
SumVg: Total Heritability Explained by All Variants in Genome-Wide Association Studies Based on Summary Statistics with Standard Error Estimates


Abstract
1. Introduction
2. Results
2.1. Overview of Methods
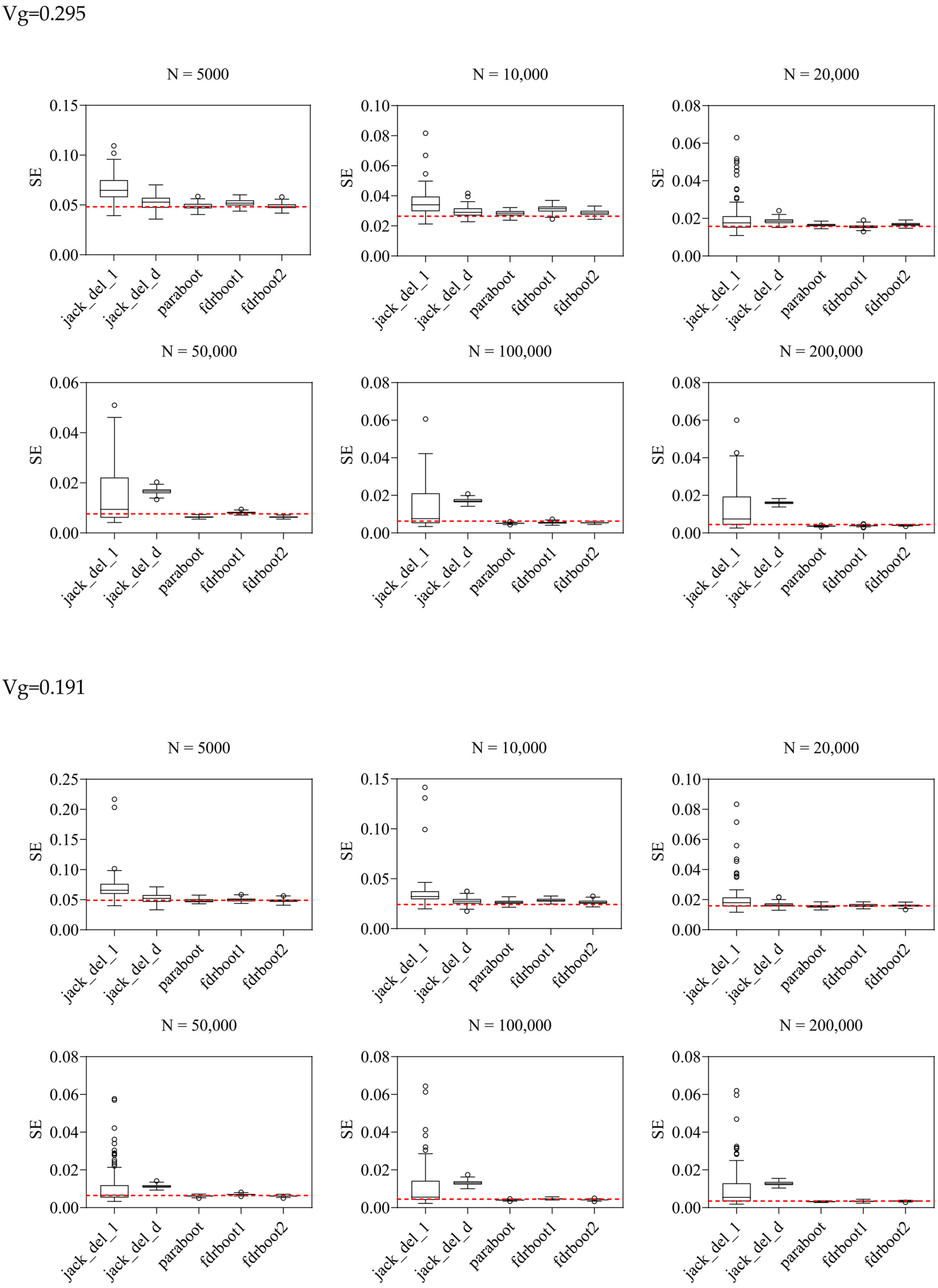
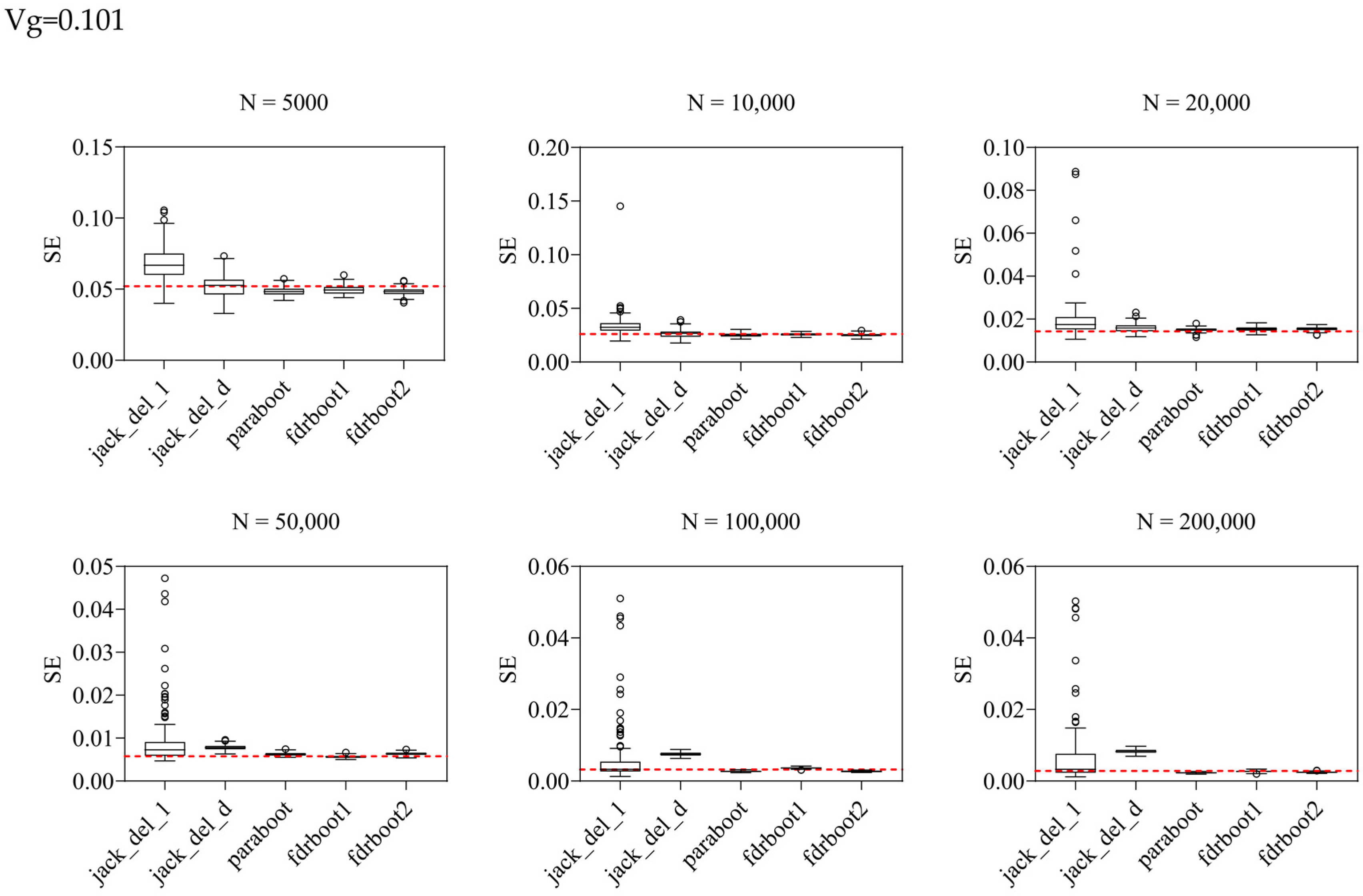
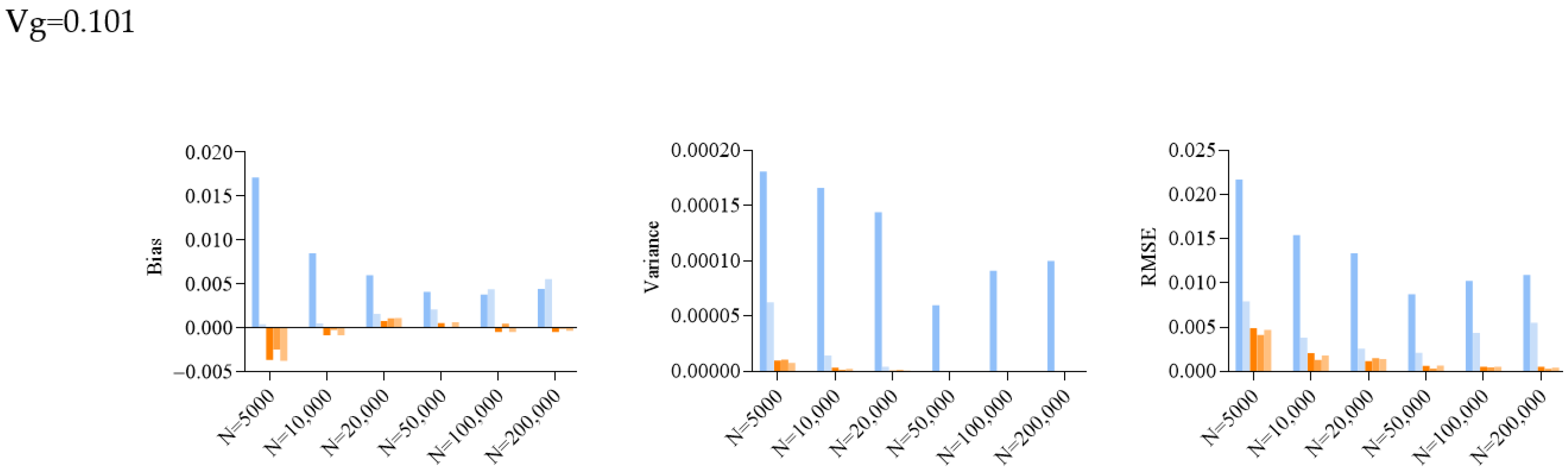
2.2. Simulation Results for SE Estimation
2.3. Performance of Different CI Construction Methods
2.4. Results on Immune Traits
2.5. R Package Implementation
3. Discussion
4. Materials and Methods
4.1. Estimation of the Total Heritability Explained (Vg)
4.1.1. Estimation of Total Vg Based on Tweedie’s Formula
4.1.2. Conversion of z-Statistics to Vg
4.1.3. Assumptions
4.1.4. An Alternative Conditional Estimator
4.2. Estimation of the Standard Error (SE) of Vg
4.2.1. Standard and Delete-d-Jackknife to Estimate SE
4.2.2. Parametric Bootstrap Approaches for Estimating SE
4.3. Construction of Confidence Intervals (CIs): An Exploratory Analysis
4.3.1. Normal Approximation (Standard Approach)
4.3.2. Percentile Approach
4.3.3. Union CI
- Normal approximation (standard approach), without bias correction (one estimator) or with bootstrap bias correction (3 estimators), then take the union of CIs;
- Percentile approach, without bias correction (3 estimators) and with bias correction (3 estimators), then take the union of CIs;
- Union of the final CI obtained from 1 and 2.
4.4. Simulation Studies
4.5. Application to Immune Traits
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Speed, D.; Hemani, G.; Johnson, M.R.; Balding, D.J. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012, 91, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Speed, D.; Balding, D.J. SumHer better estimates the SNP heritability of complex traits from summary statistics. Nat. Genet. 2019, 51, 277–284. [Google Scholar] [CrossRef]
- Zhu, H.; Zhou, X. Statistical methods for SNP heritability estimation and partition: A review. Comput. Struct. Biotechnol. J. 2020, 18, 1557–1568. [Google Scholar] [CrossRef]
- Barry, C.-J.S.; Walker, V.M.; Cheesman, R.; Davey Smith, G.; Morris, T.T.; Davies, N.M. How to estimate heritability: A guide for genetic epidemiologists. Int. J. Epidemiol. 2023, 52, 624–632. [Google Scholar] [CrossRef]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef]
- Brandes, N.; Weissbrod, O.; Linial, M. Open problems in human trait genetics. Genome Biol. 2022, 23, 131. [Google Scholar] [CrossRef] [PubMed]
- Young, A.I. Solving the missing heritability problem. PLoS Genet. 2019, 15, e1008222. [Google Scholar] [CrossRef]
- So, H.C.; Li, M.; Sham, P.C. Uncovering the total heritability explained by all true susceptibility variants in a genome-wide association study. Genet. Epidemiol. 2011, 35, 447–456. [Google Scholar] [CrossRef]
- Robbins, H. An empirical Bayes approach to statistics. In Proceedings of the Third Berkeley Symposium on Mathematical Statistics and Probability, Cambridge, UK, 26–31 December 1954, July and August 1955; University of California Press: Berkeley, CA, USA; Los Angeles, CA, USA, 1956; Volume 1, pp. 157–163. [Google Scholar]
- Brown, L.D. Admissible estimators, recurrent diffusions, and insoluble boundary value problems. Ann. Math. Stat. 1971, 42, 855–903. [Google Scholar] [CrossRef]
- Efron, B. Empirical Bayes estimates for large-scale prediction problems. J. Am. Stat. Assoc. 2009, 104, 1015–1028. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, Y.; Jiang, W.; Ye, Y.; Lu, Q.; Zhao, H. Comparison of methods for estimating genetic correlation between complex traits using GWAS summary statistics. Brief. Bioinform. 2021, 22, bbaa442. [Google Scholar] [CrossRef] [PubMed]
- Benke, K.S.; Nivard, M.G.; Velders, F.P.; Walters, R.K.; Pappa, I.; Scheet, P.A.; Xiao, X.; Ehli, E.A.; Palmer, L.J.; Whitehouse, A.J.; et al. A genome-wide association meta-analysis of preschool internalizing problems. J. Am. Acad. Child. Adolesc. Psychiatry 2014, 53, 667–676.e667. [Google Scholar] [CrossRef] [PubMed]
- Lubke, G.H.; Hottenga, J.J.; Walters, R.; Laurin, C.; de Geus, E.J.; Willemsen, G.; Smit, J.H.; Middeldorp, C.M.; Penninx, B.W.; Vink, J.M.; et al. Estimating the genetic variance of major depressive disorder due to all single nucleotide polymorphisms. Biol. Psychiatry 2012, 72, 707–709. [Google Scholar] [CrossRef]
- van Beek, J.H.; Lubke, G.H.; de Moor, M.H.; Willemsen, G.; de Geus, E.J.; Hottenga, J.J.; Walters, R.K.; Smit, J.H.; Penninx, B.W.; Boomsma, D.I. Heritability of liver enzyme levels estimated from genome-wide SNP data. Eur. J. Hum. Genet. 2014, 23, 1223–1228. [Google Scholar] [CrossRef]
- Hibar, D.P.; Stein, J.L.; Renteria, M.E.; Arias-Vasquez, A.; Desrivieres, S.; Jahanshad, N.; Toro, R.; Wittfeld, K.; Abramovic, L.; Andersson, M.; et al. Common genetic variants influence human subcortical brain structures. Nature 2015, 520, 224–229. [Google Scholar] [CrossRef]
- Paternoster, L.; Standl, M.; Waage, J.; Baurecht, H.; Hotze, M.; Strachan, D.P.; Curtin, J.A.; Bonnelykke, K.; Tian, C.; Takahashi, A.; et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015, 47, 1449–1456. [Google Scholar] [CrossRef]
- Lo, M.T.; Hinds, D.A.; Tung, J.Y.; Franz, C.; Fan, C.C.; Wang, Y.; Smeland, O.B.; Schork, A.; Holland, D.; Kauppi, K.; et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat. Genet. 2017, 49, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Minica, C.C.; Verweij, K.J.H.; van der Most, P.J.; Mbarek, H.; Bernard, M.; van Eijk, K.R.; Lind, P.A.; Liu, M.Z.; Maciejewski, D.F.; Palviainen, T.; et al. Genome-wide association meta-analysis of age at first cannabis use. Addiction 2018, 113, 2073–2086. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, T.S.; Prins, B.P.; Abdollahi, M.; Armstrong, N.J.; Aslibekyan, S.; Bain, L.; Jefferis, B.; Baumert, J.; Beekman, M.; Ben-Shlomo, Y.; et al. Genome-wide association study of circulating interleukin 6 levels identifies novel loci. Hum. Mol. Genet. 2021, 30, 393–409. [Google Scholar] [CrossRef]
- Shin, S.H.; Park, S.; Wright, C.; D’Astous, V.A.; Kim, G. The Role of Polygenic Score and Cognitive Activity in Cognitive Functioning Among Older Adults. Gerontologist 2021, 61, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Ahola-Olli, A.V.; Würtz, P.; Havulinna, A.S.; Aalto, K.; Pitkänen, N.; Lehtimäki, T.; Kähönen, M.; Lyytikäinen, L.P.; Raitoharju, E.; Seppälä, I.; et al. Genome-wide Association Study Identifies 27 Loci Influencing Concentrations of Circulating Cytokines and Growth Factors. Am. J. Hum. Genet. 2017, 100, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Steinsaltz, D.; Dahl, A.; Wachter, K.W. On Negative Heritability and Negative Estimates of Heritability. Genetics 2020, 215, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Wied, D.; Weißbach, R. Consistency of the kernel density estimator: A survey. Stat. Pap. 2012, 53, 1–21. [Google Scholar] [CrossRef][Green Version]
- Efron, B. Tweedie’s formula and selection bias. J. Am. Stat. Assoc. 2011, 106, 1602. [Google Scholar] [CrossRef] [PubMed]
- Carry, P.M.; Vanderlinden, L.A.; Dong, F.; Buckner, T.; Litkowski, E.; Vigers, T.; Norris, J.M.; Kechris, K. Inverse probability weighting is an effective method to address selection bias during the analysis of high dimensional data. Genet. Epidemiol. 2021, 45, 593–603. [Google Scholar] [CrossRef]
- Horowitz, J.L. Bootstrap methods in econometrics. Annu. Rev. Econ. 2019, 11, 193–224. [Google Scholar] [CrossRef]
- Shao, J.; Wu, C.J. A general theory for jackknife variance estimation. Ann. Stat. 1989, 17, 1176–1197. [Google Scholar] [CrossRef]
- Zhong, H.; Prentice, R.L. Bias-reduced estimators and confidence intervals for odds ratios in genome-wide association studies. Biostatistics 2008, 9, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Bull, S.B. Reduction of selection bias in genomewide studies by resampling. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2005, 28, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, S.; Pritchard, J.K. Overcoming the winner’s curse: Estimating penetrance parameters from case-control data. Am. J. Hum. Genet. 2007, 80, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Gillett, A.C.; Vassos, E.; Lewis, C.M. Transforming summary statistics from logistic regression to the liability scale: Application to genetic and environmental risk scores. Hum. Hered. 2019, 83, 210–224. [Google Scholar] [CrossRef]
- Efron, B.; Tibshirani, R.; Storey, J.D.; Tusher, V. Empirical Bayes analysis of a microarray experiment. J. Am. Stat. Assoc. 2001, 96, 1151–1160. [Google Scholar] [CrossRef]
- Miller, R.G. The jackknife—A review. Biometrika 1974, 61, 1–15. [Google Scholar]
- Chatterjee, S. Another look at the jackknife: Further examples of generalized bootstrap. Stat. Probab. Lett. 1998, 40, 307–319. [Google Scholar] [CrossRef]
- Efron, B.; Tibshirani, R.J. An Introduction to the Bootstrap; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Conley, T.G.; Hansen, C.B.; Rossi, P.E. Plausibly exogenous. Rev. Econ. Stat. 2012, 94, 260–272. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sum_of_Vg | Sample_Size | Mean_Est | TRUE_SE | SE | ||||
|---|---|---|---|---|---|---|---|---|
| jack_del_1 | jack_del_d | paraboot | fdrboot1 | fdrboot2 | ||||
| 0.295 | 5000 | 0.232 | 0.0482 | 0.0672 | 0.0524 | 0.0488 | 0.0519 | 0.0489 |
| 10,000 | 0.210 | 0.0265 | 0.0353 | 0.0295 | 0.0285 | 0.0312 | 0.0287 | |
| 20,000 | 0.244 | 0.0158 | 0.0208 | 0.0185 | 0.0165 | 0.0156 | 0.0168 | |
| 50,000 | 0.283 | 0.0076 | 0.0149 | 0.0167 | 0.0063 | 0.0081 | 0.0063 | |
| 0.312 | 0.0063 | 0.0143 | 0.0172 | 0.0051 | 0.0055 | 0.0054 | ||
| 0.321 | 0.0045 | 0.0134 | 0.0161 | 0.0036 | 0.0038 | 0.0041 | ||
| 0.191 | 5000 | 0.207 | 0.0491 | 0.0706 | 0.0523 | 0.0486 | 0.0500 | 0.0485 |
| 10,000 | 0.147 | 0.0242 | 0.0357 | 0.0274 | 0.0263 | 0.0285 | 0.0265 | |
| 20,000 | 0.158 | 0.0159 | 0.0208 | 0.0166 | 0.0156 | 0.0162 | 0.0160 | |
| 50,000 | 0.174 | 0.0064 | 0.0113 | 0.0113 | 0.0061 | 0.0070 | 0.0061 | |
| 0.195 | 0.0045 | 0.0110 | 0.0131 | 0.0040 | 0.0047 | 0.0041 | ||
| 0.207 | 0.0035 | 0.0103 | 0.0128 | 0.0031 | 0.0034 | 0.0035 | ||
| 0.101 | 5000 | 0.197 | 0.0521 | 0.0692 | 0.0524 | 0.0484 | 0.0496 | 0.0483 |
| 10,000 | 0.116 | 0.0260 | 0.0345 | 0.0265 | 0.0251 | 0.0257 | 0.0251 | |
| 20,000 | 0.098 | 0.0143 | 0.0202 | 0.0159 | 0.0150 | 0.0153 | 0.0154 | |
| 50,000 | 0.091 | 0.0058 | 0.0098 | 0.0078 | 0.0063 | 0.0057 | 0.0063 | |
| 0.094 | 0.0032 | 0.0069 | 0.0076 | 0.0027 | 0.0036 | 0.0027 | ||
| 0.107 | 0.0028 | 0.0072 | 0.0083 | 0.0023 | 0.0027 | 0.0025 | ||
| Sum_Vg | N | Bias of the Estimator for SE | Variance of the Estimator for SE | RMSE of the Estimator for SE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| jack_del_1 | jack_del_d | paraboot | fdrboot1 | fdrboot2 | jack_del_1 | jack_del_d | paraboot | fdrboot1 | fdrboot2 | jack_del_1 | jack_del_d | paraboot | fdrboot1 | fdrboot2 | ||
| 0.295 | 5000 | 1.91 | 4.26 | 5.92 × 10−4 | 3.73 | 7.16 | 1.77 | 5.14 | 1.26 | 1.15 | 9.30 × 10−6 | 2.32 | 8.34 | 3.59 | 5.04 | 3.13 × 10−3 |
| 10,000 | 8.81 | 2.98 | 2.00 × 10−3 | 4.66 | 2.25 | 7.34 | 1.21 | 3.87 | 4.99 | 3.62 × 10−6 | 1.23 | 4.58 | 2.80 × 10−3 | 5.17 | 2.94 | |
| 20,000 | 5.04 | 2.78 | 7.37 | −1.46 × 10−4 | 1.07 | 9.06 | 2.27 | 8.33 × 10−7 | 1.12 | 1.00 | 1.08 | 3.16 | 1.17 | 1.07 × 10−3 | 1.47 | |
| 50,000 | 7.25 | 9.03 | −1.32 | 4.68 × 10−4 | −1.30 | 1.29 | 1.45 | 1.36 | 1.93 | 1.30 × 10−7 | 1.35 | 9.11 | 1.37 | 6.42 × 10−4 | 1.35 | |
| 7.97 | 1.09 | −1.20 | −8.78 × 10−4 | −9.34 | 1.41 | 1.52 | 8.11 × 10−8 | 3.48 | 1.00 | 1.43 | 1.10 | 1.23 | 1.06 | 9.86 × 10−4 | ||
| 8.92 | 1.16 | −8.57 | −6.32 | −3.72 × 10−4 | 1.37 | 8.70 | 3.49 × 10−8 | 1.30 | 4.06 | 1.47 | 1.16 | 8.77 | 7.27 | 4.23 × 10−4 | ||
| 0.191 | 5000 | 2.16 | 3.21 | −5.02 × 10−4 | 9.69 | −5.23 | 5.53 | 5.41 | 1.07 | 1.02 | 8.58 × 10−6 | 3.19 | 8.03 | 3.31 | 3.34 | 2.98 × 10−3 |
| 10,000 | 1.15 | 3.16 | 2.04 × 10−3 | 4.22 | 2.29 | 2.80 | 1.43 | 4.88 | 2.98 × 10−6 | 5.03 | 2.03 | 4.93 | 3.01 × 10−3 | 4.56 | 3.20 | |
| 20,000 | 4.96 | 7.22 | −2.41 | 3.15 | 9.85 × 10−5 | 1.19 | 2.70 | 1.09 | 1.31 | 7.64 × 10−7 | 1.20 | 1.79 | 1.07 | 1.19 | 8.80 × 10−4 | |
| 50,000 | 4.90 | 4.83 | −2.92 × 10−4 | 5.76 | −2.97 | 1.10 | 8.17 | 1.66 | 1.50 | 1.24 × 10−7 | 1.16 | 4.91 | 5.01 | 6.94 | 4.60 × 10−4 | |
| 6.45 | 8.56 | −5.40 | 1.30 × 10−4 | −4.33 | 1.32 | 1.68 | 6.30 × 10−8 | 1.32 | 8.37 | 1.32 | 8.65 | 5.96 | 3.85 × 10−4 | 5.21 | ||
| 6.85 | 9.28 | −3.57 | −1.41 | −3.31 × 10−5 | 1.28 | 1.06 | 3.04 × 10−8 | 1.54 | 3.78 | 1.32 | 9.34 | 3.97 | 4.17 | 1.97 × 10−4 | ||
| 0.101 | 5000 | 1.71 | 3.38 × 10−4 | −3.72 | −2.54 | −3.81 | 1.81 | 6.26 | 1.01 | 1.07 | 7.70 × 10−6 | 2.17 | 7.92 | 4.90 | 4.14 × 10−3 | 4.71 |
| 10,000 | 8.45 | 4.62 | −8.81 | −2.84 × 10−4 | −9.03 | 1.66 | 1.45 | 3.49 | 1.66 × 10−6 | 2.46 | 1.54 | 3.83 | 2.07 | 1.32 × 10−3 | 1.81 | |
| 20,000 | 5.92 | 1.56 | 7.21 × 10−4 | 1.04 | 1.08 | 1.44 | 4.20 | 8.80 | 1.22 | 8.17 × 10−7 | 1.34 | 2.57 | 1.18 × 10−3 | 1.52 | 1.41 | |
| 50,000 | 4.03 | 2.04 | 4.91 | −1.02 × 10−4 | 5.85 | 6.00 | 4.31 | 1.61 | 1.08 × 10−7 | 1.26 | 8.73 | 2.15 | 6.34 | 3.44 × 10−4 | 6.85 | |
| 3.73 | 4.34 | −5.10 | 4.31 × 10−4 | −5.17 | 9.13 | 3.03 | 2.83 | 4.34 | 2.05 × 10−8 | 1.03 | 4.38 | 5.37 | 4.79 × 10−4 | 5.36 | ||
| 4.38 | 5.48 | −5.31 | −1.61 × 10−4 | −3.81 | 1.00 | 3.41 | 2.22 × 10−8 | 7.06 | 2.88 | 1.09 | 5.51 | 5.52 | 3.11 × 10−4 | 4.17 | ||
| N | Union CI Type | Coverage (Vg = 0.295) | Coverage (Vg = 0.191) | Coverage (Vg = 0.101) |
|---|---|---|---|---|
| 5000 | Standard | 0.75 | 0.97 | 0.77 |
| Percentile | 1 | 1 | 0.78 | |
| Standard + Percentile | 1 | 1 | 0.78 | |
| 10,000 | Standard | 0.6 | 0.67 | 0.94 |
| Percentile | 0.99 | 1 | 1 | |
| Standard + Percentile | 0.99 | 1 | 1 | |
| 20,000 | Standard | 0.89 | 0.84 | 0.96 |
| Percentile | 0.91 | 1 | 1 | |
| Standard + Percentile | 0.97 | 1 | 1 | |
| 50,000 | Standard | 1 | 1 | 0.9 |
| Percentile | 1 | 1 | 1 | |
| Standard + Percentile | 1 | 1 | 1 | |
| Standard | 1 | 1 | 1 | |
| Percentile | 1 | 1 | 1 | |
| Standard + Percentile | 1 | 1 | 1 | |
| Standard | 0.96 | 1 | 1 | |
| Percentile | 0.13 | 0.66 | 1 | |
| Standard+Percentile | 0.96 | 1 | 1 |
| Trait | Abbreviation | GWAS ID | N | SNP_h2 (LDSC) | SNP_h2_se (LDSC) |
|---|---|---|---|---|---|
| Stem cell factor | SCF | ebi-a-GCST004429 | 8290 | −0.06 | 0.055 |
| Interleukin-4 | IL4 | ebi-a-GCST004453 | 8124 | −0.0446 | 0.0595 |
| Interleukin-17 | IL17 | ebi-a-GCST004442 | 7760 | −0.0407 | 0.0623 |
| Hepatocyte growth factor | HGF | ebi-a-GCST004449 | 8292 | −0.0311 | 0.0579 |
| Basic fibroblast growth factor | FGFBasic | ebi-a-GCST004459 | 7565 | −0.0159 | 0.0597 |
| Stromal cell-derived factor-1 alpha (CXCL12) | SDF1a | ebi-a-GCST004427 | 5998 | −0.0116 | 0.0713 |
| Interleukin-6 | IL6 | ebi-a-GCST004446 | 8189 | −0.0071 | 0.0568 |
| Platelet derived growth factor BB | PDGFbb | ebi-a-GCST004432 | 8293 | −0.0043 | 0.0624 |
| TNF-related apoptosis inducing ligand | TRAIL | ebi-a-GCST004424 | 8186 | 0.0125 | 0.0613 |
| Interferon-gamma | IFNg | ebi-a-GCST004456 | 7701 | 0.0134 | 0.0624 |
| Granulocyte colony-stimulating factor | GCSF | ebi-a-GCST004458 | 7904 | 0.0173 | 0.0601 |
| Interleukin-10 | IL10 | ebi-a-GCST004444 | 7681 | 0.0186 | 0.0691 |
| Trait | N | LDSC | SumVg | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| h2 | se | h2 | r2 | n_pruned_snp | se_jack1 | se_jack_del_d | se_paraboot | se_fdrboot1 | se_fdrboot2 | ||
| SCF | 8290 | −0.06 | 0.055 | 0.333 | 0.1 | 428,593 | 0.0926 | 0.0822 | 0.0679 | 0.0443 | 0.0514 |
| 0.185 | 0.05 | 251,008 | 0.0526 | 0.0456 | 0.0467 | 0.0502 | 0.0517 | ||||
| 0.105 | 0.025 | 127,908 | 0.0307 | 0.0313 | 0.0272 | 0.0397 | 0.0335 | ||||
| 0.100 | 0.01 | 61,938 | 0.0310 | 0.0200 | 0.0220 | 0.0252 | 0.0265 | ||||
| 0.092 | 0.005 | 51,370 | 0.0229 | 0.0169 | 0.0201 | 0.0235 | 0.0230 | ||||
| 0.101 | 0.002 | 48,088 | 0.0319 | 0.0153 | 0.0220 | 0.0226 | 0.0198 | ||||
| 0.102 | 0.001 | 47,108 | 0.0316 | 0.0155 | 0.0223 | 0.0216 | 0.0188 | ||||
| IL4 | 8124 | −0.0446 | 0.0595 | 0.503 | 0.1 | 427,005 | 0.1218 | 0.1133 | 0.0616 | 0.0563 | 0.0569 |
| 0.377 | 0.05 | 249,710 | 0.1000 | 0.0823 | 0.0484 | 0.0445 | 0.0453 | ||||
| 0.302 | 0.025 | 127,248 | 0.0650 | 0.0594 | 0.0318 | 0.0365 | 0.0336 | ||||
| 0.235 | 0.01 | 61,685 | 0.0529 | 0.0313 | 0.0247 | 0.0240 | 0.0236 | ||||
| 0.215 | 0.005 | 51,196 | 0.0472 | 0.0278 | 0.0227 | 0.0217 | 0.0221 | ||||
| 0.197 | 0.002 | 47,878 | 0.0571 | 0.0273 | 0.0228 | 0.0225 | 0.0253 | ||||
| 0.187 | 0.001 | 46,911 | 0.0482 | 0.0244 | 0.0198 | 0.0226 | 0.0242 | ||||
| IL17 | 7760 | −0.0407 | 0.0623 | 0.352 | 0.1 | 427,226 | 0.1240 | 0.0946 | 0.0692 | 0.0625 | 0.0609 |
| 0.228 | 0.05 | 250,259 | 0.0683 | 0.0668 | 0.0499 | 0.0495 | 0.0495 | ||||
| 0.299 | 0.025 | 127,479 | 0.0877 | 0.0568 | 0.0360 | 0.0380 | 0.0323 | ||||
| 0.234 | 0.01 | 61,756 | 0.0485 | 0.0340 | 0.0239 | 0.0267 | 0.0256 | ||||
| 0.196 | 0.005 | 51,215 | 0.0475 | 0.0295 | 0.0237 | 0.0190 | 0.0249 | ||||
| 0.195 | 0.002 | 47,887 | 0.0634 | 0.0231 | 0.0231 | 0.0226 | 0.0210 | ||||
| 0.188 | 0.001 | 46,931 | 0.0568 | 0.0242 | 0.0183 | 0.0211 | 0.0215 | ||||
| HGF | 8292 | −0.0311 | 0.0579 | 0.366 | 0.1 | 428,318 | 0.0917 | 0.0864 | 0.0569 | 0.0642 | 0.0593 |
| 0.242 | 0.05 | 250,843 | 0.0812 | 0.0722 | 0.0483 | 0.0492 | 0.0491 | ||||
| 0.205 | 0.025 | 127,850 | 0.0657 | 0.0488 | 0.0327 | 0.0326 | 0.0357 | ||||
| 0.098 | 0.01 | 61,906 | 0.0379 | 0.0224 | 0.0225 | 0.0260 | 0.0242 | ||||
| 0.115 | 0.005 | 51,301 | 0.0347 | 0.0199 | 0.0224 | 0.0230 | 0.0203 | ||||
| 0.111 | 0.002 | 47,878 | 0.0414 | 0.0162 | 0.0189 | 0.0211 | 0.0215 | ||||
| 0.108 | 0.001 | 46,934 | 0.0312 | 0.0171 | 0.0221 | 0.0208 | 0.0211 | ||||
| FGFBasic | 7565 | −0.0159 | 0.0597 | 0.269 | 0.1 | 427,284 | 0.0835 | 0.0902 | 0.0656 | 0.0530 | 0.0577 |
| 0.217 | 0.05 | 249,930 | 0.0891 | 0.0604 | 0.0473 | 0.0504 | 0.0468 | ||||
| 0.117 | 0.025 | 127,587 | 0.0452 | 0.0431 | 0.0340 | 0.0363 | 0.0358 | ||||
| 0.133 | 0.01 | 61,911 | 0.0408 | 0.0301 | 0.0232 | 0.0239 | 0.0275 | ||||
| 0.135 | 0.005 | 51,259 | 0.0376 | 0.0243 | 0.0242 | 0.0267 | 0.0219 | ||||
| 0.143 | 0.002 | 47,874 | 0.0362 | 0.0218 | 0.0185 | 0.0233 | 0.0245 | ||||
| 0.126 | 0.001 | 46,914 | 0.0392 | 0.0206 | 0.0227 | 0.0214 | 0.0208 | ||||
| SDF1a | 5998 | −0.0116 | 0.0713 | 0.395 | 0.1 | 425,165 | 0.1120 | 0.1068 | 0.0731 | 0.0757 | 0.0870 |
| 0.256 | 0.05 | 248,727 | 0.0872 | 0.0750 | 0.0580 | 0.0565 | 0.0631 | ||||
| 0.213 | 0.025 | 126,986 | 0.0707 | 0.0462 | 0.0431 | 0.0472 | 0.0468 | ||||
| 0.163 | 0.01 | 61,680 | 0.0497 | 0.0380 | 0.0359 | 0.0349 | 0.0324 | ||||
| 0.190 | 0.005 | 51,092 | 0.0708 | 0.0318 | 0.0250 | 0.0297 | 0.0270 | ||||
| 0.165 | 0.002 | 47,702 | 0.0447 | 0.0270 | 0.0294 | 0.0304 | 0.0301 | ||||
| 0.159 | 0.001 | 46,789 | 0.0512 | 0.0232 | 0.0279 | 0.0258 | 0.0308 | ||||
| IL6 | 8189 | −0.0071 | 0.0568 | 0.422 | 0.1 | 427,566 | 0.0878 | 0.0896 | 0.0510 | 0.0575 | 0.0594 |
| 0.227 | 0.05 | 250,247 | 0.0620 | 0.0713 | 0.0402 | 0.0463 | 0.0468 | ||||
| 0.158 | 0.025 | 127,503 | 0.0672 | 0.0457 | 0.0372 | 0.0300 | 0.0360 | ||||
| 0.139 | 0.01 | 61,931 | 0.0606 | 0.0258 | 0.0220 | 0.0247 | 0.0220 | ||||
| 0.114 | 0.005 | 51,332 | 0.0288 | 0.0176 | 0.0196 | 0.0227 | 0.0236 | ||||
| 0.115 | 0.002 | 47,930 | 0.0302 | 0.0164 | 0.0191 | 0.0226 | 0.0202 | ||||
| 0.117 | 0.001 | 46,944 | 0.0319 | 0.0175 | 0.0227 | 0.0209 | 0.0211 | ||||
| PDGFbb | 8293 | −0.0043 | 0.0624 | 0.432 | 0.1 | 427,743 | 0.0907 | 0.0993 | 0.0726 | 0.0653 | 0.0676 |
| 0.341 | 0.05 | 250,325 | 0.0670 | 0.0808 | 0.0600 | 0.0496 | 0.0576 | ||||
| 0.307 | 0.025 | 127,567 | 0.0735 | 0.0554 | 0.0370 | 0.0334 | 0.0326 | ||||
| 0.154 | 0.01 | 61,789 | 0.0372 | 0.0250 | 0.0213 | 0.0245 | 0.0243 | ||||
| 0.125 | 0.005 | 51,140 | 0.0310 | 0.0226 | 0.0234 | 0.0230 | 0.0221 | ||||
| 0.120 | 0.002 | 47,822 | 0.0258 | 0.0205 | 0.0214 | 0.0208 | 0.0233 | ||||
| 0.117 | 0.001 | 46,853 | 0.0392 | 0.0192 | 0.0201 | 0.0209 | 0.0226 | ||||
| TRAIL | 8186 | 0.0125 | 0.0613 | 0.559 | 0.1 | 423,391 | 0.0613 | 0.1018 | 0.0785 | 0.0790 | 0.0750 |
| 0.304 | 0.05 | 247,717 | 0.0543 | 0.1190 | 0.0526 | 0.0503 | 0.0439 | ||||
| 0.242 | 0.025 | 126,350 | 0.0607 | 0.0647 | 0.0321 | 0.0362 | 0.0370 | ||||
| 0.128 | 0.01 | 61,114 | 0.0316 | 0.0251 | 0.0242 | 0.0229 | 0.0277 | ||||
| 0.127 | 0.005 | 50,633 | 0.0298 | 0.0231 | 0.0255 | 0.0239 | 0.0268 | ||||
| 0.128 | 0.002 | 47,359 | 0.0332 | 0.0216 | 0.0233 | 0.0215 | 0.0266 | ||||
| 0.121 | 0.001 | 46,415 | 0.0358 | 0.0195 | 0.0222 | 0.0229 | 0.0256 | ||||
| IFNg | 7701 | 0.0134 | 0.0624 | 0.393 | 0.1 | 426,740 | 0.0946 | 0.0811 | 0.0528 | 0.0590 | 0.0594 |
| 0.241 | 0.05 | 249,818 | 0.0655 | 0.0628 | 0.0553 | 0.0520 | 0.0509 | ||||
| 0.244 | 0.025 | 127,514 | 0.0734 | 0.0582 | 0.0330 | 0.0406 | 0.0320 | ||||
| 0.138 | 0.01 | 61,890 | 0.0289 | 0.0303 | 0.0267 | 0.0239 | 0.0257 | ||||
| 0.138 | 0.005 | 51,314 | 0.0424 | 0.0201 | 0.0222 | 0.0248 | 0.0293 | ||||
| 0.141 | 0.002 | 47,918 | 0.0321 | 0.0204 | 0.0251 | 0.0248 | 0.0286 | ||||
| 0.137 | 0.001 | 46,934 | 0.0253 | 0.0183 | 0.0223 | 0.0246 | 0.0233 | ||||
| GCSF | 7904 | 0.0173 | 0.0601 | 0.246 | 0.1 | 427,393 | 0.0707 | 0.0820 | 0.0620 | 0.0604 | 0.0580 |
| 0.198 | 0.05 | 250,222 | 0.0636 | 0.0607 | 0.0402 | 0.0436 | 0.0486 | ||||
| 0.164 | 0.025 | 127,583 | 0.0501 | 0.0415 | 0.0302 | 0.0360 | 0.0327 | ||||
| 0.142 | 0.01 | 61,846 | 0.0434 | 0.0257 | 0.0280 | 0.0239 | 0.0257 | ||||
| 0.122 | 0.005 | 51,266 | 0.0379 | 0.0196 | 0.0205 | 0.0247 | 0.0238 | ||||
| 0.120 | 0.002 | 47,919 | 0.0413 | 0.0183 | 0.0219 | 0.0236 | 0.0201 | ||||
| 0.112 | 0.001 | 46,939 | 0.0312 | 0.0159 | 0.0234 | 0.0207 | 0.0202 | ||||
| IL10 | 7681 | 0.0186 | 0.0691 | 0.331 | 0.1 | 427,218 | 0.0621 | 0.1019 | 0.0584 | NA | NA |
| 0.310 | 0.05 | 250,109 | 0.0670 | 0.0858 | 0.0448 | NA | NA | ||||
| 0.198 | 0.025 | 127,543 | 0.0566 | 0.0463 | 0.0356 | 0.0382 | 0.0406 | ||||
| 0.130 | 0.01 | 61,944 | 0.0328 | 0.0225 | 0.0251 | 0.0268 | 0.0258 | ||||
| 0.141 | 0.005 | 51,257 | 0.0400 | 0.0220 | 0.0237 | 0.0282 | 0.0238 | ||||
| 0.148 | 0.002 | 47,880 | 0.0433 | 0.0183 | 0.0204 | 0.0271 | 0.0231 | ||||
| 0.142 | 0.001 | 46,898 | 0.0317 | 0.0194 | 0.0219 | 0.0261 | 0.0228 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
So, H.-C.; Xue, X.; Ma, Z.; Sham, P.-C. SumVg: Total Heritability Explained by All Variants in Genome-Wide Association Studies Based on Summary Statistics with Standard Error Estimates. Int. J. Mol. Sci. 2024, 25, 1347. https://doi.org/10.3390/ijms25021347
So H-C, Xue X, Ma Z, Sham P-C. SumVg: Total Heritability Explained by All Variants in Genome-Wide Association Studies Based on Summary Statistics with Standard Error Estimates. International Journal of Molecular Sciences. 2024; 25(2):1347. https://doi.org/10.3390/ijms25021347
Chicago/Turabian StyleSo, Hon-Cheong, Xiao Xue, Zhijie Ma, and Pak-Chung Sham. 2024. "SumVg: Total Heritability Explained by All Variants in Genome-Wide Association Studies Based on Summary Statistics with Standard Error Estimates" International Journal of Molecular Sciences 25, no. 2: 1347. https://doi.org/10.3390/ijms25021347
APA StyleSo, H.-C., Xue, X., Ma, Z., & Sham, P.-C. (2024). SumVg: Total Heritability Explained by All Variants in Genome-Wide Association Studies Based on Summary Statistics with Standard Error Estimates. International Journal of Molecular Sciences, 25(2), 1347. https://doi.org/10.3390/ijms25021347