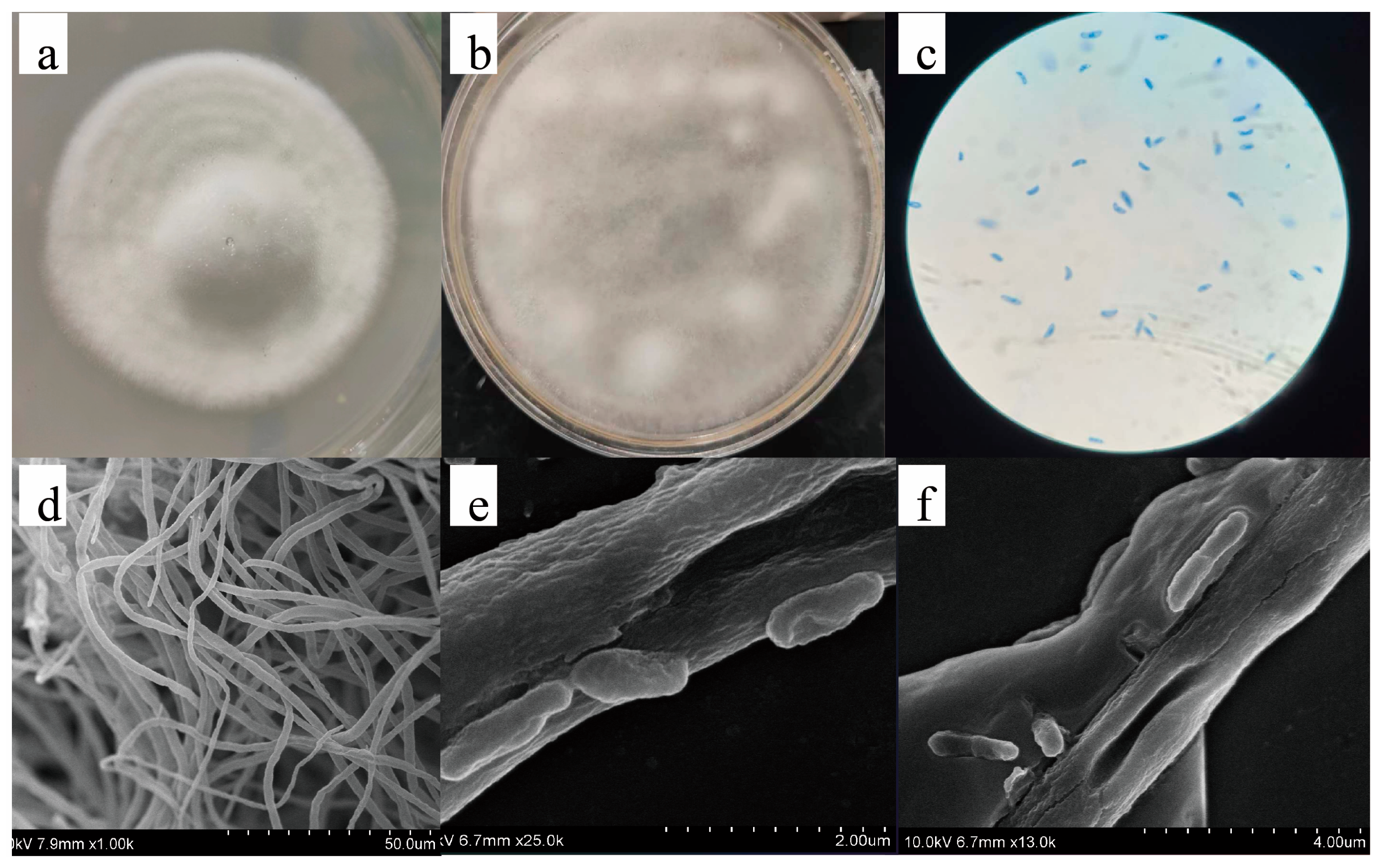
2.1. Isolation and Identification of Strains
2.2. Structural Elucidation of Compounds 1–4
2.3. Anti-Tumor Activity Assays of Compounds
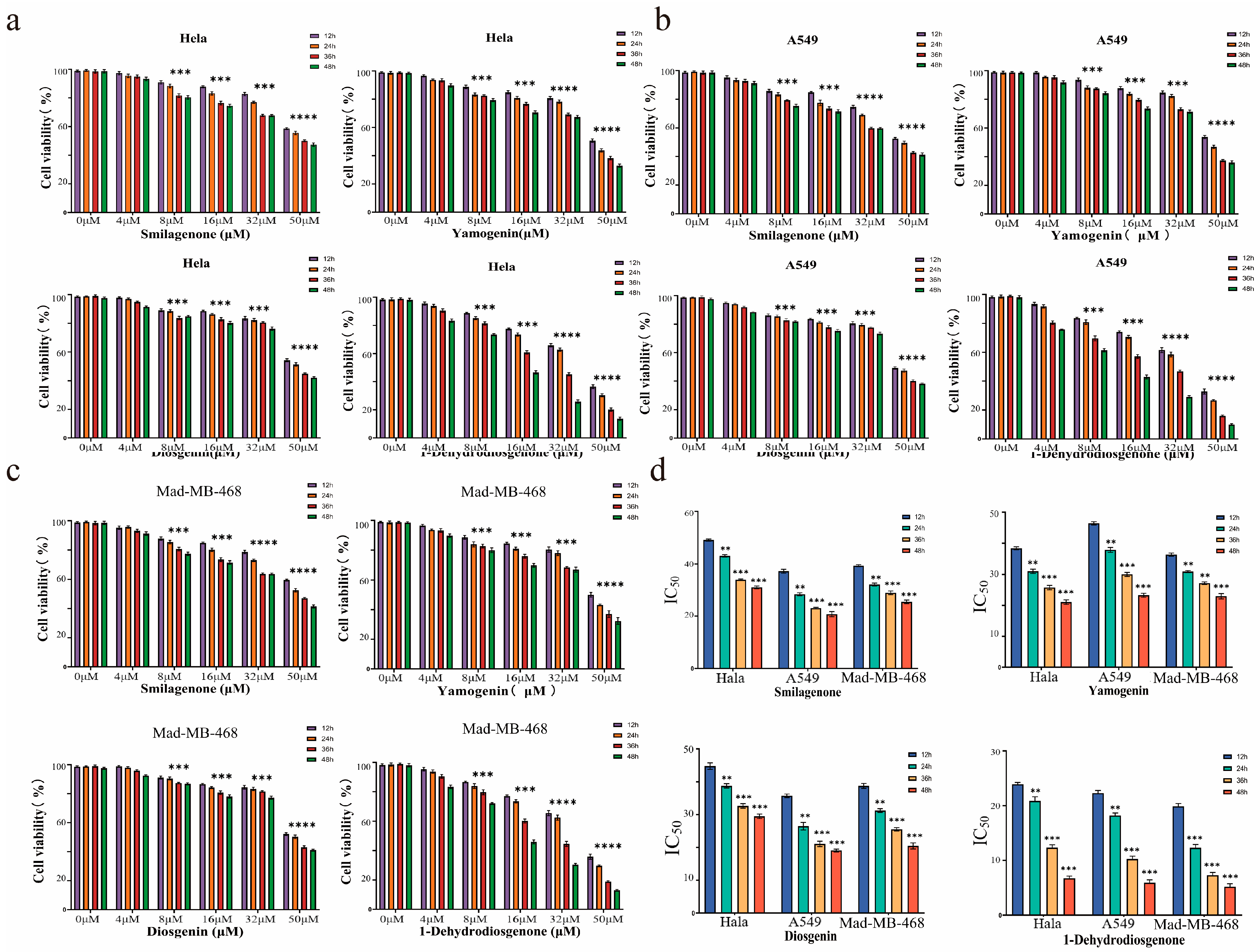
2.3.1. Compounds 1–4 Inhibit Proliferation of Three Cancer Cell Lines In Vitro
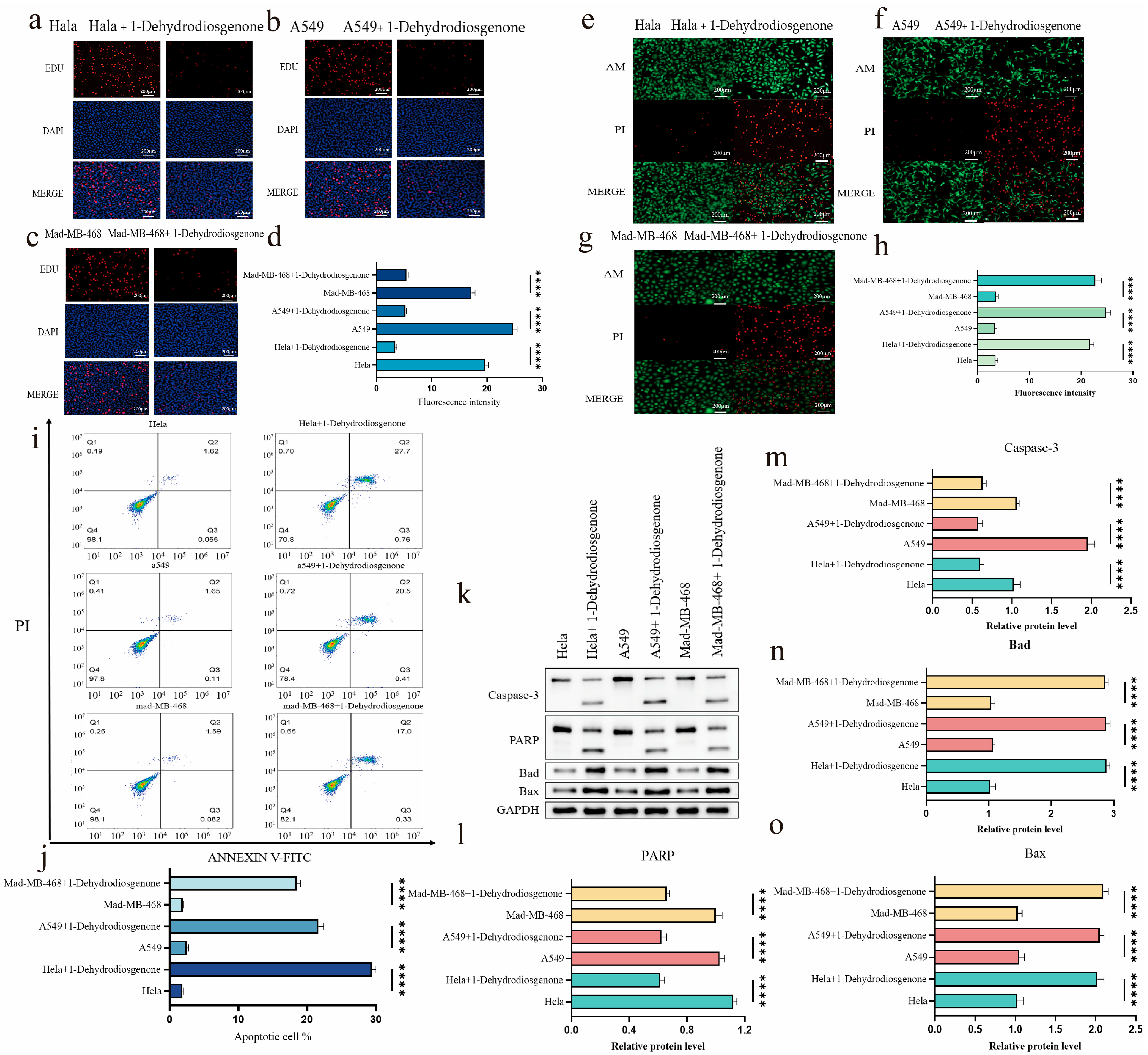
2.3.2. Effect of 1-Dehydrodiosgenone on Tumor Cell Proliferation
2.3.3. Inhibitory Effect of 1-Dehydrodiosgenone on Tumor Cells
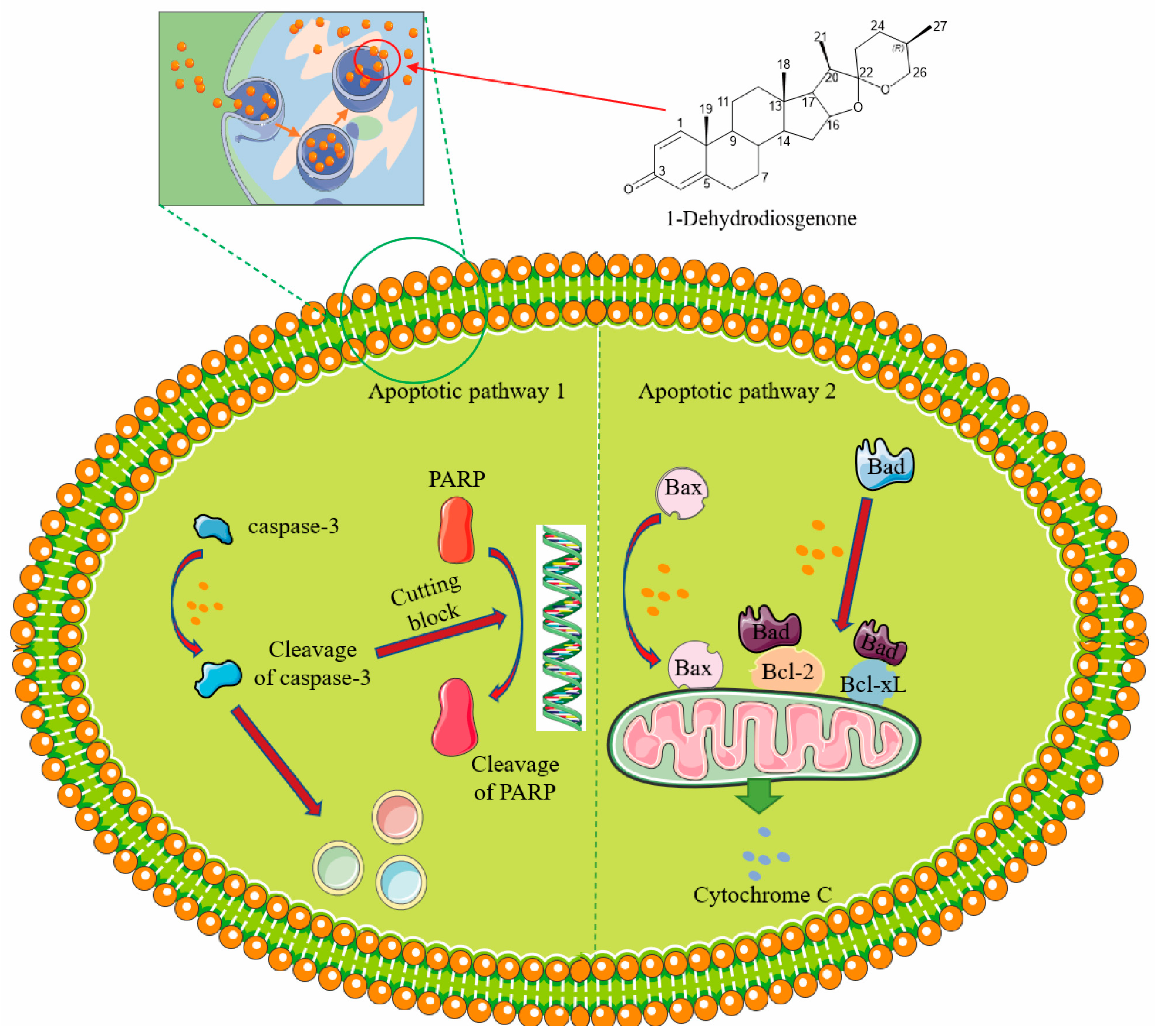
2.3.4. 1-Dehydrodiosgenone Upregulates Apoptosis-Related Proteins to Inhibit Cell Proliferation
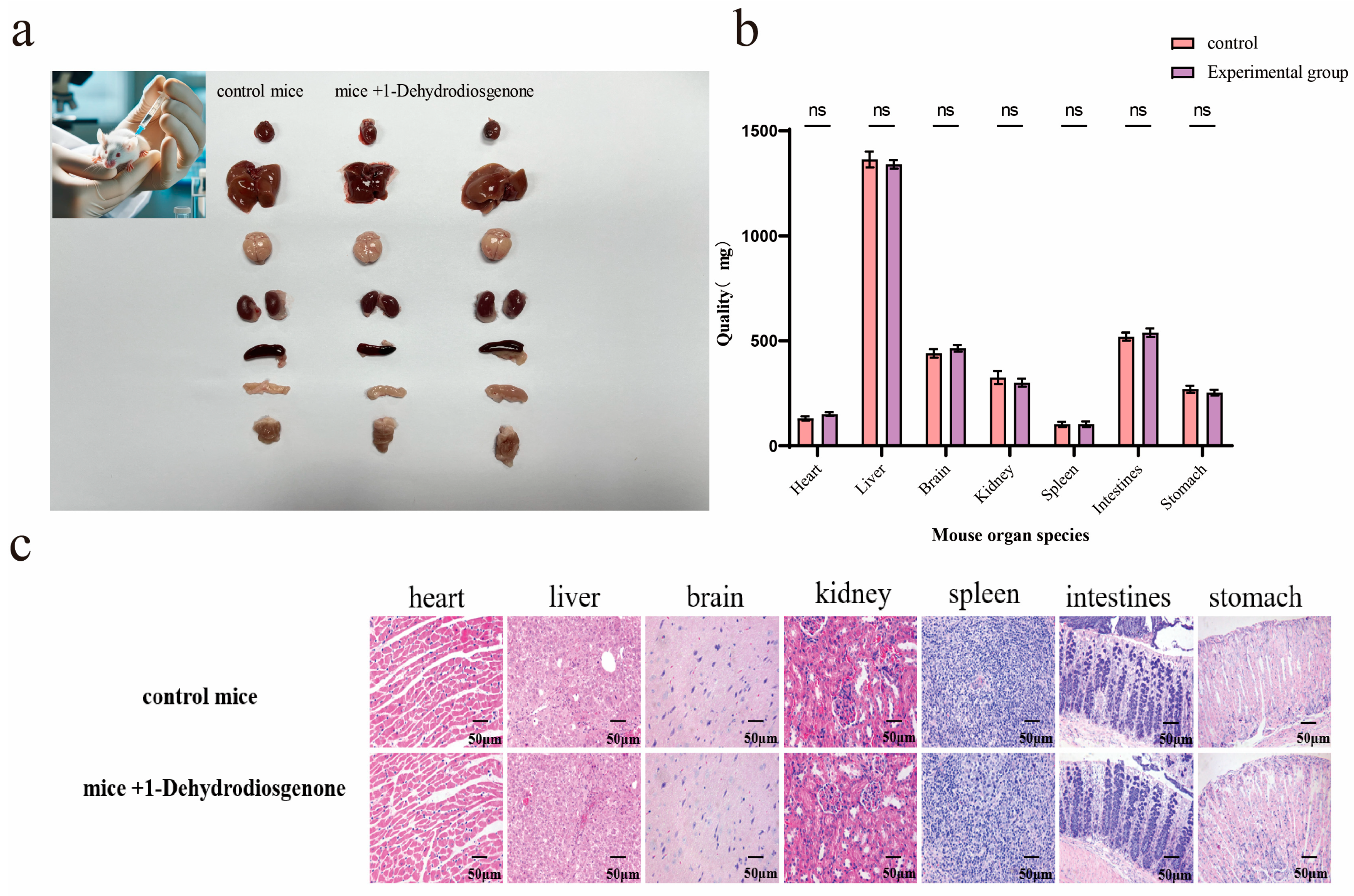
2.4. Histopathological Analysis of Mouse Organs
3. Discussion
4. Materials and Methods
4.1. Reagents and Strains Materials
4.2. Solid-State Fermentation and Extraction
4.3. Identification of Compounds
4.4. Single Crystal Analysis of Compounds 3 and 4
4.5. Anti-Tumor Activity Assay of Compounds
4.5.1. Cell Culture
4.5.2. Cytotoxicity Assay
4.5.3. Cell Proliferation Assay
4.5.4. Calcein AM/Propidium Iodide (AM/PI) Dual Staining
4.5.5. Apoptosis Detection
4.5.6. Western Blotting
4.6. Histopathological Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanderson, J.T. The Steroid Hormone Biosynthesis Pathway as a Target for Endocrine-Disrupting Chemicals. Toxicol. Sci. 2006, 94, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cabezon, L.; Galan, B.; Garcia, J.L. New Insights on Steroid Biotechnology. Front. Microbiol. 2018, 9, 958. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.A.; Carvalho, J.F.; Neves, M.A.; Silvestre, S.M.; Leitao, A.J.; Silva, M.M.; e Melo, M.L.S. Anticancer Steroids: Linking Natural and Semi-Synthetic Compounds. Nat. Prod. Rep. 2013, 30, 324–374. [Google Scholar] [CrossRef] [PubMed]
- Minorics, R.; Zupko, I. Steroidal Anticancer Agents: An Overview of Estradiol-Related Compounds. Anti-Cancer Agents Med. Chem. 2018, 18, 652–666. [Google Scholar] [CrossRef]
- Slominski, A.T.; Mahata, B.; Raman, C.; Bereshchenko, O. Editorial: Steroids and Secosteroids in the Modulation of Inflammation and Immunity. Front. Immunol. 2021, 12, 825577. [Google Scholar] [CrossRef]
- Kuo, T.; McQueen, A.; Chen, T.C.; Wang, J.C. Regulation of Glucose Homeostasis by Glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar]
