

Key Proteins of Replication Stress Response and Cell Cycle Control as Cancer Therapy Targets

 , ,
, ,
Abstract
1. Introduction
2. Replication Stress Mechanisms in Cancer
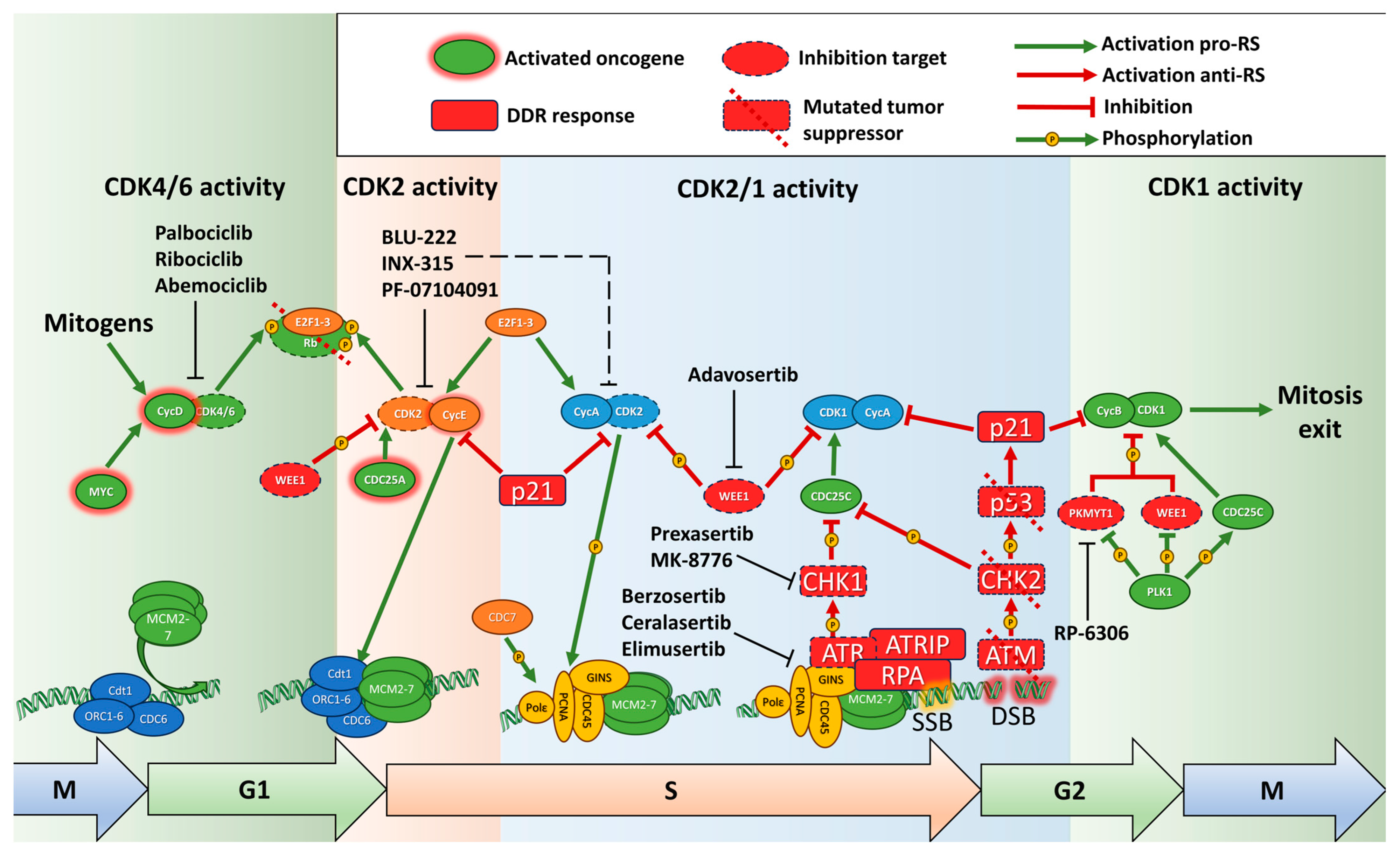
2.1. Overview of Replication Control
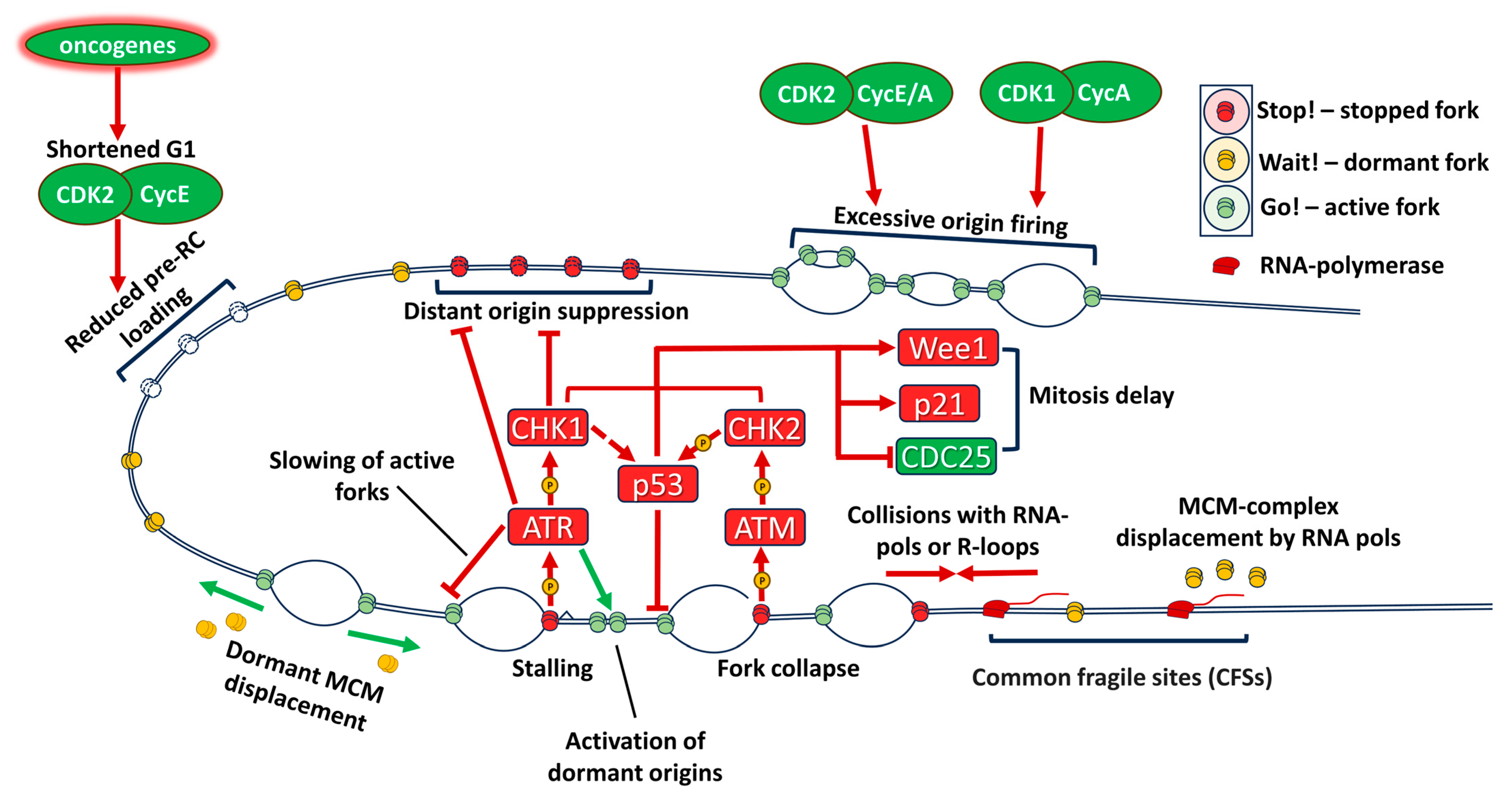
2.2. Oncogenic Transformation Leads to Replication Stress
2.3. Transcription-Replication Conflicts Are Increased in Cancers
2.4. Rb/E2F Pathway, Transition to S Phase and Replication Stress
2.5. Replication in G2 Phase and Transition to Mitosis
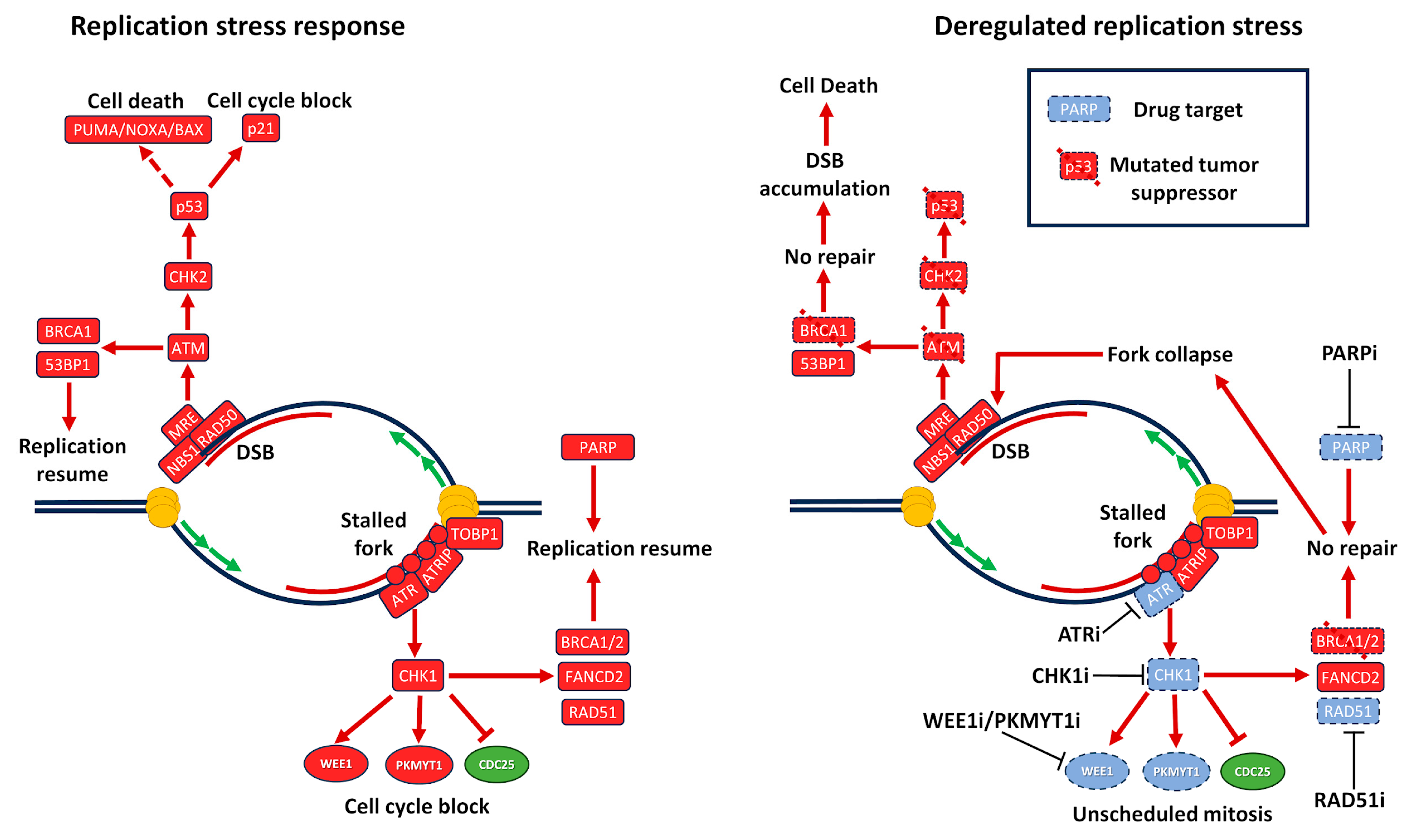
2.6. DNA Damage Response and Replication Stress
2.7. P53 and RS
3. Small Molecule Inducers of Replication Stress
3.1. ATR-CHK1 Inhibitors

{kind=link}
{kind=link}
{kind=link}
| Compound | Study Phases | Key Indications | References |
|---|---|---|---|
| Berzosertib (M6620, VX-970) | Phase 2 (7 trials) | Different types of cancer, including DDR deficient and TP53 mutant tumors | NCT02595892 [107] NCT04266912 NCT03517969 NCT02567409 [145] NCT03896503 NCT04216316 NCT03641313 NCT03718091 (completed) NCT02487095 [146,147] |
| Phase 1 (9 trials) | Different types of cancer, including DDR solid tumors | NCT02723864 NCT02589522 NCT02595931 NCT05246111 NCT04266912 NCT02567422 NCT02627443 NCT04216316 NCT04052555 NCT02157792 [101,102,103,108] | |
| Ceralasertib (AZD 6738) | NSCLC | NCT05450692 | |
| Phase 2 (28 trials) | Different types of solid tumors, including NSCLC, breast and ovarian cancers | NCT02264678 NCT04417062 NCT05061134 NCT05941897 NCT04564027 NCT05582538 NCT03801369 NCT04699838 NCT04090567 NCT03579316 NCT03878095 (suspended) NCT04239014 (withdrawn) NCT03334617 NCT03330847 NCT02937818 NCT03833440 NCT02813135 NCT04298021 NCT04298008 NCT03462342 NCT03428607 (completed) NCT03780608 NCT04065269 NCT04361825 NCT02576444 (terminated) NCT03740893 NCT03182634 NCT02664935 | |
| Phase 1 (13 trials) | Different types of solid tumors and leukemias | NCT05469919 NCT02264678 NCT05514132 NCT03328273 NCT03022409 (completed) NCT04704661 NCT03669601 NCT02630199 NCT03770429 NCT02223923 NCT01955668 (completed) NCT03527147 (completed) | |
| Elimusertib (BAY1895344) | Phase 2 (1 trial) | Relapsed or refractory solid tumors | NCT05071209 |
| Phase 1 (10 trials) | Different types of carcinomas and lymphomas | NCT05010096 (withdrawn) NCT03188965 (completed) NCT04095273 (completed) NCT05071209 NCT04616534 NCT04267939 NCT04491942 NCT04535401 NCT04576091 NCT04514497 | |
| Gartisertib (M4344, VX-803) | Phase 2 (1 trial) | Advanced breast cancer with DDR mutations | NCT04655183 (withdrawn) |
| Phase 1 (1 trial) | Solid tumors | NCT02278250 (completed) | |
| Camonsertib (RP-3500) | Phase 1/2 (2 trials) | Advanced solid tumors | NCT04972110 NCT04497116 |
| ART0380 | Phase 2 (1 trial) | Advanced tumors | NCT05798611 |
| Phase 1/2 (1 trial) | Advanced tumors | NCT04657068 |
| Compound | Study Phases | Key Indications | References |
|---|---|---|---|
| Prexasertib (LY2606368, ACR-368) | Phase 2 (7 trials) | Different types of tumors, including small cell lung cancer, ovarian cancer, etc. | NCT02735980 (completed) [159] NCT03414047 (completed) NCT02203513 (terminated) NCT02873975 (completed) NCT04095221 NCT04032080 (completed) NCT05548296 |
| Phase 1 (14 trials) | Different types of solid tumors and leukemias | NCT02778126 (completed) NCT02514603 (completed) NCT03495323 (completed) NCT02860780 (completed) NCT01115790 (completed) NCT03057145 (completed) NCT04095221 NCT02808650 (completed) NCT04023669 NCT05548296 NCT03735446 (terminated) NCT02649764 (completed) NCT02124148 (completed) NCT02555644 (completed) | |
| SRA 737 | Phase 1/2 (2 trials) | Advanced solid tumors or non-Hodgkin’s lymphoma | NCT02797964 [159,160] NCT02797977 |
| MK-8776 (SCH 900776) | Phase 2 (1 trial) | Leukemias | NCT00907517 (terminated) [161] |
| Phase 1 (2 trials) | Solid tumors, leukemias and lymphomas | NCT00779584 (completed) |
3.2. PARP Inhibitors
| Compound | Study Phases | Key Indications | References |
|---|---|---|---|
| Olaparib | Phase 4 (4 trials) | Ovarian cancer (3 trials), prostate cancer (1 trial), metastatic breast cancer (1 trial) | [175,176,177,178] |
| Phase 3 (37 trials) | Ovarian cancer (more than 30 trials), breast cancer (13 trials), prostate cancer (4 trials) | ||
| Phase 2 (more than 200 trials) | Ovarian cancer (more than 30 trials), breast cancer (more than 30 trials), prostate cancer (more than 20 trials), lung cancer (more than 20 trials) | ||
| Phase 1 (more than 100 trials) | Ovarian cancer (more than 30 trials), breast cancer (more than 20 trials), prostate cancer (12 trials), lung cancer (10 trials) | ||
| Niraparib | Phase 4 (3 trials) | Ovarian cancer (3 trials) | [179,180,181,182] |
| Phase 3 (23 trials) | Ovarian cancer (12 trials), fallopian tube cancer (5 trials), prostate cancer (3 trials), breast cancer (2 trials), | ||
| Phase 2 (more than 100 trials) | Ovarian cancer (more than 30 trials), breast cancer (15 trials), fallopian tube cancer (9 trials) | ||
| Phase 1 (62 trials) | Ovarian cancer (19 trials), breast cancer (13 trials), prostate cancer (7 trials) | ||
| Rucaparib | Phase 3 (8 trials) | Ovarian cancer (4 trials), fallopian tube cancer (4 trials), prostate cancer 2 trials) | [183,184,185,186] |
| Phase 2 (40 trials) | Ovarian cancer (8 trials), prostate cancer (7 trials), breast cancer (4 trials), | ||
| Phase 1 (24 trials) | Ovarian cancer (7 trials), breast cancer (4 trials), prostate cancer (4 trials) | ||
| Talazoparib | Phase 3 (5 trials) | Ovarian cancer (2 trials), breast cancer (1 trial), prostate cancer (1 trial) | [187,188,189,190] |
| Phase 2 (64 trials) | Breast cancer (17 trials), prostate cancer (8 trials), ovarian cancer (4 trials) | ||
| Phase 1 (51 trials) | Breast cancer (14 trials), prostate cancer (5 trials), ovarian cancer (4 trials) |
3.3. WEE1 and PKMYT1 Inhibitors
| Compound | Study Phases | Key Indications | References |
|---|---|---|---|
| Adavosertib (AZD1775) | Phase 2 (32 trials, including 7 terminated and 1 withdrawn) | Solid tumors, harboring CCNE1 amplification, ovarian (2 studies), neuroblastoma, medulloblastoma, and rhabdomyosarcoma | [194,195,208,209,210,211,212,213,214,215] |
| Phase 1 (34 trials, including 6 terminated and 1 withdrawn) | HNSCC, uterine cancers, TNBC, pancreatic cancer, acute myeloid leukemia, glioblastoma | [192,193,216,217,218,219,220,221,222,223,224,225,226,227,228] |
3.4. CDK Inhibitors and RS
| Compound | Study Phases | Key Indications | References |
|---|---|---|---|
| BLU-222 | Phase 1/2 (1 trial) | Solid tumors, including CCNE1-amplified, ovarian carcinoma, breast cancer, endometrial and gastric cancer | NCT05252416 |
| INX-315 | Phase 1/2 (1 trial) | Solid tumors, including breast cancer who progressed on a prior CDK4/6i regimen, and CCNE1-amplified solid tumors | NCT05735080 |
| PF-07104091 | Phase 1/2 (2 trials) | Small cell lung cancer, ovarian cancer, breast cancer | NCT04553133NCT05262400 |
| ARTS-021 | Phase 1/2 (1 trial) | CCNE1-amplified solid tumors | NCT05867251 |
| INCB123667 | Phase 1 (1 trial) | Solid tumors | NCT05238922 |
3.5. Biomarkers of Replication Stress and Response to RS-Inducing Drugs
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Anti-PD-L1 | anti-programmed death ligand 1 |
| ATM | ataxia telangiectasia mutated |
| ATMi | ATM inhibitor(s) |
| ATR | ataxia telangiectasia and Rad3 related protein |
| ATRi | ATR inhibitor(s) |
| BER | base excision repair |
| CDK(s) | Cyclin dependent kinase(s) |
| CHK | checkpoint kinase |
| CHK1i | CHK1 inhibitor(s) |
| DLBCL | diffuse large B-cell lymphoma |
| DDR | DNA damage response |
| DSB(s) | double-strand DNA break(s) |
| HGSOC | high-grade serous ovarian cancer |
| HNSCC | head and neck squamous cell carcinoma |
| HPV | human papilloma virus |
| HR | homologous recombination |
| KO | knockout |
| LMW-E | low-molecular-weight Cyclin E |
| NHEJ | non-homologous end-joining repair |
| NSCLC | non-small cell lung cancer |
| PARP | poly (ADP-ribose) polymerase |
| PARPi | PARP inhibitor(s) |
| PDAC | pancreatic ductal adenocarcinoma |
| PDX | patient-derived xenograft model(s) |
| PKMYT1i | PKMYT1 inhibitor(s) |
| RPA | replication protein A |
| RS | replication stress |
| SSB | single-strand DNA breaks |
| TNBC | triple-negative breast cancer |
| WEE1i | WEE1 inhibitor(s) |
| WT | wild type |
References
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication Stress and Cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Zou, L. Hallmarks of DNA Replication Stress. Mol. Cell 2022, 82, 2298–2314. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.-C.; Kearsey, S.E. A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes 2017, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Brison, O.; El-Hilali, S.; Azar, D.; Koundrioukoff, S.; Schmidt, M.; Nähse, V.; Jaszczyszyn, Y.; Lachages, A.-M.; Dutrillaux, B.; Thermes, C.; et al. Transcription-Mediated Organization of the Replication Initiation Program across Large Genes Sets Common Fragile Sites Genome-Wide. Nat. Commun. 2019, 10, 5693. [Google Scholar] [CrossRef]
- Brown, V.E.; Moore, S.L.; Chen, M.; House, N.; Ramsden, P.; Wu, H.-J.; Ribich, S.; Grassian, A.R.; Choi, Y.J. CDK2 Regulates Collapsed Replication Fork Repair in CCNE1-Amplified Ovarian Cancer Cells via Homologous Recombination. NAR Cancer 2023, 5, zcad039. [Google Scholar] [CrossRef]
- Myers, K.; Gagou, M.E.; Zuazua-Villar, P.; Rodriguez, R.; Meuth, M. ATR and Chk1 Suppress a Caspase-3-Dependent Apoptotic Response Following DNA Replication Stress. PLoS Genet. 2009, 5, e1000324. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic Lethality and Cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Zhang, J.; Chan, D.W.; Lin, S.-Y. Exploiting DNA Replication Stress as a Therapeutic Strategy for Breast Cancer. Biomedicines 2022, 10, 2775. [Google Scholar] [CrossRef]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective Killing of ATM- or p53-Deficient Cancer Cells through Inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma with ATM Loss-of-Function. Mol. Cancer Res. 2015, 13, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Nakata, D.; Kakoi, Y.; Kunitomo, M.; Murai, S.; Ebara, S.; Hata, A.; Hara, T. CDK8/19 Inhibition Induces Premature G1/S Transition and ATR-Dependent Cell Death in Prostate Cancer Cells. Oncotarget 2018, 9, 13474–13487. [Google Scholar] [CrossRef] [PubMed]
- Crozier, L.; Foy, R.; Mouery, B.L.; Whitaker, R.H.; Corno, A.; Spanos, C.; Ly, T.; Gowen Cook, J.; Saurin, A.T. CDK4/6 Inhibitors Induce Replication Stress to Cause Long-Term Cell Cycle Withdrawal. EMBO J. 2022, 41, e108599. [Google Scholar] [CrossRef]
- Arias, E.E.; Walter, J.C. Strength in Numbers: Preventing Rereplication via Multiple Mechanisms in Eukaryotic Cells. Genes Dev. 2007, 21, 497–518. [Google Scholar] [CrossRef]
- Brison, O.; Gnan, S.; Azar, D.; Koundrioukoff, S.; Melendez-Garcia, R.; Kim, S.-J.; Schmidt, M.; El-Hilali, S.; Jaszczyszyn, Y.; Lachages, A.-M.; et al. Mistimed Origin Licensing and Activation Stabilize Common Fragile Sites under Tight DNA-Replication Checkpoint Activation. Nat. Struct. Mol. Biol. 2023, 30, 539–550. [Google Scholar] [CrossRef]
- Heller, R.C.; Kang, S.; Lam, W.M.; Chen, S.; Chan, C.S.; Bell, S.P. Eukaryotic Origin-Dependent DNA Replication In Vitro Reveals Sequential Action of DDK and S-CDK Kinases. Cell 2011, 146, 80–91. [Google Scholar] [CrossRef]
- Suski, J.M.; Ratnayeke, N.; Braun, M.; Zhang, T.; Strmiska, V.; Michowski, W.; Can, G.; Simoneau, A.; Snioch, K.; Cup, M.; et al. CDC7-Independent G1/S Transition Revealed by Targeted Protein Degradation. Nature 2022, 605, 357–365. [Google Scholar] [CrossRef]
- Kim, S.; Leong, A.; Kim, M.; Yang, H.W. CDK4/6 Initiates Rb Inactivation and CDK2 Activity Coordinates Cell-Cycle Commitment and G1/S Transition. Sci. Rep. 2022, 12, 16810. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How Dormant Origins Promote Complete Genome Replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef]
- Lemmens, B.; Hegarat, N.; Akopyan, K.; Sala-Gaston, J.; Bartek, J.; Hochegger, H.; Lindqvist, A. DNA Replication Determines Timing of Mitosis by Restricting CDK1 and PLK1 Activation. Mol. Cell 2018, 71, 117–128.e3. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Fikaris, A.J.; Lewis, A.E.; Abulaiti, A.; Tsygankova, O.M.; Meinkoth, J.L. Ras Triggers Ataxia-Telangiectasia-Mutated and Rad-3-Related Activation and Apoptosis through Sustained Mitogenic Signaling. J. Biol. Chem. 2006, 281, 34759–34767. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Murcia, L.; Clemente-Ruiz, M.; Pierre-Elies, P.; Royou, A.; Milán, M. Selective Killing of RAS-Malignant Tissues by Exploiting Oncogene-Induced DNA Damage. Cell Rep. 2019, 28, 119–131.e4. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef]
- Klotz-Noack, K.; Klinger, B.; Rivera, M.; Bublitz, N.; Uhlitz, F.; Riemer, P.; Lüthen, M.; Sell, T.; Kasack, K.; Gastl, B.; et al. SFPQ Depletion Is Synthetically Lethal with BRAFV600E in Colorectal Cancer Cells. Cell Rep. 2020, 32, 108184. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased Global Transcription Activity as a Mechanism of Replication Stress in Cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Struve, N.; Hoffer, K.; Weik, A.-S.; Riepen, B.; Krug, L.; Cetin, M.H.; Burmester, J.; Ott, L.; Liebing, J.; Gatzemeier, F.; et al. Increased Replication Stress and R-Loop Accumulation in EGFRvIII-Expressing Glioblastoma Present New Therapeutic Opportunities. Neurooncol. Adv. 2022, 4, vdab180. [Google Scholar] [CrossRef]
- Tort, F.; Bartkova, J.; Sehested, M.; Orntoft, T.; Lukas, J.; Bartek, J. Retinoblastoma Pathway Defects Show Differential Ability to Activate the Constitutive DNA Damage Response in Human Tumorigenesis. Cancer Res. 2006, 66, 10258–10263. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated Cyclin E Induces Chromosome Instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Indjeian, V.B.; McManus, M.; Wang, L.; Dynlacht, B.D. CP110, a Cell Cycle-Dependent CDK Substrate, Regulates Centrosome Duplication in Human Cells. Dev. Cell 2002, 3, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, R.; Teixeira, L.K. Cyclin E/CDK2: DNA Replication, Replication Stress and Genomic Instability. Front. Cell Dev. Biol. 2021, 9, 774845. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased Replication Initiation and Conflicts with Transcription Underlie Cyclin E-Induced Replication Stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Kok, Y.P.; Guerrero Llobet, S.; Schoonen, P.M.; Everts, M.; Bhattacharya, A.; Fehrmann, R.S.N.; van den Tempel, N.; van Vugt, M.A.T.M. Overexpression of Cyclin E1 or Cdc25A Leads to Replication Stress, Mitotic Aberrancies, and Increased Sensitivity to Replication Checkpoint Inhibitors. Oncogenesis 2020, 9, 88. [Google Scholar] [CrossRef]
- Nanos-Webb, A.; Jabbour, N.A.; Multani, A.S.; Wingate, H.; Oumata, N.; Galons, H.; Joseph, B.; Meijer, L.; Hunt, K.K.; Keyomarsi, K. Targeting Low Molecular Weight Cyclin E (LMW-E) in Breast Cancer. Breast Cancer Res. Treat. 2012, 132, 575–588. [Google Scholar] [CrossRef]
- Li, M.; Tsavachidis, S.; Wang, F.; Bui, T.; Nguyen, T.D.T.; Luo, L.; Multani, A.S.; Bondy, M.L.; Hunt, K.K.; Keyomarsi, K. Low-Molecular-Weight Cyclin E Deregulates DNA Replication and Damage Repair to Promote Genomic Instability in Breast Cancer. Oncogene 2022, 41, 5331–5346. [Google Scholar] [CrossRef]
- Akli, S.; Zheng, P.-J.; Multani, A.S.; Wingate, H.F.; Pathak, S.; Zhang, N.; Tucker, S.L.; Chang, S.; Keyomarsi, K. Tumor-Specific Low Molecular Weight Forms of Cyclin E Induce Genomic Instability and Resistance to p21, p27, and Antiestrogens in Breast Cancer. Cancer Res. 2004, 64, 3198–3208. [Google Scholar] [CrossRef]
- Sheaff, R.J.; Groudine, M.; Gordon, M.; Roberts, J.M.; Clurman, B.E. Cyclin E-CDK2 Is a Regulator of p27Kip1. Genes Dev. 1997, 11, 1464–1478. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Low, K.-H.; Alexander, A.; Jiang, Y.; Karakas, C.; Hess, K.R.; Carey, J.P.W.; Bui, T.N.; Vijayaraghavan, S.; Evans, K.W.; et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin. Cancer Res. 2018, 24, 6594–6610. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, D.; Carey, J.P.W.; Karakas, C.; Albarracin, C.; Sahin, A.A.; Arun, B.K.; Guray Durak, M.; Li, M.; Kohansal, M.; et al. Targeting Replicative Stress and DNA Repair by Combining PARP and Wee1 Kinase Inhibitors Is Synergistic in Triple Negative Breast Cancers with Cyclin E or BRCA1 Alteration. Cancers 2021, 13, 1656. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Young, J.T.F.; Fourtounis, J.; Martino, G.; Álvarez-Quilón, A.; Bernier, C.; Duffy, N.M.; Papp, R.; Roulston, A.; Stocco, R.; et al. CCNE1 Amplification Is Synthetic Lethal with PKMYT1 Kinase Inhibition. Nature 2022, 604, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; da Costa, A.A.B.A.; Gulhan, D.; Lee, E.K.; Cheng, S.-C.; Hendrickson, A.E.W.; Kochupurakkal, B.; Kolin, D.L.; Kohn, E.C.; Liu, J.F.; et al. A Replication Stress Biomarker Is Associated with Response to Gemcitabine versus Combined Gemcitabine and ATR Inhibitor Therapy in Ovarian Cancer. Nat. Commun. 2021, 12, 5574. [Google Scholar] [CrossRef]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.-C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 Activation Defines a Hepatocellular Carcinoma Subclass with a Rearrangement Signature of Replication Stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef]
- Sailo, B.L.; Banik, K.; Girisa, S.; Bordoloi, D.; Fan, L.; Halim, C.E.; Wang, H.; Kumar, A.P.; Zheng, D.; Mao, X.; et al. FBXW7 in Cancer: What Has Been Unraveled Thus Far? Cancers 2019, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. C-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef]
- Mirkin Ekaterina, V.; Mirkin Sergei, M. Replication Fork Stalling at Natural Impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef]
- Smirnov, E.; Borkovec, J.; Kováčik, L.; Svidenská, S.; Schröfel, A.; Skalníková, M.; Švindrych, Z.; Křížek, P.; Ovesný, M.; Hagen, G.M.; et al. Separation of Replication and Transcription Domains in Nucleoli. J. Struct. Biol. 2014, 188, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Ohkawa, Y.; Fujita, M. Genome-Wide Analysis of the Spatiotemporal Regulation of Firing and Dormant Replication Origins in Human Cells. Nucleic Acids Res. 2018, 46, 6683–6696. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.M.; Gall, A.-L.; Zheng, N.; Pavletich, N.P. Structure of the Rb C-Terminal Domain Bound to E2F1-DP1: A Mechanism for Phosphorylation-Induced E2F Release. Cell 2005, 123, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Hura, G.L.; Rubin, S.M. Structures of Inactive Retinoblastoma Protein Reveal Multiple Mechanisms for Cell Cycle Control. Genes Dev. 2012, 26, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Doan, A.; Arand, J.; Gong, D.; Drainas, A.P.; Shue, Y.T.; Lee, M.C.; Zhang, S.; Walter, D.M.; Chaikovsky, A.C.; Feldser, D.M.; et al. RB Depletion Is Required for the Continuous Growth of Tumors Initiated by Loss of RB. PLoS Genet. 2021, 17, e1009941. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Ishak, C.A.; Coschi, C.H.; Roes, M.V.; Dick, F.A. Disruption of CDK-Resistant Chromatin Association by pRB Causes DNA Damage, Mitotic Errors, and Reduces Condensin II Recruitment. Cell Cycle 2017, 16, 1430–1439. [Google Scholar] [CrossRef][Green Version]
- Braden, W.A.; Lenihan, J.M.; Lan, Z.; Luce, K.S.; Zagorski, W.; Bosco, E.; Reed, M.F.; Cook, J.G.; Knudsen, E.S. Distinct Action of the Retinoblastoma Pathway on the DNA Replication Machinery Defines Specific Roles for Cyclin-Dependent Kinase Complexes in Prereplication Complex Assembly and S-Phase Progression. Mol. Cell. Biol. 2006, 26, 7667–7681. [Google Scholar] [CrossRef]
- Manickavinayaham, S.; Velez-Cruz, R.; Biswas, A.K.; Chen, J.; Guo, R.; Johnson, D.G. The E2F1 Transcription Factor and RB Tumor Suppressor Moonlight as DNA Repair Factors. Cell Cycle 2020, 19, 2260–2269. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-Phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000.e6. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Zamalloa, L.G.; Pruitt, M.M.; Hermance, N.M.; Gali, H.; Flynn, R.L.; Manning, A.L. RB Loss Sensitizes Cells to Replication-Associated DNA Damage after PARP Inhibition by Trapping. Life Sci. Alliance 2023, 6. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669. [Google Scholar] [CrossRef] [PubMed]
- Donker, L.; Houtekamer, R.; Vliem, M.; Sipieter, F.; Canever, H.; Gómez-González, M.; Bosch-Padrós, M.; Pannekoek, W.-J.; Trepat, X.; Borghi, N.; et al. A Mechanical G2 Checkpoint Controls Epithelial Cell Division through E-Cadherin-Mediated Regulation of Wee1-Cdk1. Cell Rep. 2022, 41, 111475. [Google Scholar] [CrossRef] [PubMed]
- Serpico, A.F.; Febbraro, F.; Pisauro, C.; Grieco, D. Compartmentalized Control of Cdk1 Drives Mitotic Spindle Assembly. Cell Rep. 2022, 38, 110305. [Google Scholar] [CrossRef]
- Timofeev, O.; Cizmecioglu, O.; Settele, F.; Kempf, T.; Hoffmann, I. Cdc25 Phosphatases Are Required for Timely Assembly of CDK1-Cyclin B at the G2/M Transition. J. Biol. Chem. 2010, 285, 16978–16990. [Google Scholar] [CrossRef]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Qian, C.; Sugitani, N.; Osmanbeyoglu, H.U.; Bakkenist, C.J. WEE1 Kinase Inhibitor AZD1775 Induces CDK1 Kinase-Dependent Origin Firing in Unperturbed G1- and S-Phase Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 23891–23893. [Google Scholar] [CrossRef]
- Elbæk, C.R.; Petrosius, V.; Benada, J.; Erichsen, L.; Damgaard, R.B.; Sørensen, C.S. WEE1 Kinase Protects the Stability of Stalled DNA Replication Forks by Limiting CDK2 Activity. Cell Rep. 2022, 38, 110261. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA Damage Checkpoint Kinases in Cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the Biology and Clinical Potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in Cell Biology and Cancer Therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef]
- Okita, N.; Minato, S.; Ohmi, E.; Tanuma, S.-I.; Higami, Y. DNA Damage-Induced CHK1 Autophosphorylation at Ser296 Is Regulated by an Intramolecular Mechanism. FEBS Lett. 2012, 586, 3974–3979. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Seraia, E.; Hatch, S.B.; Wan, X.; Ebner, D.V.; Aroldi, F.; Jiang, Y.; Ryan, A.J.; Bogenrieder, T.; Weyer-Czernilofsky, U.; et al. CHK1 Inhibition Exacerbates Replication Stress Induced by IGF Blockade. Oncogene 2022, 41, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, B.C.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. Poly(ADP-Ribose) Polymerase-1 Modulates DNA Repair Capacity and Prevents Formation of DNA Double Strand Breaks. DNA Repair 2008, 7, 932–940. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku Compete for Repair of DNA Double Strand Breaks by Distinct NHEJ Pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA Damage Response Factors from Diverse Pathways, Including DNA Crosslink Repair, Mediate Alternative End Joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-Ribose) Polymerase-1 Antagonizes DNA Resection at Double-Strand Breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [PubMed]
- Gralewska, P.; Gajek, A.; Rybaczek, D.; Marczak, A.; Rogalska, A. The Influence of PARP, ATR, CHK1 Inhibitors on Premature Mitotic Entry and Genomic Instability in High-Grade Serous BRCAMUT and BRCAWT Ovarian Cancer Cells. Cells 2022, 11, 1889. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell Cycle Regulation: p53-p21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 Checkpoint Control: Regulation of 14-3-3 Protein Binding by Phosphorylation of Cdc25C on Serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Wang, X.W.; Zhan, Q.; Coursen, J.D.; Khan, M.A.; Kontny, H.U.; Yu, L.; Hollander, M.C.; O’Connor, P.M.; Fornace, A.J., Jr.; Harris, C.C. GADD45 Induction of a G2/M Cell Cycle Checkpoint. Proc. Natl. Acad. Sci. USA 1999, 96, 3706–3711. [Google Scholar] [CrossRef]
- Zeng, J.; Hills, S.A.; Ozono, E.; Diffley, J.F.X. Cyclin E-Induced Replicative Stress Drives p53-Dependent Whole-Genome Duplication. Cell 2023, 186, 528–542.e14. [Google Scholar] [CrossRef]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 Modulates Homologous Recombination by Transcriptional Regulation of the RAD51 Gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid Recruitment of p53 to DNA Damage Sites Directs DNA Repair Choice and Integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119. [Google Scholar] [CrossRef]
- Poletto, M.; Legrand, A.J.; Fletcher, S.C.; Dianov, G.L. p53 Coordinates Base Excision Repair to Prevent Genomic Instability. Nucleic Acids Res. 2016, 44, 3165–3175. [Google Scholar] [CrossRef]
- Yeo, C.Q.X.; Alexander, I.; Lin, Z.; Lim, S.; Aning, O.A.; Kumar, R.; Sangthongpitag, K.; Pendharkar, V.; Ho, V.H.B.; Cheok, C.F. p53 Maintains Genomic Stability by Preventing Interference between Transcription and Replication. Cell Rep. 2016, 15, 132–146. [Google Scholar] [CrossRef]
- Hampp, S.; Kiessling, T.; Buechle, K.; Mansilla, S.F.; Thomale, J.; Rall, M.; Ahn, J.; Pospiech, H.; Gottifredi, V.; Wiesmüller, L. DNA Damage Tolerance Pathway Involving DNA Polymerase ι and the Tumor Suppressor p53 Regulates DNA Replication Fork Progression. Proc. Natl. Acad. Sci. USA 2016, 113, E4311–E4319. [Google Scholar] [CrossRef]
- Roy, S.; Tomaszowski, K.-H.; Luzwick, J.W.; Park, S.; Li, J.; Murphy, M.; Schlacher, K. p53 Orchestrates DNA Replication Restart Homeostasis by Suppressing Mutagenic RAD52 and POLθ Pathways. eLife 2018, 7, e31723. [Google Scholar] [CrossRef] [PubMed]
- Klusmann, I.; Rodewald, S.; Müller, L.; Friedrich, M.; Wienken, M.; Li, Y.; Schulz-Heddergott, R.; Dobbelstein, M. p53 Activity Results in DNA Replication Fork Processivity. Cell Rep. 2016, 17, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Kranz, D.; Dobbelstein, M. Nongenotoxic p53 Activation Protects Cells against S-Phase-Specific Chemotherapy. Cancer Res. 2006, 66, 10274–10280. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Saini, P.; Sriraman, A.; Dobbelstein, M. Mdm2 Inhibition Confers Protection of p53-Proficient Cells from the Cytotoxic Effects of Wee1 Inhibitors. Oncotarget 2015, 6, 32339–32352. [Google Scholar] [CrossRef] [PubMed]
- Segeren, H.A.; van Liere, E.A.; Riemers, F.M.; de Bruin, A.; Westendorp, B. Oncogenic RAS Sensitizes Cells to Drug-Induced Replication Stress via Transcriptional Silencing of P53. Oncogene 2022, 41, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR Suppression with Oncogenic Ras Synergistically Increases Genomic Instability, Causing Synthetic Lethality or Tumorigenesis in a Dosage-Dependent Manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef]
- Gorecki, L.; Andrs, M.; Rezacova, M.; Korabecny, J. Discovery of ATR Kinase Inhibitor Berzosertib (VX-970, M6620): Clinical Candidate for Cancer Therapy. Pharmacol. Ther. 2020, 210, 107518. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 Study of the ATR Inhibitor Berzosertib (formerly M6620, VX-970) Combined with Gemcitabine ± Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 Study of the ATR Inhibitor Berzosertib in Combination with Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Baschnagel, A.M.; Elnaggar, J.H.; VanBeek, H.J.; Kromke, A.C.; Skiba, J.H.; Kaushik, S.; Abel, L.; Clark, P.A.; Longhurst, C.A.; Nickel, K.P.; et al. ATR Inhibitor M6620 (VX-970) Enhances the Effect of Radiation in Non-Small Cell Lung Cancer Brain Metastasis Patient-Derived Xenografts. Mol. Cancer Ther. 2021, 20, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Takahashi, N.; Rajapakse, V.N.; Zhang, X.; Sun, Y.; Ceribelli, M.; Wilson, K.M.; Zhang, Y.; Beck, E.; Sciuto, L.; et al. Therapeutic Targeting of ATR Yields Durable Regressions in Small Cell Lung Cancers with High Replication Stress. Cancer Cell 2021, 39, 566–579.e7. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.D.; Li, Z.; Lustberg, M.; Grenade, C.; Wesolowski, R. Remarkable Response to a Novel ATR Inhibitor in a Patient with Poorly Differentiated Neuroendocrine Carcinoma. Cancer Treat. Res. Commun. 2018, 16, 9–12. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus Gemcitabine versus Gemcitabine Alone in Platinum-Resistant High-Grade Serous Ovarian Cancer: A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Plummer, R.; Dean, E.; Arkenau, H.-T.; Redfern, C.; Spira, A.I.; Melear, J.M.; Chung, K.Y.; Ferrer-Playan, J.; Goddemeier, T.; Locatelli, G.; et al. A Phase 1b Study Evaluating the Safety and Preliminary Efficacy of Berzosertib in Combination with Gemcitabine in Patients with Advanced Non-Small Cell Lung Cancer. Lung Cancer 2022, 163, 19–26. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e11. [Google Scholar] [CrossRef]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-Yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-Yl}-1H-Indole (AZ20): A Potent and Selective Inhibitor of ATR Protein Kinase with Monotherapy in Vivo Antitumor Activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ge, Y.; Wang, T.; Edwards, H.; Ren, Q.; Jiang, Y.; Quan, C.; Wang, G. Inhibition of ATR Potentiates the Cytotoxic Effect of Gemcitabine on Pancreatic Cancer Cells through Enhancement of DNA Damage and Abrogation of Ribonucleotide Reductase Induction by Gemcitabine. Oncol. Rep. 2017, 37, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, X.; Su, Y.; Zhao, J.; Luedtke, D.A.; Epshteyn, V.; Edwards, H.; Wang, G.; Wang, Z.; Chu, R.; et al. Mechanisms Responsible for the Synergistic Antileukemic Interactions between ATR Inhibition and Cytarabine in Acute Myeloid Leukemia Cells. Sci. Rep. 2017, 7, 41950. [Google Scholar] [CrossRef]
- Jones, C.D.; Blades, K.; Foote, K.M.; Guichard, S.M.; Jewsbury, P.J.; McGuire, T.; Nissink, J.W.; Odedra, R.; Tam, K.; Thommes, P.; et al. Abstract 2348: Discovery of AZD6738, a Potent and Selective Inhibitor with the Potential to Test the Clinical Efficacy of ATR Kinase Inhibition in Cancer Patients. Cancer Res. 2013, 73, 2348. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef] [PubMed]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The Orally Active and Bioavailable ATR Kinase Inhibitor AZD6738 Potentiates the Anti-Tumor Effects of Cisplatin to Resolve ATM-Deficient Non-Small Cell Lung Cancer in Vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef] [PubMed]
- Dunne, V.; Ghita, M.; Small, D.M.; Coffey, C.B.M.; Weldon, S.; Taggart, C.C.; Osman, S.O.; McGarry, C.K.; Prise, K.M.; Hanna, G.G.; et al. Inhibition of Ataxia Telangiectasia Related-3 (ATR) Improves Therapeutic Index in Preclinical Models of Non-Small Cell Lung Cancer (NSCLC) Radiotherapy. Radiother. Oncol. 2017, 124, 475–481. [Google Scholar] [CrossRef]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.-B.; Fornari, C.; Yates, J.W.T.; Bernaldo de Quirós Fernández, S.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef]
- Leonard, B.C.; Lee, E.D.; Bhola, N.E.; Li, H.; Sogaard, K.K.; Bakkenist, C.J.; Grandis, J.R.; Johnson, D.E. ATR Inhibition Sensitizes HPV− and HPV+ Head and Neck Squamous Cell Carcinoma to Cisplatin. Oral Oncol. 2019, 95, 35–42. [Google Scholar] [CrossRef]
- Bristol, M.L.; Das, D.; Morgan, I.M. Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling. Viruses 2017, 9, 268. [Google Scholar] [CrossRef]
- Kim, H.-J.; Min, A.; Im, S.-A.; Jang, H.; Lee, K.H.; Lau, A.; Lee, M.; Kim, S.; Yang, Y.; Kim, J.; et al. Anti-Tumor Activity of the ATR Inhibitor AZD6738 in HER2 Positive Breast Cancer Cells. Int. J. Cancer 2017, 140, 109–119. [Google Scholar] [CrossRef]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, C.R.; Wallez, Y.; Johnson, T.I.; Bernaldo de Quirós Fernández, S.; Durant, S.T.; Cadogan, E.B.; Lau, A.; Richards, F.M.; Jodrell, D.I. Complete Loss of ATM Function Augments Replication Catastrophe Induced by ATR Inhibition and Gemcitabine in Pancreatic Cancer Models. Br. J. Cancer 2020, 123, 1424–1436. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Konstantinopoulos, P.A. From Checkpoint to Checkpoint: DNA Damage ATR/Chk1 Checkpoint Signalling Elicits PD-L1 Immune Checkpoint Activation. Br. J. Cancer 2018, 118, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef]
- Szydzik, J.; Lind, D.E.; Arefin, B.; Kurhe, Y.; Umapathy, G.; Siaw, J.T.; Claeys, A.; Gabre, J.L.; Van den Eynden, J.; Hallberg, B.; et al. ATR Inhibition Enables Complete Tumour Regression in ALK-Driven NB Mouse Models. Nat. Commun. 2021, 12, 6813. [Google Scholar] [CrossRef]
- Harold, J.; Bellone, S.; Manavella, D.D.; Mutlu, L.; McNamara, B.; Hartwich, T.M.P.; Zipponi, M.; Yang-Hartwich, Y.; Demirkiran, C.; Verzosa, M.S.; et al. Elimusertib (BAY1895344), a Novel ATR Inhibitor, Demonstrates in Vivo Activity in ATRX Mutated Models of Uterine Leiomyosarcoma. Gynecol. Oncol. 2023, 168, 157–165. [Google Scholar] [CrossRef]
- Manavella, D.D.; McNamara, B.; Harold, J.; Bellone, S.; Hartwich, T.M.P.; Yang-Hartwich, Y.; Mutlu, L.; Zipponi, M.; Demirkiran, C.; Verzosa, M.S.; et al. Ovarian and Uterine Carcinosarcomas Are Sensitive in Vitro and in Vivo to Elimusertib, a Novel Ataxia-Telangiectasia and Rad3-Related (ATR) Kinase Inhibitor. Gynecol. Oncol. 2023, 169, 98–105. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Fu, H.; Yao, Q.; Wang, L.; Lou, L. Combined Inhibition of PARP and ATR Synergistically Potentiates the Antitumor Activity of HER2-Targeting Antibody-Drug Conjugate in HER2-Positive Cancers. Am. J. Cancer Res. 2023, 13, 161–175. [Google Scholar]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef] [PubMed]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells with Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Seidel, P.; Rubarth, A.; Zodel, K.; Peighambari, A.; Neumann, F.; Federkiel, Y.; Huang, H.; Hoefflin, R.; Adlesic, M.; Witt, C.; et al. ATR Represents a Therapeutic Vulnerability in Clear Cell Renal Cell Carcinoma. JCI Insight 2022, 7, e156087. [Google Scholar] [CrossRef] [PubMed]
- Turchick, A.; Zimmermann, A.; Chiu, L.-Y.; Dahmen, H.; Elenbaas, B.; Zenke, F.T.; Blaukat, A.; Vassilev, L.T. Selective Inhibition of ATM-Dependent Double-Strand Break Repair and Checkpoint Control Synergistically Enhances the Efficacy of ATR Inhibitors. Mol. Cancer Ther. 2023, 22, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Fontana, E.; Lee, E.K.; Spigel, D.R.; Højgaard, M.; Lheureux, S.; Mettu, N.B.; Carneiro, B.A.; Carter, L.; Plummer, R.; et al. Camonsertib in DNA Damage Response-Deficient Advanced Solid Tumors: Phase 1 Trial Results. Nat. Med. 2023, 29, 1400–1411. [Google Scholar] [CrossRef]
- Roulston, A.; Zimmermann, M.; Papp, R.; Skeldon, A.; Pellerin, C.; Dumas-Bérube, É.; Dumais, V.; Dorich, S.; