

Mitochondrial and Cellular Function in Fibroblasts, Induced Neurons, and Astrocytes Derived from Case Study Patients: Insights into Major Depression as a Mitochondria-Associated Disease

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
1.1. Epidemiology of MDD
1.2. Pathophysiological Hypotheses
1.3. Mitochondrial Dysfunction in MDD
1.4. Mitochondrial Diseases
1.5. Mitochondrial Diseases and Psychiatric Illnesses
1.6. Novel Cellular Model Approach
1.7. Complementing Cohort Studies with Case Studies
1.8. Presentation of the Case Study Patients
2. Results
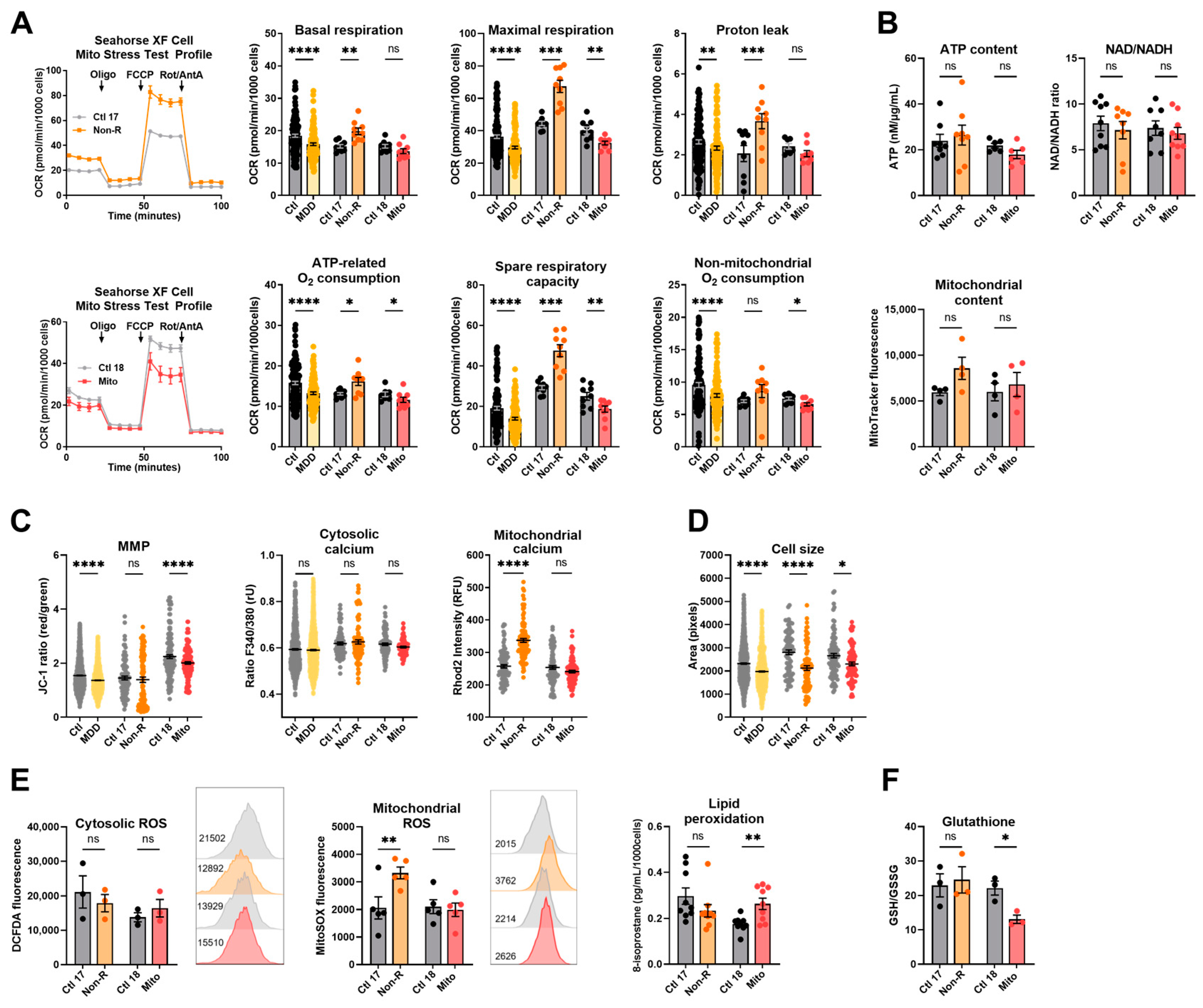
2.1. Fibroblasts from the Antidepressant Non-Responder (Non-R) and the Mitochondriopathy Patient (Mito) Show Altered Bioenergetic Properties and Calcium Homeostasis
2.1.1. Bioenergetic Properties
2.1.2. Calcium Homeostasis and Cell Size
2.2. Redox Homeostasis Is Partly Affected in AD Non-R and Mito Patients’ Fibroblasts
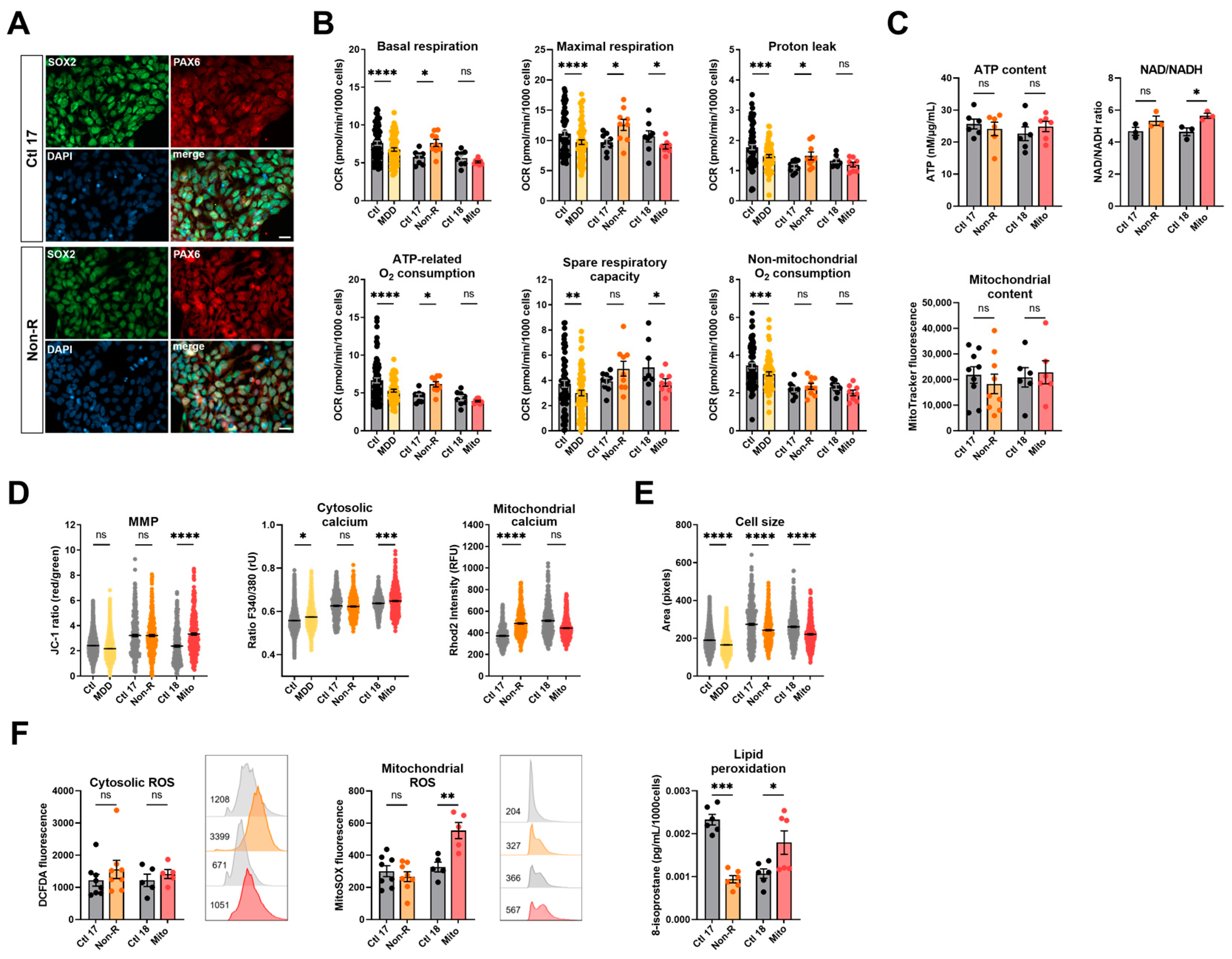
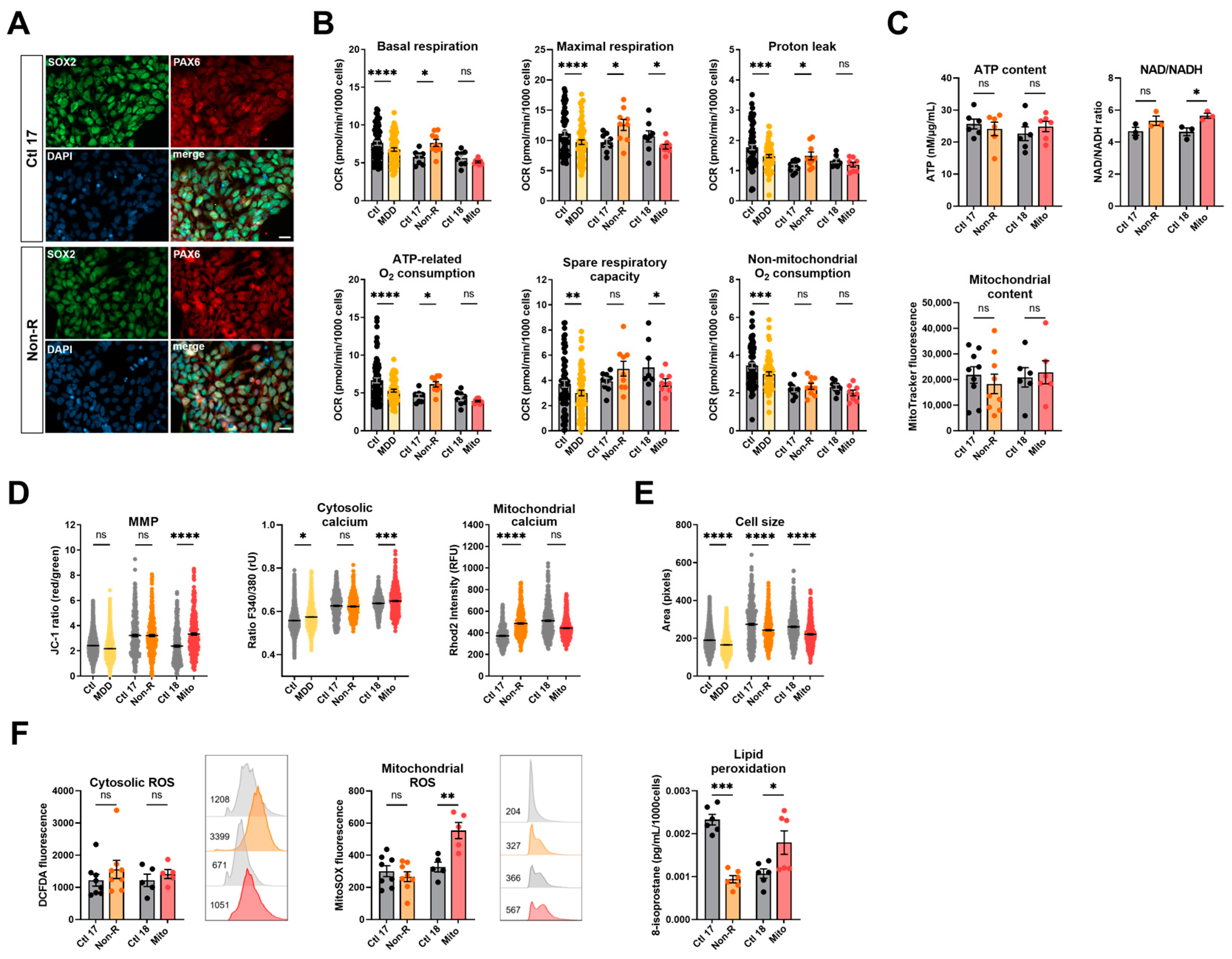
2.3. Induced Neural Progenitor Cells of Patients Show Alterations in Bioenergetic Properties
2.3.1. Bioenergetic Properties
2.3.2. Calcium Homeostasis and Cell Size
2.3.3. Redox Homeostasis
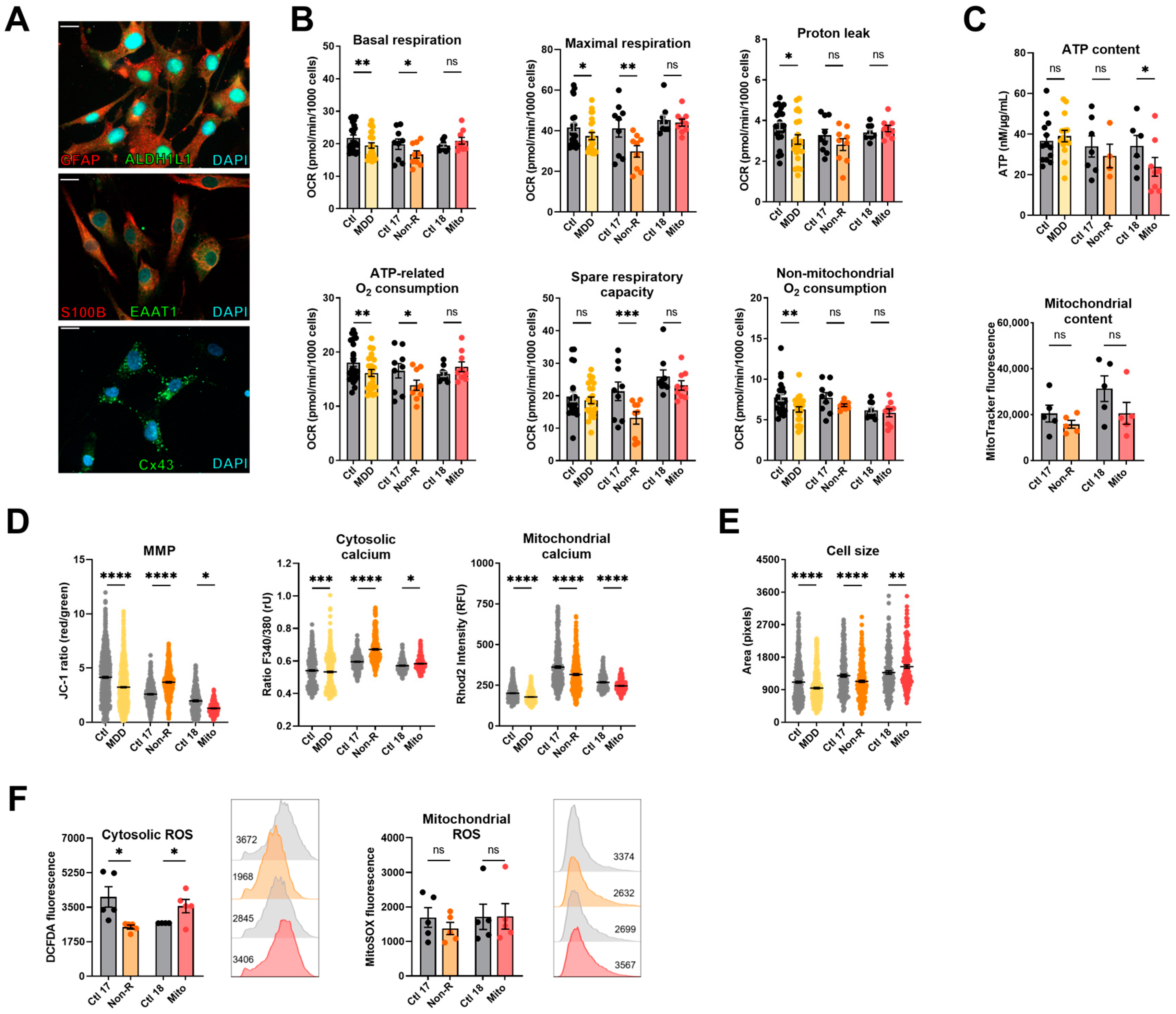
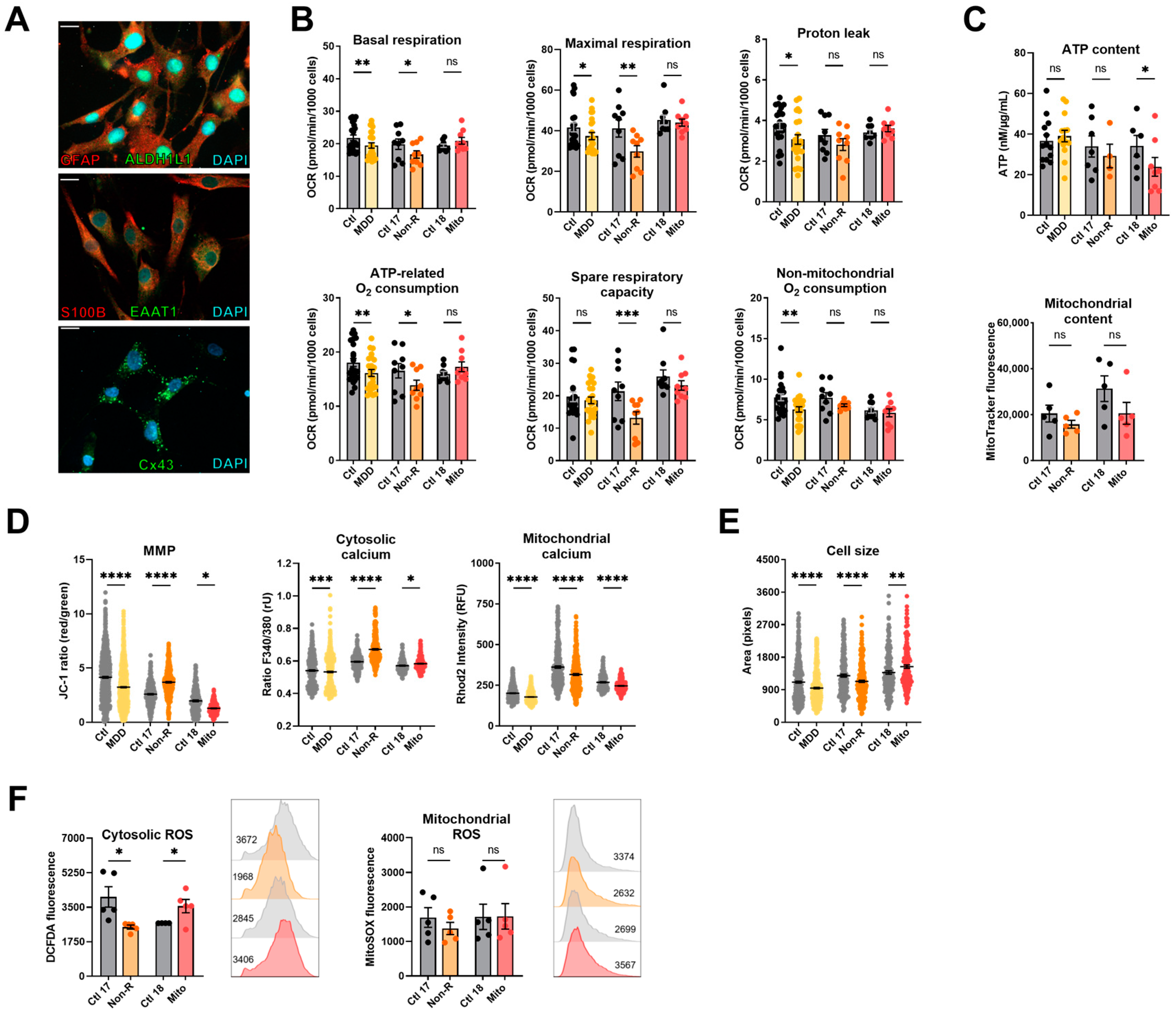
2.4. Induced Astrocytes of Patients Show Altered Bioenergetic Properties and Oxidative Stress
2.4.1. Bioenergetic Properties
2.4.2. Calcium Homeostasis and Cell Size
2.4.3. Redox Homeostasis
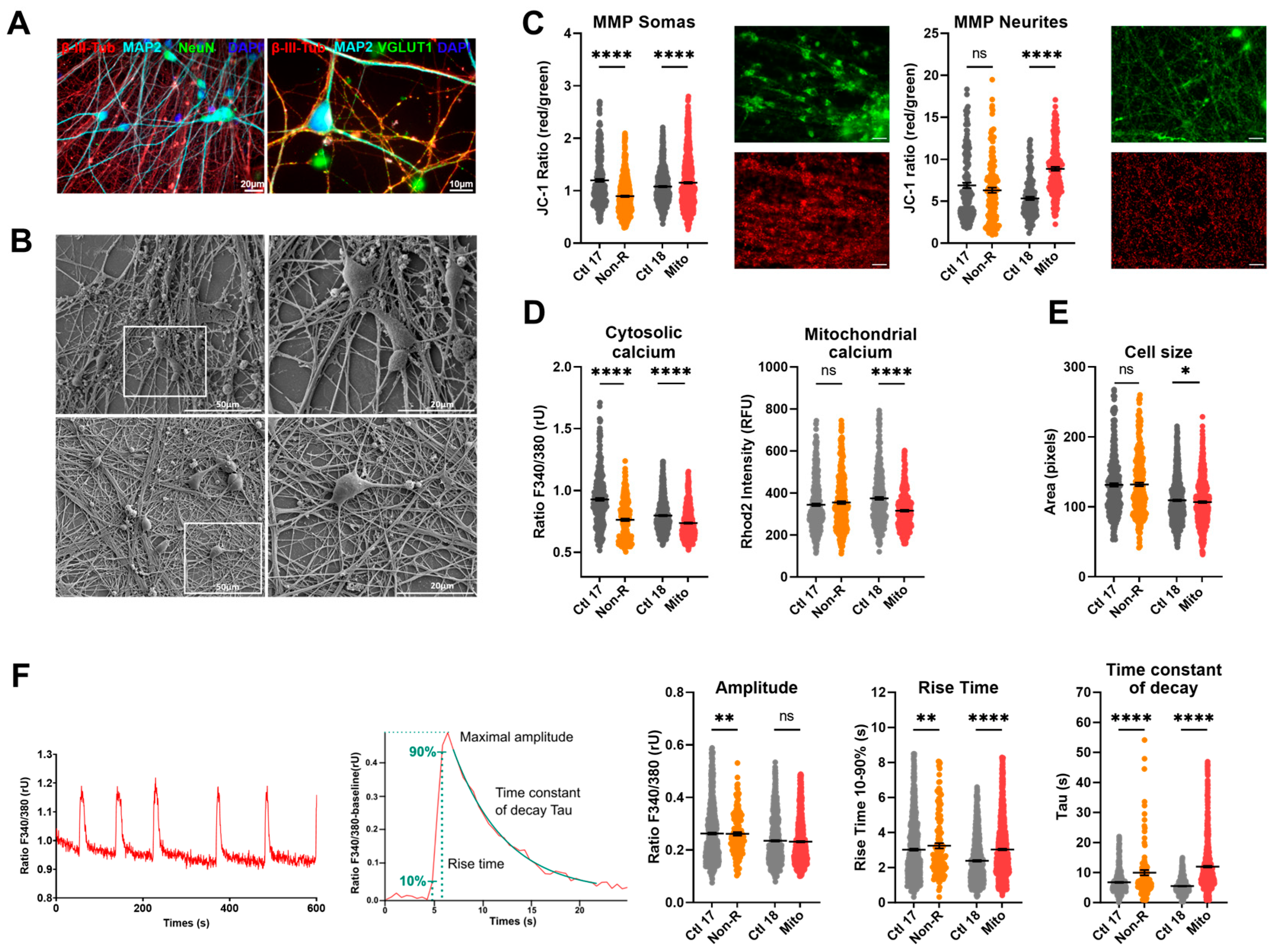
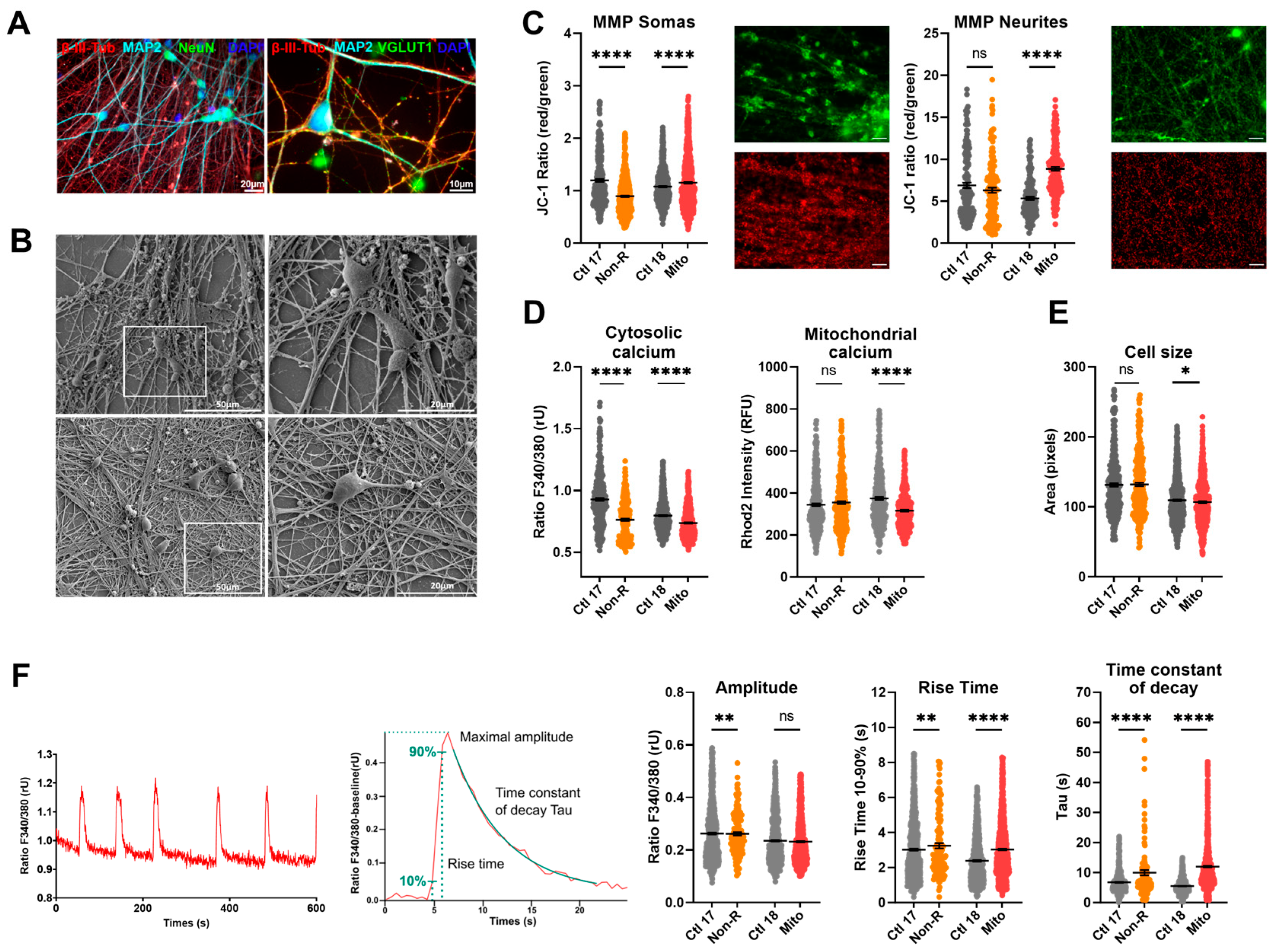
2.5. Neurons of Patients Show Altered Mitochondrial Membrane Potential and Calcium Homeostasis
2.5.1. Characterization of Neuronal Identity
2.5.2. Mitochondrial Membrane Potential
2.5.3. Calcium Homeostasis
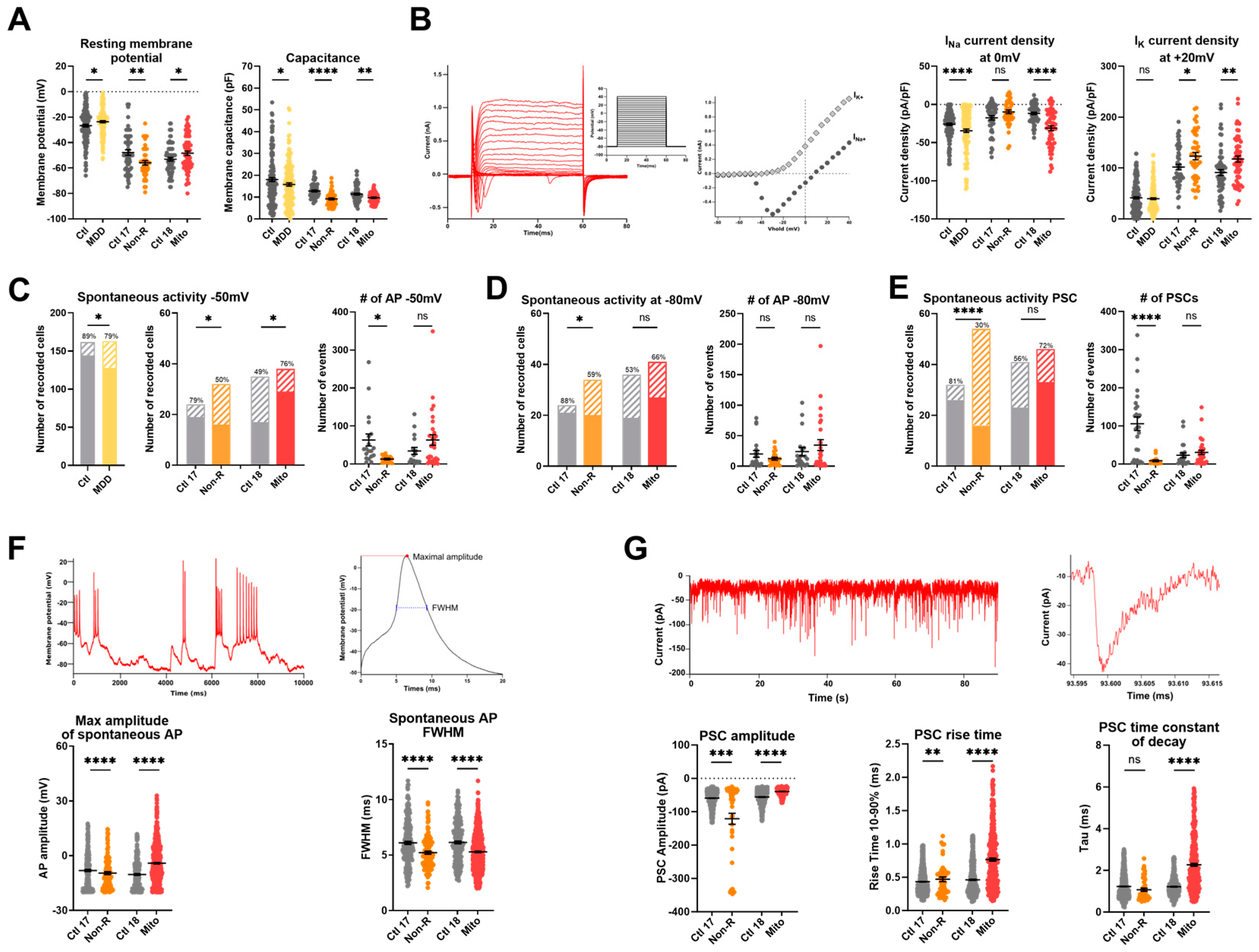
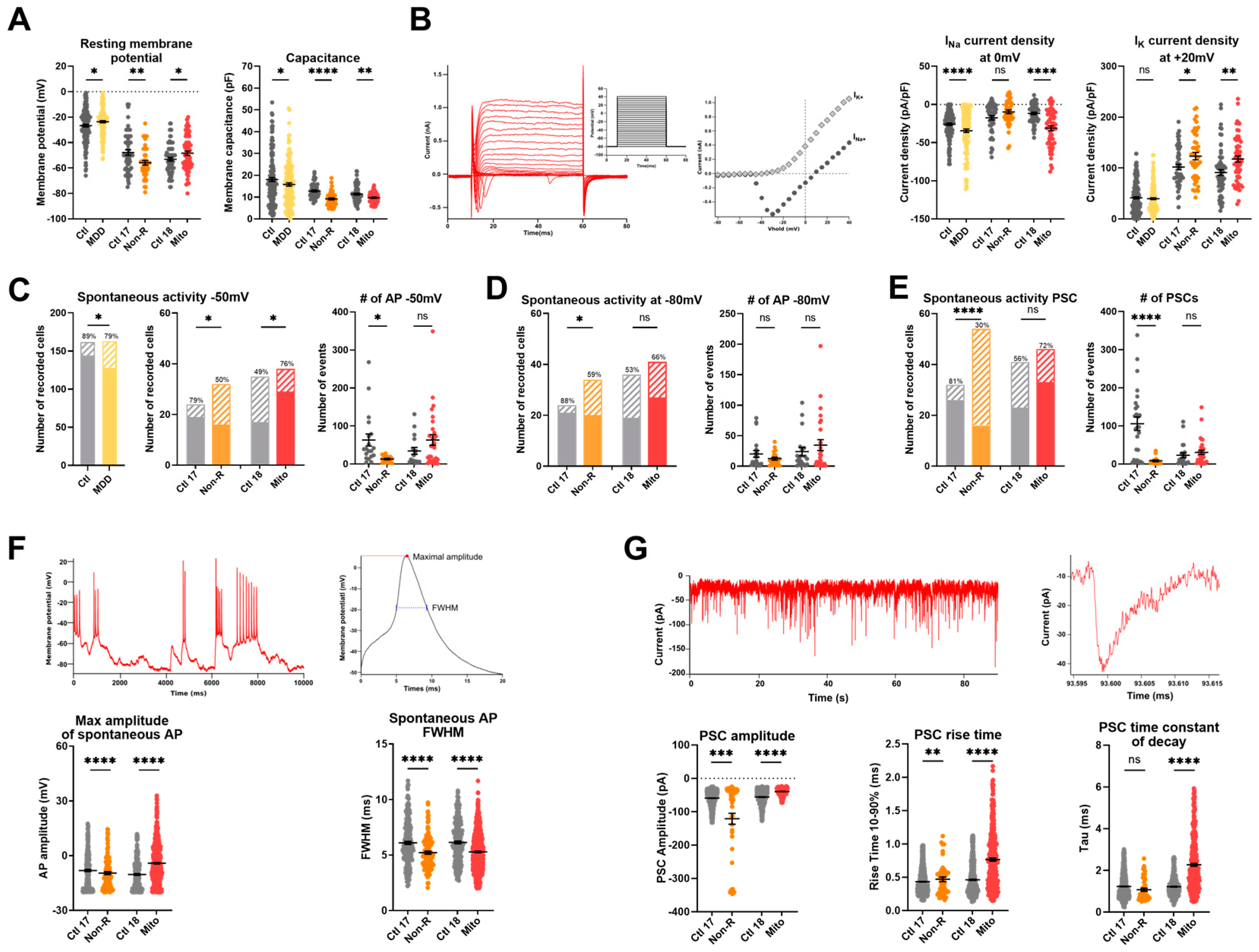
2.6. Functional Properties and the Activity of Patient-Derived Neurons Are Altered
2.6.1. Resting Membrane Potential and Capacitance
2.6.2. Sodium and Potassium Current Densities
2.6.3. Spontaneous Action Potentials
2.6.4. Spontaneous Post-Synaptic Currents
2.6.5. Spontaneous Calcium Activity
3. Discussion
4. Limitations
5. Materials and Methods
5.1. Generation of Control and MDD Patient iPSCs from Fibroblasts
5.2. iPSC Differentiation to NPCs and Neuron Differentiation
5.3. Astrocyte Differentiation
5.4. Analysis of Mitochondrial Respiration
5.5. Immunofluorescence
5.6. Luminescent Assay for ATP Content
5.7. Luminescent Assay for NAD/NADH Ratio
5.8. Luminescent Assay for GSH/GSSG Ratio
5.9. Lipid Peroxidation ELISA
5.10. Imaging of Mitochondrial Membrane Potential (JC-1) Cytosolic Ca2+ (Fura-2/AM) and Mitochondrial Ca2+ (Rhod-2/AM)
5.11. Flow Cytometry (MitoTracker Green, DCFDA, MitoSOX)
5.12. Electrophysiology
5.13. Statistical Analysis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Mental Health Report: Transforming Mental Health for All; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Lopizzo, N.; Bocchio Chiavetto, L.; Cattane, N.; Plazzotta, G.; Tarazi, F.I.; Pariante, C.M.; Riva, M.A.; Cattaneo, A. Gene-environment interaction in major depression: Focus on experience-dependent biological systems. Front. Psychiatry 2015, 6, 68. [Google Scholar] [CrossRef]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e19. [Google Scholar] [CrossRef]
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The Role of Neural Plasticity in Depression: From Hippocampus to Prefrontal Cortex. Neural Plast. 2017, 2017, 6871089. [Google Scholar] [CrossRef]
- Fava, M.; Rush, A.J. Current status of augmentation and combination treatments for major depressive disorder: A literature review and a proposal for a novel approach to improve practice. Psychother. Psychosom. 2006, 75, 139–153. [Google Scholar] [CrossRef]
- El-Hage, W.; Leman, S.; Camus, V.; Belzung, C. Mechanisms of antidepressant resistance. Front. Pharmacol. 2013, 4, 146. [Google Scholar] [CrossRef]
- Sluzewska, A. Indicators of Immune Activation in Depressed Patients. In Cytokines, Stress, and Depression; Dantzer, R., Wollman, E.E., Yirmiya, R., Eds.; Springer: New York, NY, USA, 1999; pp. 59–73. [Google Scholar]
- Maydych, V. The Interplay Between Stress, Inflammation, and Emotional Attention: Relevance for Depression. Front. Neurosci. 2019, 13, 384. [Google Scholar] [CrossRef]
- Vreeburg, S.A.; Hoogendijk, W.J.G.; van Pelt, J.; DeRijk, R.H.; Verhagen, J.C.M.; van Dyck, R.; Smit, J.H.; Zitman, F.G.; Penninx, B.W.J.H. Major Depressive Disorder and Hypothalamic-Pituitary-Adrenal Axis Activity: Results from a Large Cohort Study. Arch. Gen. Psychiatry 2009, 66, 617–626. [Google Scholar] [CrossRef]
- Stokes, P.E. The potential role of excessive cortisol induced by HPA hyperfunction in the pathogenesis of depression. Eur. Neuropsychopharmacol. 1995, 5, 77–82. [Google Scholar] [CrossRef]
- Moretti, A.; Gorini, A.; Villa, R.F. Affective disorders, antidepressant drugs and brain metabolism. Mol. Psychiatry 2003, 8, 773–785. [Google Scholar] [CrossRef]
- Klinedinst, N.J.; Regenold, W.T. A mitochondrial bioenergetic basis of depression. J. Bioenerg. Biomembr. 2015, 47, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef]
- Gardner, A.; Boles, R.G. Beyond the serotonin hypothesis: Mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 730–743. [Google Scholar] [CrossRef]
- Monzel, A.S.; Enriquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef]
- Berndt, N.; Holzhutter, H.G. The high energy demand of neuronal cells caused by passive leak currents is not a waste of energy. Cell Biochem. Biophys. 2013, 67, 527–535. [Google Scholar] [CrossRef]
- Schaefer, A.M.; McFarland, R.; Blakely, E.L.; He, L.; Whittaker, R.G.; Taylor, R.W.; Chinnery, P.F.; Turnbull, D.M. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 2008, 63, 35–39. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Koenig, M.K. Presentation and diagnosis of mitochondrial disorders in children. Pediatr. Neurol. 2008, 38, 305–313. [Google Scholar] [CrossRef]
- Finsterer, J. Central nervous system manifestations of mitochondrial disorders. Acta Neurol. Scand. 2006, 114, 217–238. [Google Scholar] [CrossRef]
- Morava, E.; Gardeitchik, T.; Kozicz, T.; de Boer, L.; Koene, S.; de Vries, M.C.; McFarland, R.; Roobol, T.; Rodenburg, R.J.; Verhaak, C.M. Depressive behaviour in children diagnosed with a mitochondrial disorder. Mitochondrion 2010, 10, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Fattal, O.; Link, J.; Quinn, K.; Cohen, B.H.; Franco, K. Psychiatric comorbidity in 36 adults with mitochondrial cytopathies. CNS Spectr. 2007, 12, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Anglin, R.E.; Tarnopolsky, M.A.; Mazurek, M.F.; Rosebush, P.I. The psychiatric presentation of mitochondrial disorders in adults. J. Neuropsychiatry Clin. Neurosci. 2012, 24, 394–409. [Google Scholar] [CrossRef]
- Riquin, E.; Duverger, P.; Cariou, C.; Barth, M.; Prouteau, C.; Van Bogaert, P.; Bonneau, D.; Roy, A. Neuropsychological and Psychiatric Features of Children and Adolescents Affected With Mitochondrial Diseases: A Systematic Review. Front. Psychiatry 2020, 11, 747. [Google Scholar] [CrossRef]
- Chan, S.T.; McCarthy, M.J.; Vawter, M.P. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr. Res. 2020, 217, 136–147. [Google Scholar] [CrossRef]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Dobeln, U.; Hagenfeldt, L.; Hallstrom, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef]
- Hroudova, J.; Fisar, Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regen. Res. 2013, 8, 363–375. [Google Scholar]
- Sjovall, F.; Ehinger, J.K.; Marelsson, S.E.; Morota, S.; Frostner, E.A.; Uchino, H.; Lundgren, J.; Arnbjornsson, E.; Hansson, M.J.; Fellman, V.; et al. Mitochondrial respiration in human viable platelets—Methodology and influence of gender, age and storage. Mitochondrion 2013, 13, 7–14. [Google Scholar] [CrossRef]
- Karabatsiakis, A.; Bock, C.; Salinas-Manrique, J.; Kolassa, S.; Calzia, E.; Dietrich, D.E.; Kolassa, I.T. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl. Psychiatry 2014, 4, e397. [Google Scholar] [CrossRef]
- Garbett, K.A.; Vereczkei, A.; Kalman, S.; Wang, L.; Korade, Z.; Shelton, R.C.; Mirnics, K. Fibroblasts from patients with major depressive disorder show distinct transcriptional response to metabolic stressors. Transl. Psychiatry 2015, 5, e523. [Google Scholar] [CrossRef]
- Kuffner, K.; Triebelhorn, J.; Meindl, K.; Benner, C.; Manook, A.; Sudria-Lopez, D.; Siebert, R.; Nothdurfter, C.; Baghai, T.C.; Drexler, K.; et al. Major Depressive Disorder is Associated with Impaired Mitochondrial Function in Skin Fibroblasts. Cells 2020, 9, 884. [Google Scholar] [CrossRef] [PubMed]
- Triebelhorn, J.; Cardon, I.; Kuffner, K.; Bader, S.; Jahner, T.; Meindl, K.; Rothhammer-Hampl, T.; Riemenschneider, M.J.; Drexler, K.; Berneburg, M.; et al. Induced neural progenitor cells and iPS-neurons from major depressive disorder patients show altered bioenergetics and electrophysiological properties. Mol. Psychiatry 2022. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Berlim, M.T.; Turecki, G. What is the meaning of treatment resistant/refractory major depression (TRD)? A systematic review of current randomized trials. Eur. Neuropsychopharmacol. 2007, 17, 696–707. [Google Scholar] [CrossRef]
- Pitkanen, S.; Robinson, B.H. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J. Clin. Investig. 1996, 98, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Pitkanen, S.; Kassovska-Bratinova, S.; Robinson, B.H.; Lehotay, D.C. Excessive formation of hydroxyl radicals and aldehydic lipid peroxidation products in cultured skin fibroblasts from patients with complex I deficiency. J. Clin. Investig. 1997, 99, 2877–2882. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, C.T.; Chen, J.; Pankratz, M.T.; Xi, J.; Li, J.; Yang, Y.; Lavaute, T.M.; Li, X.J.; Ayala, M.; et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 2010, 7, 90–100. [Google Scholar] [CrossRef]
- Mecocci, P.; Fano, G.; Fulle, S.; MacGarvey, U.; Shinobu, L.; Polidori, M.C.; Cherubini, A.; Vecchiet, J.; Senin, U.; Beal, M.F. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 1999, 26, 303–308. [Google Scholar] [CrossRef]
- Pratico, D. Lipid peroxidation and the aging process. Sci. Aging Knowl. Environ. 2002, 2002, re5. [Google Scholar] [CrossRef]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2004, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Roskams, A.J.I.; Cai, X.; Ronnett, G.V. Expression of neuron-specific beta-III tubulin during olfactory neurogenesis in the embryonic and adult rat. Neuroscience 1998, 83, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebratesxs. Development 1992, 116, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Martineau, M.; Guzman, R.E.; Fahlke, C.; Klingauf, J. VGLUT1 functions as a glutamate/proton exchanger with chloride channel activity in hippocampal glutamatergic synapses. Nat. Commun. 2017, 8, 2279. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.C. Dendritic integration of excitatory synaptic input. Nat. Rev. Neurosci. 2000, 1, 181–190. [Google Scholar] [CrossRef]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef]
- Neher, E.; Sakaba, T. Multiple Roles of Calcium Ions in the Regulation of Neurotransmitter Release. Neuron 2008, 59, 861–872. [Google Scholar] [CrossRef]
- Arun, S.; Liu, L.; Donmez, G. Mitochondrial Biology and Neurological Diseases. Curr. Neuropharmacol. 2016, 14, 143–154. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P. Mitochondrial bioenergetics and neurodegeneration: A paso doble. Neural Regen. Res. 2021, 16, 686–687. [Google Scholar] [CrossRef]
- Brand, M.D.; Pakay, J.L.; Ocloo, A.; Kokoszka, J.; Wallace, D.C.; Brookes, P.S.; Cornwall, E.J. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 2005, 392 Pt 2, 353–362. [Google Scholar] [CrossRef]
- Drahota, Z.; Endlicher, R.; Stankova, P.; Rychtrmoc, D.; Milerova, M.; Cervinkova, Z. Characterization of calcium, phosphate and peroxide interactions in activation of mitochondrial swelling using derivative of the swelling curves. J. Bioenerg. Biomembr. 2012, 44, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Huntington, T.E.; Srinivasan, R. Astrocytic mitochondria in adult mouse brain slices show spontaneous calcium influx events with unique properties. Cell Calcium 2021, 96, 102383. [Google Scholar] [CrossRef] [PubMed]
- Parnis, J.; Montana, V.; Delgado-Martinez, I.; Matyash, V.; Parpura, V.; Kettenmann, H.; Sekler, I.; Nolte, C. Mitochondrial exchanger NCLX plays a major role in the intracellular Ca2+ signaling, gliotransmission, and proliferation of astrocytes. J. Neurosci. 2013, 33, 7206–7219. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.K.; Craven, L.; Hoogewijs, K.; Russell, O.M.; Lightowlers, R.N. Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem. 2018, 62, 455–465. [Google Scholar] [PubMed]
- Liemburg-Apers, D.C.; Schirris, T.J.; Russel, F.G.; Willems, P.H.; Koopman, W.J. Mitoenergetic Dysfunction Triggers a Rapid Compensatory Increase in Steady-State Glucose Flux. Biophys. J. 2015, 109, 1372–1386. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Rose, J.; Brian, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Mitochondrial Metabolism in Astrocytes Regulates Brain Bioenergetics, Neurotransmission and Redox Balance. Front. Neurosci. 2020, 14, 536682. [Google Scholar] [CrossRef]
- Hyslop, P.A.; Hinshaw, D.B.; Halsey, W.A.; Schraufstätter, I.U.; Sauerheber, R.D.; Spragg, R.G.; Jackson, J.H.; Cochrane, C.G. Mechanisms of oxidant-mediated cell injury. The glycolytic and mitochondrial pathways of ADP phosphorylation are major intracellular targets inactivated by hydrogen peroxide. J. Biol. Chem. 1988, 263, 1665–1675. [Google Scholar] [CrossRef]
- Pizzo, P.; Pozzan, T. Mitochondria–endoplasmic reticulum choreography: Structure and signaling dynamics. Trends Cell Biol. 2007, 17, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Mankad, P.; James, A.; Siriwardena, A.K.; Elliott, A.C.; Bruce, J.I.E. Insulin Protects Pancreatic Acinar Cells from Cytosolic Calcium Overload and Inhibition of Plasma Membrane Calcium Pump. J. Biol. Chem. 2012, 287, 1823–1836. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Trumbeckaite, S.; Nguyen, H.P.; Pallas, T.; Arandarcikaite, O.; Vielhaber, S.; Seppet, E.; Striggow, F. The regulation of OXPHOS by extramitochondrial calcium. Biochim. Biophys. Acta 2010, 1797, 1018–1027. [Google Scholar] [CrossRef]
- Reeg, S.; Jung, T.; Castro, J.P.; Davies, K.J.A.; Henze, A.; Grune, T. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic. Biol. Med. 2016, 99, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Bader, S.; Wurfel, T.; Jahner, T.; Nothdurfter, C.; Rupprecht, R.; Milenkovic, V.M.; Wetzel, C.H. Impact of Translocator Protein 18 kDa (TSPO) Deficiency on Mitochondrial Function and the Inflammatory State of Human C20 Microglia Cells. Cells 2023, 12, 954. [Google Scholar] [CrossRef]
- Banasr, M.; Dwyer, J.M.; Duman, R.S. Cell atrophy and loss in depression: Reversal by antidepressant treatment. Curr. Opin. Cell Biol. 2011, 23, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Vose, L.R.; Stanton, P.K. Synaptic Plasticity, Metaplasticity and Depression. Curr. Neuropharmacol. 2017, 15, 71–86. [Google Scholar] [CrossRef]
- Vadodaria, K.C.; Ji, Y.; Skime, M.; Paquola, A.; Nelson, T.; Hall-Flavin, D.; Fredlender, C.; Heard, K.J.; Deng, Y.; Le, A.T.; et al. Serotonin-induced hyperactivity in SSRI-resistant major depressive disorder patient-derived neurons. Mol. Psychiatry 2019, 24, 795–807. [Google Scholar] [CrossRef]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Development 2016, 143, 1981–1992. [Google Scholar]
- Sproule, D.M.; Kaufmann, P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: Basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann. N. Y. Acad. Sci. 2008, 1142, 133–158. [Google Scholar] [CrossRef]
- Lorenzoni, P.J.; Scola, R.H.; Kay, C.S.; Silvado, C.E.; Werneck, L.C. When should MERRF (myoclonus epilepsy associated with ragged-red fibers) be the diagnosis? Arq. Neuropsiquiatr. 2014, 72, 803–811. [Google Scholar] [CrossRef] [PubMed]
- De La Rossa, A.; Laporte, M.H.; Astori, S.; Marissal, T.; Montessuit, S.; Sheshadri, P.; Ramos-Fernández, E.; Mendez, P.; Khani, A.; Quairiaux, C.; et al. Paradoxical neuronal hyperexcitability in a mouse model of mitochondrial pyruvate import deficiency. eLife 2022, 11, e72595. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.M.; Kubota-Sakashita, M.; Fujimori-Tonou, N.; Saitow, F.; Fuke, S.; Masuda, A.; Itohara, S.; Suzuki, H.; Kato, T. Ant1 mutant mice bridge the mitochondrial and serotonergic dysfunctions in bipolar disorder. Mol. Psychiatry 2018, 23, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Tian, J.; Du, H. Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.; Martinez, J.H.; Young, D.; Chelminski, I.; Dalrymple, K. Severity classification on the Hamilton depression rating scale. J. Affect. Disord. 2013, 150, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Yan, Y.; Shin, S.; Jha, B.S.; Liu, Q.; Sheng, J.; Li, F.; Zhan, M.; Davis, J.; Bharti, K.; Zeng, X.; et al. Efficient and rapid derivation of primitive neural stem cells and generation of brain subtype neurons from human pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 862–870. [Google Scholar] [CrossRef]
- Milenkovic, V.M.; Slim, D.; Bader, S.; Koch, V.; Heinl, E.S.; Alvarez-Carbonell, D.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. CRISPR-Cas9 Mediated TSPO Gene Knockout alters Respiration and Cellular Metabolism in Human Primary Microglia Cells. Int. J. Mol. Sci. 2019, 20, 3359. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Responder Patient | |||
|---|---|---|---|
| Age, Sex | 43, Male | ||
| Clinical findings | Symptoms | Ratings | |
|
| ||
| Treatments | Antidepressants
| ||
| Mitochondriopathy Patient | |||
| Age, Sex | 18, Female | ||
| Clinical findings | Symptoms | Excluded pathologies | |
|
| ||
| Blood tests relevant to diagnosis | Measured parameters | Alterations | Descriptions |
| LDH2 | Decreased | Enzymes involved in lactate metabolism | |
| LDH4 | Increased | ||
| Citrate | Starkly decreased | Enzymes involved in tricarboxylic acid cycle | |
| Cis-aconitate | Starkly decreased | ||
| Isocitrate | Starkly decreased | ||
| Pyruvate kinase M2 | Increased | Enzyme catalyzing the last step of glycolysis | |
| Vitamin D3 | Decreased | Regulator of mitochondrial respiration and oxidative stress | |
| Coenzyme Q10 | Decreased | Electron carrier in the electron transport chain | |
| Urine test | 5-Hydroxyindolacetatic acid | Starkly decreased | Serotonin metabolite, low levels linked with depression |
| Non-R | Mito | MDD Cohort | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Fibroblasts | NPCs | Astrocytes | Fibroblasts | NPCs | Astrocytes | Fibroblasts | NPCs | Astrocytes | |
| Respiration | ↗ | ↗ | ↘ | ↘ | ↘ | = | ↘ | ↘ | ↘ |
| ATP | = | = | = | = | = | ↘ * | ↘ * | = | = |
| MMP | = | = | ↗ **** | ↘ **** | ↗ **** | ↘ * | ↘ **** | = | ↘ **** |
| Cytosolic Ca2+ | = | = | ↗ **** | = | ↗ *** | ↗ * | = | ↗ * | ↘ *** |
| Mitochondrial Ca2+ | ↗ **** | ↗ **** | ↘ **** | = | = (↘) | ↘ **** | ↘ **** | ||
| Cell size | ↘ **** | ↘**** | ↘ **** | ↘ * | ↘ **** | ↗ ** | ↘ **** | ↘**** | ↘ **** |
| Neurons | |||||||||
| MMP somas | ↘ **** | ↗ **** | |||||||
| MMP neurites | = | ↗ **** | |||||||
| Cytosolic Ca2+ | ↘ **** | ↘ **** | |||||||
| Mitochondrial Ca2+ | = | ↘ **** | |||||||
| RMP | ↗ ** (hyperpolarized) | ↘ * (depolarized) | ↘ * (depolarized) | ||||||
| Capacitance | ↘ **** | ↘ ** | ↘ * | ||||||
| INa+ 0 mV curr. density | = | ↘ **** | ↘ **** | ||||||
| IK+ 20 mV curr. density | ↗ * | ↗ ** | = | ||||||
| Spontaneous activity | −50 mV | −80 mV | PSCs | −50 mV | −80 mV | PSCs | −50 mV | ||
| ↘* | ↘* | ↘**** | ↗ * | = | = | ↘* | |||
| Spont. AP amplitude | ↘ **** | ↗ **** | |||||||
| Spont. AP FWHM | ↘ **** | ↘ **** | |||||||
| PSC amplitude | ↗ *** | ↘ **** | |||||||
| PSC rise time | ↗ ** | ↗ **** | |||||||
| PSC decay time (Tau) | = | ↗ **** | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardon, I.; Grobecker, S.; Kücükoktay, S.; Bader, S.; Jahner, T.; Nothdurfter, C.; Koschitzki, K.; Berneburg, M.; Weber, B.H.F.; Stöhr, H.; et al. Mitochondrial and Cellular Function in Fibroblasts, Induced Neurons, and Astrocytes Derived from Case Study Patients: Insights into Major Depression as a Mitochondria-Associated Disease. Int. J. Mol. Sci. 2024, 25, 963. https://doi.org/10.3390/ijms25020963
Cardon I, Grobecker S, Kücükoktay S, Bader S, Jahner T, Nothdurfter C, Koschitzki K, Berneburg M, Weber BHF, Stöhr H, et al. Mitochondrial and Cellular Function in Fibroblasts, Induced Neurons, and Astrocytes Derived from Case Study Patients: Insights into Major Depression as a Mitochondria-Associated Disease. International Journal of Molecular Sciences. 2024; 25(2):963. https://doi.org/10.3390/ijms25020963
Chicago/Turabian StyleCardon, Iseline, Sonja Grobecker, Selin Kücükoktay, Stefanie Bader, Tatjana Jahner, Caroline Nothdurfter, Kevin Koschitzki, Mark Berneburg, Bernhard H. F. Weber, Heidi Stöhr, and et al. 2024. "Mitochondrial and Cellular Function in Fibroblasts, Induced Neurons, and Astrocytes Derived from Case Study Patients: Insights into Major Depression as a Mitochondria-Associated Disease" International Journal of Molecular Sciences 25, no. 2: 963. https://doi.org/10.3390/ijms25020963
APA StyleCardon, I., Grobecker, S., Kücükoktay, S., Bader, S., Jahner, T., Nothdurfter, C., Koschitzki, K., Berneburg, M., Weber, B. H. F., Stöhr, H., Höring, M., Liebisch, G., Braun, F., Rothammer-Hampl, T., Riemenschneider, M. J., Rupprecht, R., Milenkovic, V. M., & Wetzel, C. H. (2024). Mitochondrial and Cellular Function in Fibroblasts, Induced Neurons, and Astrocytes Derived from Case Study Patients: Insights into Major Depression as a Mitochondria-Associated Disease. International Journal of Molecular Sciences, 25(2), 963. https://doi.org/10.3390/ijms25020963