- Sari, D.A.; Samodra, G.; Kusuma, I.Y. Molecular Mechanism of Glucocorticoid-Induced Hyperglycemia. Pharm. Rep. 2021, 1, 1. [Google Scholar] [CrossRef]
- Leite, P.M.; Amorim, J.M.; Castilho, R.O. Immunomodulatory Role of Terpenoids and Phytosteroids. In Plants and Phytomolecules for Immunomodulation; Springer Nature: Singapore, 2022; pp. 321–360. [Google Scholar]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Bizuayehu, H.M.; Ahmed, K.Y.; Kibret, G.D.; Dadi, A.F.; Belachew, S.A.; Bagade, T.; Tegegne, T.K.; Venchiarutti, R.L.; Kibret, K.T.; Hailegebireal, A.H.; et al. Global Disparities of Cancer and Its Projected Burden in 2050. JAMA Netw. Open 2024, 7, e2443198. [Google Scholar] [CrossRef]
- Shrestha, S.; Shankar, P.R.; Palaian, S.; Singaraju, M. Safety Profile and Toxicity Amelioration Strategies of Common Adverse Effects Associated with Anticancer Medications. J. Pharm. Res. Int. 2020, 32, 18–30. [Google Scholar]
- Beato, M.; Klug, J. Steroid Hormone Receptors: An Update. Hum. Reprod. Update 2000, 6, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Kundu, A.; Chakraborty, H.J.; Kar, B.; Choi, W.S.; Lee, B.M.; Bhakta, T.; Atanasov, A.G.; Kim, H.S. Therapeutic Value of Steroidal Alkaloids in Cancer: Current Trends and Future Perspectives. Int. J. Cancer 2019, 145, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Gahtori, R.; Tripathi, A.H.; Kumari, A.; Negi, N.; Paliwal, A.; Tripathi, P.; Joshi, P.; Rai, R.C.; Upadhyay, S.K. Anticancer Plant-Derivatives: Deciphering Their Oncopreventive and Therapeutic Potential in Molecular Terms. Future J. Pharm. Sci. 2023, 9, 14. [Google Scholar] [CrossRef]
- Upadhyay, S.; Jeena, G.S.; Shikha; Shukla, R.K. Recent Advances in Steroidal Saponins Biosynthesis and in Vitro Production. Planta 2018, 248, 519–544. [Google Scholar] [CrossRef]
- Pang, X.; Huang, H.Z.; Zhao, Y.; Xiong, C.-Q.; Yu, L.Y.; Ma, B.-P. Conversion of Furostanol Saponins into Spirostanol Saponins Improves the Yield of Diosgenin from Dioscorea Zingiberensis by Acid Hydrolysis. RSC Adv. 2015, 5, 4831–4837. [Google Scholar] [CrossRef]
- Larsson, D.G. Pollution from Drug Manufacturing: Review and Perspectives. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130571. [Google Scholar] [CrossRef]
- Yazdan, M.M.S.; Kumar, R.; Leung, S.W. The Environmental and Health Impacts of Steroids and Hormones in Wastewater Effluent, as Well as Existing Removal Technologies: A Review. Ecologies 2022, 3, 206–224. [Google Scholar] [CrossRef]
- Pan, C.; Zhao, Y.; Liu, G.; Dou, G.; Ru, Z.; Zhu, K. Development and Demonstration of a Cleaner Process to Produce Diosgenin from Dioscorea Zingiberensis Based on Physical Separation. J. Clean. Prod. 2014, 76, 161–166. [Google Scholar] [CrossRef]
- Wang, P.; Ma, C.; Chen, S.; Zhu, S.; Lou, Z.; Wang, H. Conversion of Steroid Saponins into Diosgenin by Catalytic Hydrolysis Using Acid-Functionalized Ionic Liquid under Microwave Irradiation. J. Clean. Prod. 2014, 79, 265–270. [Google Scholar] [CrossRef]
- Zhao, W.-Y.; Hao, H.-Q.; Xiao, W.; Zhu, Q.-S. Subcritical Water Extraction of Diosgenin from Dioscorea Nipponica Makino and Its Antioxidant Activity. IOP Conf. Ser. Earth Environ. Sci. 2020, 559, 012004. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, F.; Zhao, M.; Pan, Z.; Cheng, Q.; Zhou, H. Synthesis and Characterization of Magnetic Solid Acid Fe3o4@Pei@So3h and Application for the Production of Diosgenin by Alcoholysis of Turmeric Saponins. Mol. Catal. 2021, 511, 111751. [Google Scholar] [CrossRef]
- He, Y.; Hu, Z.; Li, A.; Zhu, Z.; Yang, N.; Ying, Z.; He, J.; Wang, C.; Yin, S.; Cheng, S. Recent Advances in Biotransformation of Saponins. Molecules 2019, 24, 2365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Guo, Y.; Xue, P.; Xue, Z.; Zhang, Y.; Zhang, H.; Ito, Y.; Dou, J.; Guo, Z. Research Progress of Diosgenin Extraction from Dioscorea zingiberensis C. H. Wright: Inspiration of Novel Method with Environmental Protection and Efficient Characteristics. Steroids 2023, 192, 109181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, X.; Xiong, L.; Liu, K.; Liu, Y.; Xue, Z.; Han, R. Green Manufacturing of Steroids Via Mycolicbacteria: Current Status and Development Trends. Fermentation 2023, 9, 890. [Google Scholar] [CrossRef]
- Zappaterra, F.; Costa, S.; Summa, D.; Bertolasi, V.; Semeraro, B.; Pedrini, P.; Buzzi, R.; Vertuani, S. Biotransformation of Cortisone with Rhodococcus Rhodnii: Synthesis of New Steroids. Molecules 2021, 26, 1352. [Google Scholar] [CrossRef]
- Feng, J.; Wu, Q.; Zhu, D.; Ma, Y. Biotransformation Enables Innovations toward Green Synthesis of Steroidal Pharmaceuticals. ChemSusChem 2022, 15, e202102399. [Google Scholar] [CrossRef]
- Zhang, H.; Ren, J.; Wang, Y.; Sheng, C.; Wu, Q.; Diao, A.; Zhu, D. Effective Multi-Step Functional Biotransformations of Steroids by a Newly Isolated Fusarium Oxysporum Sc1301. Tetrahedron 2013, 69, 184–189. [Google Scholar] [CrossRef]
- Pandey, A. Solid-State Fermentation. Biochem. Eng. J. 2003, 13, 81–84. [Google Scholar] [CrossRef]
- Meruvu, H.; Dos Santos, J.C. Colors of Life: A Review on Fungal Pigments. Crit. Rev. Biotechnol. 2021, 41, 1153–1177. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, S.; Mariano-Silva, G.; Leite, M.O.; Mura, F.B.; Verma, M.L.; da Silva, S.S.; Chandel, A.K. Production of Fungal and Bacterial Pigments and Their Applications. In Biotechnological Production of Bioactive Compounds; Elsevier: Amsterdam, The Netherlands, 2020; pp. 327–361. [Google Scholar]
- Haraguchi, M.; Zaccarias Dos Santos, A.P.; Claudia Marx Young, M.; Paulo Chui, E. Steroidal Prosapogenins from Dioscorea olfersiana. Phytochemistry 1994, 36, 1005–1008. [Google Scholar] [CrossRef]
- Viñas-Bravo, O.; Merino-Montiel, P.; Romero-López, A.; Montiel-Smith, S.; Meza-Reyes, S.; Meléndez, F.J.; Sandoval-Ramírez, J. Epimerization of C-22 in (25r)- and (25s)-Sapogenins. Steroids 2015, 93, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Blunden, G.; Jaffer, J.A.; Jewers, K.; Griffin, W.J. Steroidal Sapogenins from Leaves of Cordyline Species. J. Nat. Prod. 2004, 44, 441–447. [Google Scholar] [CrossRef]
- Blunden, G.; Jaffer, J.A.; Jewers, K.; Griffin, W.J. Epi-Neotigogenin and Epi-Tigogenin, Two New Steroidal Sapogenins from Cordyline Cannifolia Leaves. J. Nat. Prod. 2004, 42, 478–482. [Google Scholar] [CrossRef]
- Gnoumou, E.; Kongpichitchoke, T.; Jose, A. Effect of Solid-State Fermentation (Ssf) on the Antioxidant and Hypoglycemic Activities of Jackfruit (Artocarpus Heterophyllus Lam.) by-Products Using Aspergillus Niger, Aspergillus Oryzae and Rhizopus Oryzae. Trends Sci. 2023, 20, 5648. [Google Scholar] [CrossRef]
- Slaný, O.; Klempová, T.; Marcinčák, S.; Čertík, M. Production of High-Value Bioproducts Enriched with Γ-Linolenic Acid and Β-Carotene by Filamentous Fungi Umbelopsis isabellina Using Solid-State Fermentations. Ann. Microbiol. 2020, 70, 5. [Google Scholar] [CrossRef]
- Dewi, R.T.; Artanti, N.; Mulyani, H.; Lotulung, P.D.N.; Minarti, M. Production of Lovastatin and Sulochrin by Aspergillus Terreus Using Solid State Fermentation. Makara Technol. Ser. 2011, 15, 1. [Google Scholar] [CrossRef]
- Liu, W.; Xiang, H.; Zhang, T.; Pang, X.; Su, J.; Liu, H.; Ma, B.; Yu, L. Screening and Selection of a New Medium for Diosgenin Production Via Microbial Biocatalysis of Fusarium Sp. Pharmaceuticals 2021, 14, 390. [Google Scholar] [CrossRef]
- Huang, N.; Yu, D.; Huo, J.; Wu, J.; Chen, Y.; Du, X.; Wang, X. Study of Saponin Components after Biotransformation of Dioscorea Nipponica by Endophytic Fungi C39. J. Anal. Methods Chem. 2022, 2022, 2943177. [Google Scholar] [CrossRef]
- Cai, B.; Liao, A.; Lee, K.-K.; Ban, J.-S.; Yang, H.-S.; Im, Y.J.; Chun, C. Design, Synthesis of Methotrexate-Diosgenin Conjugates and Biological Evaluation of Their Effect on Methotrexate Transport-Resistant Cells. Steroids 2016, 116, 45–51. [Google Scholar] [CrossRef]
- Webb, C. Design Aspects of Solid State Fermentation as Applied to Microbial Bioprocessing. J. Appl. Biotechnol. Bioeng. 2017, 4, 511–532. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in Cancer: From Pathogenesis to Treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Shanmugam, M.K.; Warrier, S.; Merarchi, M.; Arfuso, F.; Kumar, A.P.; Bishayee, A. Pro-Apoptotic and Anti-Cancer Properties of Diosgenin: A Comprehensive and Critical Review. Nutrients 2018, 10, 645. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Hempel, A.; Coll, N.S. Protease Signaling in Animal and Plant-Regulated Cell Death. FEBS J. 2016, 283, 2577–2598. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Ramirez, M.L.G.; Salvesen, G.S. A Primer on Caspase Mechanisms. Semin. Cell Dev. Biol. 2018, 82, 79–85. [Google Scholar] [CrossRef]
