
From the Gut to the Brain: The Role of Enteric Glial Cells and Their Involvement in the Pathogenesis of Parkinson’s Disease


Abstract
1. Introduction: Braak’s Hypothesis and the Enteric Nervous System
1.1. Parkinson’s Disease
1.2. Parkinson’s Disease and the Gastrointestinal Tract
1.3. The Enteric Nervous System
2. Methods
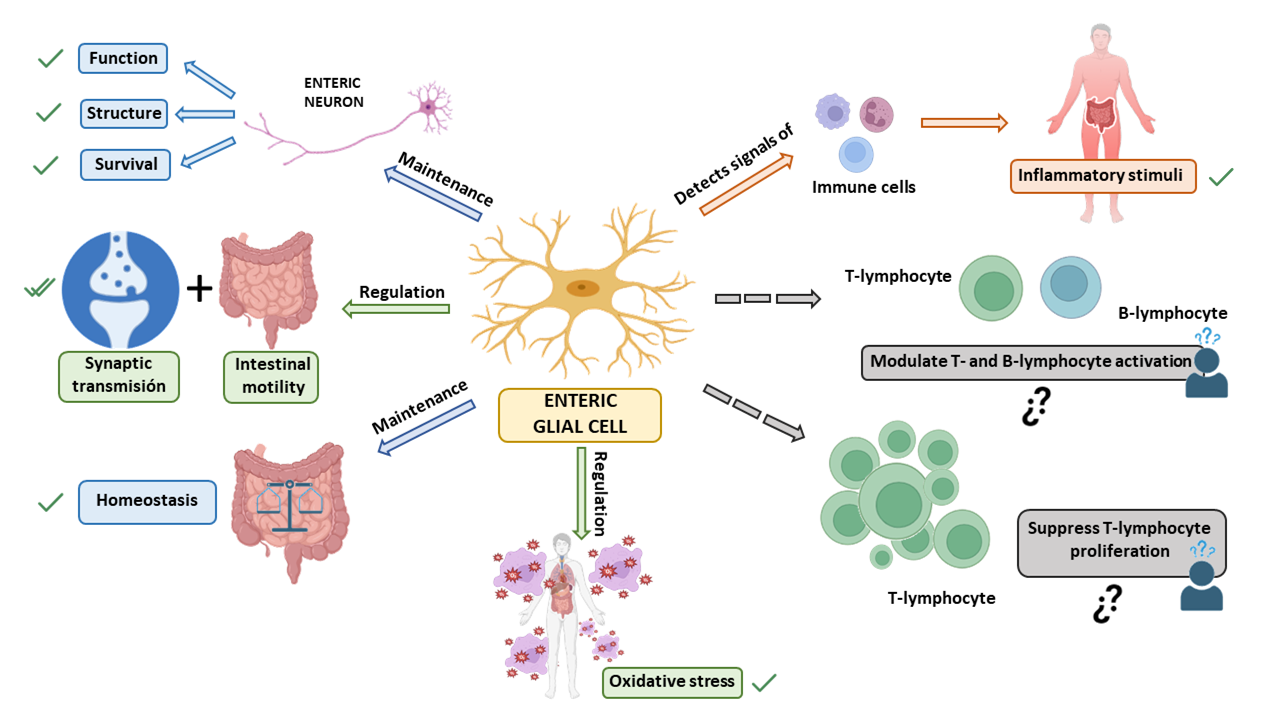
3. Enteric Glial Cells
3.1. Types of Enteric Glia
3.2. Enteric Glial Markers
3.3. Functions of the Enteric Glia and Their Role in Disease
4. Parkinson’s Disease and Enteric Glia
4.1. Studies Using Animal Models and Cell Cultures
4.1.1. Rotenone and Other Pesticides
4.1.2. MTPT1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
4.1.3. 6-Hydroxydopamine (6-OHA)
4.1.4. Adeno-Associated Virus (AAV)-α-Synuclein
4.1.5. A53 α-Synuclein Mouse Model
4.2. Studies Using Human Biopsies from PD Patients
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| 4-HNE | 4-hydroxynonenal |
| 6-OHDA | 6-hydroxydopamine |
| α-syn | α-synuclein |
| AVV | adeno-associated virus |
| CA | caffeic acid |
| CD | Crohn’s disease |
| CGA | chlorogenic acid |
| CNS | central nervous system |
| EGCs | enteric glial cells |
| ENS | enteric nervous system |
| GDNF | glial-derived neurotrophic factor |
| GFAP | glial fibrillary acidic protein |
| GI | gastrointestinal |
| IEB | intestinal epithelial barrier |
| IBD | inflammatory bowel disease |
| IBS | irritable bowel syndrome |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| IR | immunoreactivity, immunoreactive |
| KO | knockout |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| LRRK2 | leucine-rich repeat kinase 2 |
| LPS | lipopolysaccharide |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MPTP/p | MPTP/probenecid |
| MTs | metallothioneins |
| NAD | nicotine adenine dinucleotide |
| PD | Parkinson’s disease |
| p-α-syn | Phosphorylated alpha-synuclein |
| PPAR | proliferator activated receptor |
| PLP1 | proteolipid protein 1 |
| S100β | calcium binding protein B |
| TDO | tryptophan 2,3-dioxygenase |
| TNF-α | tumor necrosis factor-alpha |
| TLR | Toll-like receptors |
| UC | ulcerative colitis |
| WT | wild type |
References
- Ortega Moreno, L.; Bagues, A.; Martínez, V.; Abalo, R. New Pieces for an Old Puzzle: Approaching Parkinson’s Disease from Translatable Animal Models, Gut Microbiota Modulation, and Lipidomics. Nutrients 2023, 15, 2775. [Google Scholar] [CrossRef]
- Zhu, B.; Yin, D.; Zhao, H.; Zhang, L. The immunology of Parkinson’s disease. Semin. Immunopathol. 2022, 44, 659–672. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; Jiang, L.; Zhang, J.; Tong, X.; Chen, D.; Le, W. Intestinal Inflammation and Parkinson’s Disease. Aging Dis. 2021, 12, 2052–2068. [Google Scholar] [CrossRef]
- Fernández-Espejo, E. Agregación de alfa-sinucleína y degeneración Parkinsoniana. In Fisiología: Boletín Informativo de la SECF; Sociedad Española de Ciencias Fisiológicas: Madrid, Spain, 2013; pp. 1–3. [Google Scholar]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S36. [Google Scholar] [CrossRef]
- Hardy, J.; Lewis, P.; Revesz, T.; Lees, A.; Paisan-Ruiz, C. The genetics of Parkinson’s syndromes: A critical review. Curr. Opin. Genet. Dev. 2009, 19, 254–265. [Google Scholar] [CrossRef]
- Matsuda, N.; Tanaka, K. Does impairment of the ubiquitin-proteasome system or the autophagy-lysosome pathway predispose individuals to neurodegenerative disorders such as Parkinson’s disease? J. Alzheimer’s Dis. 2010, 19, 1–9. [Google Scholar] [CrossRef]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. neurol 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Killinger, B.A.; Madaj, Z.; Sikora, J.W.; Rey, N.; Haas, A.J.; Vepa, Y.; Lindqvist, D.; Chen, H.; Thomas, P.M.; Brundin, P.; et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5280. [Google Scholar] [CrossRef]
- Furness, J.B. The Enteric Nervous System; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Grubišić, V.; Verkhratsky, A.; Zorec, R.; Parpura, V. Enteric glia regulate gut motility in health and disease. Brain Res. Bull. 2018, 136, 109–117. [Google Scholar] [CrossRef]
- Progatzky, F.; Pachnis, V. The role of enteric glia in intestinal immunity. Curr. Opin. Immunol. 2022, 77, 102183. [Google Scholar] [CrossRef]
- Capoccia, E.; Cirillo, C.; Gigli, S.; Pesce, M.; D’Alessandro, A.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. Enteric glia: A new player in inflammatory bowel diseases. Int. J. Immunopathol. Pharmacol. 2015, 28, 443–451. [Google Scholar] [CrossRef]
- Seguella, L.; Gulbransen, B.D. Enteric glial biology, intercellular signalling and roles in gastrointestinal disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 571–587. [Google Scholar] [CrossRef]
- Drokhlyansky, E.; Smillie, C.S.; Van Wittenberghe, N.; Ericsson, M.; Griffin, G.K.; Eraslan, G.; Dionne, D.; Cuoco, M.S.; Goder-Reiser, M.N.; Sharova, T.; et al. The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 2020, 182, 1606–1622.e23. [Google Scholar] [CrossRef]
- Gershon, M.D. The enteric nervous system: A second brain. Hosp. Pract. 1999, 34, 31–52. [Google Scholar] [CrossRef]
- Boesmans, W.; Nash, A.; Tasnády, K.R.; Yang, W.; Stamp, L.; Hao, M.M. Development, Diversity, and Neurogenic Capacity of Enteric Glia. Front. Cell Dev. Biol. 2022, 19, 775102. [Google Scholar] [CrossRef]
- Yu, Y.B.; Li, Y.Q. Enteric glial cells and their role in the intestinal epithelial barrier. World J. Gastroenterol. 2014, 20, 11273–11280. [Google Scholar] [CrossRef]
- Furness, J.B. The enteric nervous system: Normal functions and enteric neuropathies. Neurogastroenterol. Motil. 2008, 20 (Suppl. S1), 32–38. [Google Scholar] [CrossRef]
- Coelho-Aguiar, J.d.M.; Bon-Frauches, A.C.; Gomes, A.L.; Veríssimo, C.P.; Aguiar, D.P.; Matias, D.; Thomasi, B.B.; Gomes, A.S.; Brito, G.A.; Moura-Neto, V. The enteric glia: Identity and functions. Glia 2015, 63, 921–935. [Google Scholar] [CrossRef]
- Holland, A.M.; Bon-Frauches, A.C.; Keszthelyi, D.; Melotte, V.; Boesmans, W. The enteric nervous system in gastrointestinal disease etiology. Cell. Mol. Life Sci. 2021, 78, 4713–4733. [Google Scholar] [CrossRef]
- Boesmans, W.; Lasrado, R.; Vanden Berghe, P.; Pachnis, V. Heterogeneity and phenotypic plasticity of glial cells in the mammalian enteric nervous system. Glia 2015, 63, 229–241. [Google Scholar] [CrossRef]
- Liu, C.; Yang, J. Enteric Glial Cells in Immunological Disorders of the Gut. Front. Cell Neurosci. 2022, 16, 895871. [Google Scholar] [CrossRef]
- López-Gómez, L.; Szymaszkiewicz, A.; Zielińska, M.; Abalo, R. Nutraceuticals and Enteric Glial Cells. Molécules 2021, 26, 3762. [Google Scholar] [CrossRef]
- Sharkey, K.A. Emerging roles for enteric glia in gastrointestinal disorders. J. Clin. Investig. 2015, 125, 918–925. [Google Scholar] [CrossRef]
- Benvenuti, L.; D’Antongiovanni, V.; Pellegrini, C.; Antonioli, L.; Bernardini, N.; Blandizzi, C.; Fornai, M. Enteric Glia at the Crossroads between Intestinal Immune System and Epithelial Barrier: Implications for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 9199. [Google Scholar] [CrossRef]
- Kim, J.; Lo, L.; Dormand, E.; Anderson, D.J. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron 2003, 8, 17–31. [Google Scholar] [CrossRef]
- Rao, M.; Nelms, B.D.; Dong, L.; Salinas-Rios, V.; Rutlin, M.; Gershon, M.D.; Corfas, G. Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 2015, 63, 2040–2057. [Google Scholar] [CrossRef]
- Guillamón-Vivancos, T.; Gómez-Pinedo, U.; Matías-Guiu, J. Astrocytes in neurodegenerative diseases (I): Function and molecular description. Neurologia 2015, 30, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, D.; Loris, E.; Maas-Omlor, S.; Huang, W.; Scheller, A.; Kirchhoff, F.; Schäfer, K.H. Enteric Glia: S100, GFAP, and Beyond. Anat. Rec. 2019, 302, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- López-Gómez, L.; Abalo, R. Modulation of Enteric Glial Cells by Nutraceuticals during Pathological Processes. In Natural Molecules in Neuroprotection and Neurotoxicity; De, O., Ed.; Academic Press: Cambridge, MA, USA, 2023. [Google Scholar]
- Rolle, U.; Nemeth, L.; Puri, P. Nitrergic innervation of the normal gut and in motility disorders of childhood. J. Pediatr. Surg. 2002, 37, 551–567. [Google Scholar] [CrossRef] [PubMed]
- McCann, C.J.; Alves, M.M.; Brosens, E.; Natarajan, D.; Perin, S.; Chapman, C.; Hofstra, R.M.; Burns, A.J.; Thapar, N. Neuronal Development and Onset of Electrical Activity in the Human Enteric Nervous System. Gastroenterology 2019, 156, 1483–1495.e6. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.T.; Costa, D.V.; Gomes, A.S.; Martins, C.S.; Silva, A.M.; Coelho-Aguiar, J.M.; Castelucci, P.; Lima-Júnior, R.C.; Leitão, R.F.; Moura-Neto, V.; et al. The involvement of mast cells in the irinotecan-induced enteric neurons loss and reactive gliosis. J. Neuroinflamm. 2017, 14, 79. [Google Scholar] [CrossRef]
- da Cunha Franceschi, R.; Nardin, P.; Machado, C.V.; Tortorelli, L.S.; Martinez-Pereira, M.A.; Zanotto, C.; Gonçalves, C.A.; Zancan, D.M. Enteric glial reactivity to systemic LPS administration: Changes in GFAP and S100B protein. Neurosci. Res. 2017, 119, 15–23. [Google Scholar] [CrossRef]
- Thacker, M.; Rivera, L.R.; Cho, H.J.; Furness, J.B. The relationship between glial distortion and neuronal changes following intestinal ischemia and reperfusion. Neurogastroenterol. Motil. 2011, 23, e500–e509. [Google Scholar] [CrossRef]
- Romero-Trujillo, J.O.; Frank-Márquez, N.; Cervantes-Bustamante, R.; Cadena-León, J.F.; Montijo-Barrios, E.; Zárate-Mondragón, F.; Cázares-Méndez, J.M.; Ramírez-Mayans, J. Enteric nervous system and gastrointestinal motility. Acta Pediátrica De México 2012, 33, 207–214. [Google Scholar]
- Ruhl, A.; Franzke, S.; Collins, S.M.; Stremmel, W. Interleukin-6 expression and regulation in rat enteric glial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1163–G1171. [Google Scholar] [CrossRef]
- Meir, M.; Kannapin, F.; Diefenbacher, M.; Ghoreishi, Y.; Kollmann, C.; Flemming, S.; Germer, C.T.; Waschke, J.; Leven, P.; Schneider, R.; et al. Intestinal Epithelial Barrier Maturation by Enteric Glial Cells Is GDNF-Dependent. Int. J. Mol. Sci. 2021, 22, 1887. [Google Scholar] [CrossRef]
- Cornet, A.; Savidge, T.C.; Cabarrocas, J.; Deng, W.L.; Colombel, J.F.; Lassmann, H.; Desreumaux, P.; Liblau, R.S. Enterocolitis induced by autoimmune targeting of enteric glial cells: A possible mechanism in Crohn’s disease? Proc. Natl. Acad. Sci. USA 2001, 98, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Sarnelli, G.; Esposito, G.; Grosso, M.; Petruzzelli, R.; Izzo, P.; Calì, G.; D’Armiento, F.P.; Rocco, A.; Nardone, G.; et al. Increased mucosal nitric oxide production in ulcerative colitis is mediated in part by the enteroglial-derived S100B protein. Neurogastroenterol. Motil. 2009, 21, 1209-e112. [Google Scholar] [CrossRef] [PubMed]
- von Boyen, G.B.; Schulte, N.; Pflüger, C.; Spaniol, U.; Hartmann, C.; Steinkamp, M. Distribution of enteric glia and GDNF during gut inflammation. BMC Gastroenterol. 2011, 11, 3. [Google Scholar] [CrossRef]
- Pochard, C.; Coquenlorge, S.; Freyssinet, M.; Naveilhan, P.; Bourreille, A.; Neunlist, M.; Rolli-Derkinderen, M. The multiple faces of inflammatory enteric glial cells: Is Crohn’s disease a gliopathy? Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G1–G11. [Google Scholar] [CrossRef] [PubMed]
- Hagbom, M.; De Faria, F.M.; Winberg, M.E.; Westerberg, S.; Nordgren, J.; Sharma, S.; Keita, Å.V.; Loitto, V.; Magnusson, K.E.; Svensson, L. Neurotrophic factors protect the intestinal barrier from rotavirus insult in mice. MBio 2020, 11, e02834–e02919. [Google Scholar] [CrossRef]
- Coquenlorge, S.; Van Landeghem, L.; Jaulin, J.; Cenac, N.; Vergnolle, N.; Duchalais, E.; Neunlist, M.; Rolli-Derkinderen, M. The arachidonic acid metabolite 11β-ProstaglandinF2α controls intestinal epithelial healing: Deficiency in patients with Crohn’s disease. Sci. Rep. 2016, 6, 25203. [Google Scholar] [CrossRef]
- Zeledón Corrales, N.; Serrano Suárez, J.A.; Fernández Agudelo, S. Irritable Bowel Syndrome. Rev. Méd. Sinerg. 2021, 6, e645. [Google Scholar] [CrossRef]
- Labanski, A.; Langhorst, J.; Engler, H.; Elsenbruch, S. Stress and the brain-gut axis in functional and chronic-inflammatory gastrointestinal diseases: A transdisciplinary challenge. Psychoneuroendocrinology 2020, 111, 104501. [Google Scholar] [CrossRef]
- Niesler, B.; Kuerten, S.; Demir, I.E.; Schäfer, K.H. Disorders of the enteric nervous system—A holistic view. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 393–410. [Google Scholar] [CrossRef]
- Rosenbaum, C.; Schick, M.A.; Wollborn, J.; Heider, A.; Scholz, C.J.; Cecil, A.; Niesler, B.; Hirrlinger, J.; Walles, H.; Metzger, M. Activation of Myenteric Glia during Acute Inflammation In Vitro and In Vivo. PLoS ONE 2016, 11, e0151335. [Google Scholar] [CrossRef]
- Voss, U.; Sand, E.; Olde, B.; Ekblad, E. Enteric neuropathy can be induced by high fat diet in vivo and palmitic acid exposure in vitro. PLoS ONE 2013, 8, e81413. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.F.; Shen, Y.Q. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s Disease. Ageing Res. Rev. 2018, 45, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, S.; Le, W. Intestinal Permeability, Dysbiosis, Inflammation and Enteric Glia Cells: The Intestinal Etiology of Parkinson’s Disease. Aging Dis. 2022, 13, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Claudino Dos Santos, J.C.; Lima, M.P.P.; Brito, G.A.C.; Viana, G.S.B. Role of enteric glia and microbiota-gut-brain axis in parkinson disease pathogenesis. Ageing Res. Rev. 2023, 84, 101812. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.K.; Grubišić, V.; Gulbransen, B.D. Enteric Glia Regulate Lymphocyte Activation via Autophagy-Mediated MHC-II Expression. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1215–1237. [Google Scholar] [CrossRef]
- Yemula, N.; Dietrich, C.; Dostal, V.; Hornberger, M. Parkinson’s Disease and the Gut: Symptoms, Nutrition, and Microbiota. J. Parkinsons Dis. 2021, 11, 1491–1505. [Google Scholar] [CrossRef]
- Clairembault, T.; Leclair-Visonneau, L.; Neunlist, M.; Derkinderen, P. Enteric glial cells: New players in Parkinson’s disease? Mov. Disord. 2015, 30, 494–498. [Google Scholar] [CrossRef]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Trichka, J.; Zou, W.Q. Modulation of Neuroinflammation by the Gut Microbiota in Prion and Prion-Like Diseases. Pathogens 2021, 10, 887. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Miyazaki, I.; Sogawa, N.; Miyoshi, K.; Asanuma, M. Neuroprotective effects of metallothionein against rotenone-induced myenteric neurodegeneration in parkinsonian mice. Neurotox. Res. 2014, 26, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Miyazaki, I.; Miyoshi, K.; Asanuma, M. Long-Term Systemic Exposure to Rotenone Induces Central and Peripheral Pathology of Parkinson’s Disease in Mice. Neurochem. Res. 2015, 40, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Wada, K.; Kikuoka, R.; Kitamura, Y.; Asanuma, M. Effects of Enteric Environmental Modification by Coffee Components on Neurodegeneration in Rotenone-Treated Mice. Cells 2019, 8, 221. [Google Scholar] [CrossRef]
- Palanisamy, B.N.; Sarkar, S.; Malovic, E.; Samidurai, M.; Charli, A.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Environmental neurotoxic pesticide exposure induces gut inflammation and enteric neuronal degeneration by impairing enteric glial mitochondrial function in pesticide models of Parkinson’s disease: Potential relevance to gut-brain axis inflammation in Parkinson’s disease pathogenesis. Int. J. Biochem. Cell Biol. 2022, 147, 106225. [Google Scholar]
- Dodiya, H.B.; Forsyth, C.B.; Voigt, R.M.; Engen, P.A.; Patel, J.; Shaikh, M.; Green, S.J.; Naqib, A.; Roy, A.; Kordower, J.H.; et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol. Dis. 2020, 135, 104352. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Grobben, Y.; Willemsen-Seegers, N.; Hartog, M.; Tutone, M.; Muller, M.; Adolfs, Y.; Pasterkamp, R.J.; Vu-Pham, D.; van Doornmalen, A.M.; et al. Pharmacological validation of TDO as a target for Parkinson’s disease. FEBS J. 2021, 288, 4311–4331. [Google Scholar] [CrossRef]
- Chaumette, T.; Lebouvier, T.; Aubert, P.; Lardeux, B.; Qin, C.; Li, Q.; Accary, D.; Bézard, E.; Bruley des Varannes, S.; Derkinderen, P.; et al. Neurochemical plasticity in the enteric nervous system of a primate animal model of experimental Parkinsonism. Neurogastroenterol. Motil. 2009, 21, 215–222. [Google Scholar] [CrossRef]
- Coletto, E.; Dolan, J.S.; Pritchard, S.; Gant, A.; Hikima, A.; Jackson, M.J.; Benham, C.D.; Chaudhuri, K.R.; Rose, S.; Jenner, P.; et al. Contractile dysfunction and nitrergic dysregulation in small intestine of a primate model of Parkinson’s disease. NPJ Park. Dis. 2019, 5, 10. [Google Scholar] [CrossRef]
- Heng, Y.; Li, Y.Y.; Wen, L.; Yan, J.Q.; Chen, N.H.; Yuan, Y.H. Gastric Enteric Glial Cells: A New Contributor to the Synucleinopathies in the MPTP-Induced Parkinsonism Mouse. Molecules 2022, 27, 7414. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; Colucci, R.; Tirotta, E.; Blandini, F.; Levandis, G.; Cerri, S.; Segnani, C.; Ippolito, C.; Bernardini, N.; et al. Alteration of colonic excitatory tachykininergic motility and enteric inflammation following dopaminergic nigrostriatal neurodegeneration. J. Neuroinflammation 2016, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Ippolito, C.; Segnani, C.; Dolfi, A.; Errede, M.; Virgintino, D.; Fornai, M.; Antonioli, L.; Garelli, F.; Nericcio, A.; et al. Pathological remodelling of colonic wall following dopaminergic nigrostriatal neurodegeneration. Neurobiol. Dis. 2020, 139, 104821. [Google Scholar] [CrossRef] [PubMed]
- Thomasi, B.B.M.; Valdetaro, L.; Ricciardi, M.C.G.; Hayashide, L.; Fernandes, A.C.M.N.; Mussauer, A.; da Silva, M.L.; da Cunha Faria-Melibeu, A.; Ribeiro, M.G.L.; de Mattos Coelho-Aguiar, J.; et al. Enteric glial cell reactivity in colonic layers and mucosal modulation in a mouse model of Parkinson’s disease induced by 6-hydroxydopamine. Brain Res. Bull. 2022, 187, 111–121. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, S.M.; Crowley, E.K.; Brown, J.R.; O’Sullivan, O.; O’Leary, O.F.; Timmons, S.; Nolan, Y.M.; Clarke, D.J.; Hyland, N.P.; Joyce, S.A.; et al. Nigral overexpression of α-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol. Motil. 2020, 32, e13726. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; D’Antongiovanni, V.; Miraglia, F.; Rota, L.; Benvenuti, L.; Di Salvo, C.; Testa, G.; Capsoni, S.; Carta, G.; Antonioli, L.; et al. Enteric α-synuclein impairs intestinal epithelial barrier through caspase-1-inflammasome signaling in Parkinson’s disease before brain pathology. NPJ Park. Dis. 2022, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar]
- Langston, J.W.; Irwin, I.; Langston, E.B.; Forno, L.S. 1-Methyl-4-phenylpyridinium ion (MPP+): Identification of a metabolite of MPTP, a toxin selective to the substantia nigra. Neurosci. Lett. 1984, 48, 87–92. [Google Scholar] [CrossRef]
- Hare, D.J.; Adlard, P.A.; Doble, P.A.; Finkelstein, D.I. Metallobiology of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Metallomics 2014, 5, 91–109. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Noelker, C.; Grünewald, A.; Vulinović, F.; Guerreiro, S.; Fuchs, J.; Lu, L.; Lombès, A.; Hirsch, E.C.; Oertel, W.H.; et al. Probenecid potentiates MPTP/MPP+ toxicity by interference with cellular energy metabolism. J. Neurochem. 2013, 127, 782–792. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Kelly, R.; Bemelmans, A.P.; Joséphine, C.; Brouillet, E.; McKernan, D.P.; Dowd, E. Time-Course of Alterations in the Endocannabinoid System after Viral-Mediated Overexpression of α-Synuclein in the Rat Brain. Molecules 2022, 27, 507. [Google Scholar] [CrossRef] [PubMed]
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl. Neurodegener 2019, 8, 5. [Google Scholar] [CrossRef]
- Clairembault, T.; Kamphuis, W.; Leclair-Visonneau, L.; Rolli-Derkinderen, M.; Coron, E.; Neunlist, M.; Hol, E.M.; Derkinderen, P. Enteric GFAP expression and phosphorylation in Parkinson’s disease. J. Neurochem. 2014, 130, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Bellini, G.; Benvenuti, L.; Ippolito, C.; Frosini, D.; Segnani, C.; Rettura, F.; Pancetti, A.; Bertani, L.; D’Antongiovanni, V.; Palermo, G.; et al. Intestinal histomorphological and molecular alterations in patients with Parkinson’s disease. Eur. J. Neurol. 2023, 30, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Du, Y.; Li, X.; Kambey, P.A.; Wang, L.; Xia, Y.; Tang, C.; Shi, M.; Li, Z.L.; Xin, Z.E.; et al. Lower GDNF Serum Level Is a Possible Risk Factor for Constipation in Patients with Parkinson Disease: A Case-Control Study. Front. Neurol. 2022, 12, 777591. [Google Scholar] [CrossRef] [PubMed]
- de Guilhem de Lataillade, A.; Caillaud, M.; Oullier, T.; Naveilhan, P.; Pellegrini, C.; Tolosa, E.; Neunlist, M.; Rolli-Derkinderen, M.; Gelpi, E.; Derkinderen, P. LRRK2 expression in normal and pathologic human gut and in rodent enteric neural cell lines. J. Neurochem. 2023, 164, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Emmi, A.; Sandre, M.; Russo, F.P.; Tombesi, G.; Garrì, F.; Campagnolo, M.; Carecchio, M.; Biundo, R.; Spolverato, G.; Macchi, V.; et al. Duodenal alpha-Synuclein Pathology and Enteric Gliosis in Advanced Parkinson’s Disease. Mov. Disord. 2023, 38, 885–894. [Google Scholar] [CrossRef]
- Barrenschee, M.; Zorenkov, D.; Böttner, M.; Lange, C.; Cossais, F.; Scharf, A.B.; Deuschl, G.; Schneider, S.A.; Ellrichmann, M.; Fritscher-Ravens, A.; et al. Distinct pattern of enteric phospho-alpha-synuclein aggregates and gene expression profiles in patients with Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 1. [Google Scholar] [CrossRef]
- Pellegrini, C.; Antonioli, L.; Colucci, R.; Ballabeni, V.; Barocelli, E.; Bernardini, N.; Blandizzi, C.; Fornai, M. Gastric motor dysfunctions in Parkinson’s disease: Current pre-clinical evidence. Park. Relat. Disord. 2015, 21, 1407–1414. [Google Scholar] [CrossRef]
- Harapan, B.N.; Frydrychowicz, C.; Classen, J.; Wittekind, C.; Gradistanac, T.; Rumpf, J.J.; Mueller, W. No enhanced (p-) α-synuclein deposition in gastrointestinal tissue of Parkinson’s disease patients. Park. Relat. Disord. 2020, 80, 82–88. [Google Scholar] [CrossRef]
- Takemura, M.; Gomi, H.; Colucci-Guyon, E.; Itohara, S. Protective role of phosphorylation in turnover of glial fibrillary acidic protein in mice. J. Neurosci. 2002, 22, 6972–6979. [Google Scholar] [CrossRef]
- Chandra, R.; Hiniker, A.; Kuo, Y.M.; Nussbaum, R.L.; Liddle, R.A. α-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2017, 2, e92295. [Google Scholar] [CrossRef]
- Hirayama, M.; Ohno, K. Parkinson’s Disease and Gut Microbiota. Ann. Nutr. Metab. 2021, 77 (Suppl. S2), 28–35. [Google Scholar] [CrossRef]
- López-Gómez, L.; Szymaszkiewicz, A.; Zielińska, M.; Abalo, R. The Enteric Glia and Its Modulation by the Endocannabinoid System, a New Target for Cannabinoid-Based Nutraceuticals? Molecules 2022, 27, 6773. [Google Scholar] [CrossRef]
- López-Gómez, L.; Alcorta, A.; Abalo, R. Probiotics and Probiotic-like Agents against Chemotherapy-Induced Intestinal Mucositis: A Narrative Review. J. Pers. Med. 2023, 13, 1487. [Google Scholar] [CrossRef]

{kind=link}
| Type | Subtype | Location | Functions |
|---|---|---|---|
| Intraganglionar | Myenteric type I | Small and extended star-shaped cells that surround the neurons in the myenteric ganglia | Modulation of enteric neuron activity |
| Oxidative stress regulation | |||
| Trophic support | |||
| Neuroinflammation regulation | |||
| Gliogenesis | |||
| Neurogenesis | |||
| Mucosal glia replenishment | |||
| Submucosal type I | Associated with neurons within submucosal ganglia | Modulation of secretory neuron activity | |
| Extraganglionar | Interganglionar type II | Located in the interganglionic fiber tracts | They propagate the signal in the glial network |
| Mucosa type III | Some follow nerve fibers, while others terminate in the mucosal epithelium | Influence the maturation of epithelial cells | |
| Potentially modulate immune responses Identification from postnatal development | |||
| Myenteric plexus/submucosal plexus type III | Located in the extraganglionic regions at the level of the myenteric and submucosal plexuses | Unknown | |
| Intramuscular type IV | Associated with nerve fibers in the circular and longitudinal muscle layers of smooth muscles | Unknown |
| Type | Characteristics | Mechanisms of Action |
|---|---|---|
| Activated | Controls the activity of surrounding cells | The enteric glial activation encoded by intracellular Ca2+ responses modulate enteric excitatory motor and secretomotor neurocircuits |
| Exerts beneficial homeostatic effects | ||
| Responds to physiological stimuli | ||
| Reactive | Responds to physiopathological disturbances of any severity | Responds to intestinal inflammation. |
| Contributes to neuronal death during acute intestinal inflammation | ||
| Changes can alter glial activities through gain or loss of functions, which can be beneficial or detrimental | Contributes to vagal anti-inflammatory effects on resident intestinal immune cells after intestinal injury | |
| Dysfunctional | Dysfunctional or maladaptive response of glial cells | Altered enteric glial networks, displaying dysfunctional responses in patients with different GI disorders, including IBD, immunological disorders of the gut or PD |
| Exerts harmful effects contributing to a disease, in addition to being permanent |
| Marker | Characteristics | Functions |
|---|---|---|
| Nuclear transcription factor (Sox-10) | Key to the development of the neuronal crest cells and the enteric glia | Crucial role in neuronal crest cells and peripheral glia differentiation and maintenance |
| Specific marker for EGCs progenitors | Controls and modulates the expression of several key genes for early ENS development | |
| Found in glial precursors and in most of the mature and immature EGCs | Promotes the expression of various transcription factors crucial for neuronal differentiation, such as Phox2b and Ascl1 | |
| Glial fibrillary acidic protein (GFAP) | It is found along neuronal plexuses. | There is an increased GFAP intensity when the tissue is inflamed or next to colonic cancer |
| Does not occur prenatally | ||
| All subtypes of enteric glia within the mouse ileum express GFAP, but at different levels. | ||
| Dynamic expression that varies depending on the glial state | ||
| GFAPκ is the main isoform in colonic EGCs relative to GFAPα and GFAPδ | ||
| Calcium-binding protein (S100β) | Expressed by both progenitors and differentiated enteric glial cells | Among other functions, this protein contributes to structural support and regulation of the immune response |
| Proteolipid protein 1 (PLP1) | In adult mice, it is expressed in both ENS plexuses, in both, the small and large intestine | Unknown |
| PD Induction | Animal | Findings | Ref |
|---|---|---|---|
| Rotenone | C57BL mice | Increased MT levels. | [65] |
| EGCs activation (GFAP-positive) | |||
| MT KO mice | Severe myenteric neuronal damage | [65] | |
| Reduced EGCs activation | |||
| Aggravation of lipid peroxidation | |||
| C57BL mice | Increased GFAP-IR in the myenteric plexus | [66] | |
| Activated EGCs before neurodegeneration in the CNS | |||
| Lack of activation of EGCs at early stages | |||
| C57BL mice | No MT presence in the intestine of mice | [67] | |
| Cell cultures | CA or CGA prevented rotenone-induced downregulation of MT in cultured cells | ||
| C57BL mice Cell cultures | Increased GFAP staining in the myenteric plexus | [68] | |
| Impaired mitochondrial bioenergetics | |||
| Activation of inflammatory pathways | |||
| C57BL/6J mice | TLR4-mediated gut inflammation | [55] | |
| TLR4-knockout mice | GFAP staining was unaffected by rotenone administration | ||
| C57BL Mice | Restraint stress exacerbated rotenone-induced activation of EGCs | [69] | |
| C57BL/6NCrl mice | A TDO inhibitor decreased rotenone-induced labelling of GFAP | [70] | |
| MPTP and MPTP/p | Macaca mulatta | No differences in the number/phenotype of Sox-10 IR EGCs | [71] |
| EGCs are not a primary target of MPTP in the ENS | |||
| The ratio of EGC to neurons was decreased by MPTP | |||
| Less protection of myenteric neurons | |||
| Marmoset | EGCs IR to Sox-10 were increased | [72] | |
| MPTP treatment led to inflammation of the ileum | |||
| C5BL/6 mice | Chronic MPTP/p increased aggregated and nitrated α-syn in GFAP-IR EGCs | [73] | |
| Acute, elevated 4-HNE in the EGCs after 3 h | |||
| EGCs could be initial contributors to synucleinopathies in the stomach | |||
| 6-OHDA | Sprague-Dawley rats | GFAP IR of EGCs was increased | [74] |
| Sprague-Dawley rats | Density of S100β IR EGCs was increased | [75] | |
| Nigrostriatal neurodegeneration leads to an increased presence of EGCs in the mucosa | |||
| C57BL/6 male mice | Dual response of EGCs: can promote inflammation or intestinal tissue protection | [76] | |
| Virus AVV | Sprague-Dawley rats | No changes in EGCs in the ileal submucosal plexus | [77] |
| Increase in glial number in the myenteric plexus | |||
| Voluntary running protected from increased EGCs | |||
| A53 α-syn mouse model | Mutant mice expressing human A53T | Increased GFAP IR EGCs in the mucosa, submucosa and myenteric plexus at 3 months of age | [78] |
| Co-localization of GFAP-IR EGCs and TLR2 IR |
| Number of Samples | Samples | Findings | Refs |
|---|---|---|---|
| Control and PD patients (2 samples each) | Ascending Colon | GFAP and Sox-10 were significantly elevated | [63] |
| No significant changes in the S100β marker | |||
| Levels of glial markers are negatively correlated with disease duration | |||
| 24 PD, 6 progressive supranuclear palsy and 6 multiple system atrophy patients | Colonic biopsies | Hypophosphorylation of GFAP in EGCs during PD | [86] |
| GFAPκ was the major isoform in colonic EGCs | |||
| 19 asymptomatic PD patients | Colonic biopsies | Increase in S100β-positive glial cells | [87] |
| Activation of enteric glial cells | |||
| Abnormal tissue repair with development of fibrosis in the mucosa of PD patients | |||
| 128 patients with PD | Serum | GDNF may act as a protective factor in the prevention of constipation | [88] |
| Reduction in GDNF affects the integrity of intestinal mucosal barrier | |||
| 16 Lewy’s body, 12 non-Lewy’s body disorders cases | Colonic biopsies | Enteric glial cells express LRRK2 | [89] |
| EGC-expressed LRRK2 could participate in the modulation of intestinal α-syn aggregation and inflammation in the gut | |||
| 18 patients with advanced PD, 4 untreated patients with early PD | Duodenal biopsies | Increased size and density of GFAP-positive EGCs suggesting reactive gliosis | [90] |
| No colocalization between markers α-syn-5G4 and GFAP antibodies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montalbán-Rodríguez, A.; Abalo, R.; López-Gómez, L. From the Gut to the Brain: The Role of Enteric Glial Cells and Their Involvement in the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 1294. https://doi.org/10.3390/ijms25021294
Montalbán-Rodríguez A, Abalo R, López-Gómez L. From the Gut to the Brain: The Role of Enteric Glial Cells and Their Involvement in the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(2):1294. https://doi.org/10.3390/ijms25021294
Chicago/Turabian StyleMontalbán-Rodríguez, Alba, Raquel Abalo, and Laura López-Gómez. 2024. "From the Gut to the Brain: The Role of Enteric Glial Cells and Their Involvement in the Pathogenesis of Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 2: 1294. https://doi.org/10.3390/ijms25021294
APA StyleMontalbán-Rodríguez, A., Abalo, R., & López-Gómez, L. (2024). From the Gut to the Brain: The Role of Enteric Glial Cells and Their Involvement in the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences, 25(2), 1294. https://doi.org/10.3390/ijms25021294