- Porter, A.G.; Janicke, R.U. Emerging Roles of Caspase-3 in Apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Lyons, A.B.; Hasbold, J.; Hodgkin, P.D. Flow Cytometric Analysis of Cell Division History Using Dilution of Carboxyfluorescein Diacetate Succinimidyl Ester, a Stably Integrated Fluorescent Probe. Methods Cell Biol. 2001, 63, 375–398. [Google Scholar]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of Poly(Adp-Ribose) Polymerase (Parp) Cleavage in Apoptosis. Caspase 3-Resistant Parp Mutant Increases Rates of Apoptosis in Transfected Cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Sayers, T.J. Targeting the Extrinsic Apoptosis Signaling Pathway for Cancer Therapy. Cancer Immunol. Immunother. 2011, 60, 1173–1180. [Google Scholar] [CrossRef]
- Alexander, B.L.; Ali, R.R.; Alton, E.W.; Bainbridge, J.W.; Braun, S.; Cheng, S.H.; Flotte, T.R.; Gaspar, H.B.; Grez, M.; Griesenbach, U.; et al. Progress and Prospects: Gene Therapy Clinical Trials (Part 1). Gene Ther. 2007, 14, 1439–1447. [Google Scholar] [PubMed]
- Hetz, C.; Vitte, P.-A.; Bombrun, A.; Rostovtseva, T.K.; Montessuit, S.; Hiver, A.; Schwarz, M.K.; Church, D.J.; Korsmeyer, S.J.; Martinou, J.-C.; et al. Bax Channel Inhibitors Prevent Mitochondrion-Mediated Apoptosis and Protect Neurons in a Model of Global Brain Ischemia. J. Biol. Chem. 2005, 280, 42960–42970. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, S.; Urooj, M.; Saleem, S.; Gillani, Z.; Shaheen, S.; Qazi, M.H.; Naseer, M.I.; Iqbal, Z.; Ansari, S.A.; Haque, A.; et al. Bad, a Proapoptotic Protein, Escapes Erk/Rsk Phosphorylation in Deguelin and Sirna-Treated Hela Cells. PLoS ONE 2016, 11, e0145780. [Google Scholar] [CrossRef] [PubMed]
- Institute of Laboratory Animal Resources (US); Committee on Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals; US Department of Health and Human Services, Public Health Service, National Institutes of Health: Bethesda, MD, USA, 1986. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound 1 | Compound 2 | Compound 3 | Compound 4 |
|---|---|---|---|---|
| 1 | 37.3 | 37.6 | 37.3 | 155.9 |
| 2 | 31.5 | 32.4 | 37.1 | 124.0 |
| 3 | 71.9 | 72.1 | 213.4 | 186.4 |
| 4 | 42.4 | 42.7 | 42.5 | 127.6 |
| 5 | 140.9 | 141.2 | 44.3 | 169.2 |
| 6 | 121.6 | 121.8 | 26.2 | 33.8 |
| 7 | 32.2 | 32.2 | 26.6 | 32.0 |
| 8 | 31.6 | 32.0 | 35.3 | 35.3 |
| 9 | 50.2 | 50.4 | 40.9 | 52.5 |
| 10 | 36.8 | 37.0 | 35.1 | 43.7 |
| 11 | 21.0 | 21.3 | 21.1 | 22.8 |
| 12 | 39.9 | 40.7 | 40.2 | 39.6 |
| 13 | 40.4 | 40.2 | 40.8 | 40.8 |
| 14 | 56.6 | 56.9 | 56.4 | 55.3 |
| 15 | 32.0 | 31.8 | 31.9 | 32.9 |
| 16 | 81.0 | 81.2 | 80.9 | 80.6 |
| 17 | 62.2 | 62.5 | 62.3 | 62.1 |
| 18 | 16.4 | 16.7 | 16.6 | 16.6 |
| 19 | 19.6 | 21.3 | 22.8 | 18.9 |
| 20 | 41.7 | 42.0 | 41.7 | 41.7 |
| 21 | 14.7 | 14.9 | 14.6 | 14.6 |
| 22 | 109.5 | 109.7 | 109.4 | 109.4 |
| 23 | 31.7 | 31.8 | 31.5 | 31.4 |
| 24 | 67.0 | 29.2 | 28.9 | 28.9 |
| 25 | 30.4 | 30.7 | 30.4 | 30.4 |
| 26 | 28.9 | 67.2 | 67.0 | 67.0 |
| 27 | 17.3 | 17.5 | 17.3 | 17.2 |
| No. | Compound 1 | Compound 2 | Compound 3 | Compound 4 |
|---|---|---|---|---|
| 1 | a 1.83 m b 1.05 m | a 0.99 m b 1.82 m | a 2.32 dd, (14.7, 5.4) b 1.38 m | 7.03 d (10.1) |
| 2 | 1.62 (m) | a 1.43 m b 1.97 m | a 2.17 m b 2.02 m | 6.06 overlap |
| 3 | 3.52 m | 3.44 m | ||
| 4 | a 2.28 m b 2.22 m | a 2.27 m b 2.21 m | a 2.68 dd (15.1, 13.3) b 2.02 m | 6.22 dd (10.1, 1.9) |
| 5 | 1.78 m | |||
| 6 | 5.33 overlap | 5.33 overlap | a 1.51 m b 1.09 m | a 1.93 m b 1.03 m |
| 7 | 1.96 m | a 1.25 m b 1.43 m | a 1.89 m b 1.25 m | a 1.99 m b 1.31 m |
| 8 | 1.64 m | 1.56 m | 1.65 m | 1.79 m |
| 9 | 0.93 m | 0.87 m | 1.46 m | 1.06 m |
| 10 | ||||
| 11 | 1.49 m | a 1.54 m b 1.61 m | 1.45 m | 1.66 m |
| 12 | a 1.72 m b 1.16 m | a 1.09 m b 1.24 m | a 1.74 m b 1.20 m | a 1.74 m b 1.15 m |
| 13 | ||||
| 14 | 1.08 m | 1.02 m | 1.20 m | 1.06 m |
| 15 | a 1.83 m b 1.27 m | a 1.64 m b 1.82 m | a 2.02 m b 1.25 m | a 2.48 tdd (13.5, 5.1, 1.6) b 2.35 ddd (13.3, 4.4, 2.5) |
| 16 | 4.40 dd (14.4, 7.8) | 4.39 dd (14.4, 7.8) | 4.41 dd (14.3, 7.9) | 4.39 dd (14.9, 7.6) |
| 17 | 1.76 m | 1.85 m | 1.78 m | 1.74 m |
| 18 | 0.78 s | 0.79 s | 0.79 s | 0.84 s |
| 19 | 1.02 s | 0.96 s | 1.03 s | 1.24 s |
| 20 | 1.85 m | 1.97 m | 1.86 m | 1.85 m |
| 21 | 0.96 d (7.0) | 0.98 d (7.0) | 0.97 d (6.9) | 0.96 d (6.9) |
| 22 | ||||
| 23 | a 1.61 m b 1.50 m | a 1.39 m b 1.59 m | 1.62 m | a 1.99 m b 1.60 m |
| 24 | a 3.46 ddd (10.9, 4.6, 2.1) b 3.36 (t, 10.9) | a 1.37 m b 1.56 m | a 1.62 m b 1.45 m | a 1.60 m b 1.11 m |
| 25 | 1.60 m | 1.61 m | 1.62 m | 1.60 m |
| 26 | a 1.60 m b 1.45 m | a 3.37 m b 3.31 m | a 3.48 ddd (10.9, 4.5, 2.1) b 3.37 t (10.9) | a 3.46 dd (11.0, 2.9) b 3.35 t (11.0) |
| 27 | 0.78 d (6.4) | 0.79 d (6.4) | 0.79 d (6.3) | 0.78 d (6.4) |
| IC50 (μM) | ||||
|---|---|---|---|---|
| Time | Compounds | Hala | A549 | Mad-MB-468 |
| 12 h | Diosgenin | 43.83 ± 1.05 | 36.24 ± 1.73 | 38.98 ± 1.35 |
| Yamogenin | 38.54 ± 0.96 | 46.58 ± 0.81 | 36.46 ± 1.17 | |
| Smilagenone | 48.93 ± 1.43 | 37.56 ± 0.54 | 39.76 ± 1.24 | |
| 1-Dehydrodiosgenone | 24.34 ± 1.12 | 22.76 ± 0.98 | 20.42 ± 0.57 | |
| 24 h | Diosgenin | 38.24 ± 1.06 | 27.76 ± 1.22 | 31.67 ± 0.53 |
| Yamogenin | 30.25 ± 2.13 | 38.76 ± 1.37 | 30.79 ± 0.71 | |
| Smilagenone | 42.87 ± 0.73 | 28.34 ± 0.76 | 31.76 ± 0.83 | |
| 1-Dehydrodiosgenone | 20.34 ± 1.45 | 18.49 ± 1.42 | 12.48 ± 1.36 | |
| 36 h | Diosgenin | 31.89 ± 1.37 | 20.19 ± 0.55 | 25.63 ± 1.75 |
| Yamogenin | 25.46 ± 2.23 | 30.76 ± 0.76 | 27.56 ± 0.85 | |
| Smilagenone | 33.76 ± 0.92 | 23.19 ± 1.19 | 28.46 ± 0.97 | |
| 1-Dehydrodiosgenone | 12.46 ± 0.74 | 10.76 ± 1.76 | 7.83 ± 0.83 | |
| 48 h | Diosgenin | 28.76 ± 0.57 | 18.75 ± 2.13 | 20.67 ± 0.73 |
| Yamogenin | 21.43 ± 1.07 | 23.57 ± 1.20 | 23.88 ± 0.49 | |
| Smilagenone | 30.43 ± 2.43 | 20.76 ± 0.59 | 25.76 ± 1.21 | |
| 1-Dehydrodiosgenone | 6.59 ± 1.75 | 5.43 ± 0.77 | 4.81 ± 1.26 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Feng, S.; Zhang, Y.; Mou, F.; Guo, T.; Qin, B.; Liu, Y. Potent Anti-Cancer Activity of 1-Dehydrodiosgenone from the Product of Microbial Transformation of Steroid Saponins. Int. J. Mol. Sci. 2024, 25, 13118. https://doi.org/10.3390/ijms252313118
Li Q, Feng S, Zhang Y, Mou F, Guo T, Qin B, Liu Y. Potent Anti-Cancer Activity of 1-Dehydrodiosgenone from the Product of Microbial Transformation of Steroid Saponins. International Journal of Molecular Sciences. 2024; 25(23):13118. https://doi.org/10.3390/ijms252313118
Chicago/Turabian StyleLi, Quanshun, Shirong Feng, Yuanyuan Zhang, Fangyuan Mou, Ting Guo, Baofu Qin, and Yihan Liu. 2024. "Potent Anti-Cancer Activity of 1-Dehydrodiosgenone from the Product of Microbial Transformation of Steroid Saponins" International Journal of Molecular Sciences 25, no. 23: 13118. https://doi.org/10.3390/ijms252313118
APA StyleLi, Q., Feng, S., Zhang, Y., Mou, F., Guo, T., Qin, B., & Liu, Y. (2024). Potent Anti-Cancer Activity of 1-Dehydrodiosgenone from the Product of Microbial Transformation of Steroid Saponins. International Journal of Molecular Sciences, 25(23), 13118. https://doi.org/10.3390/ijms252313118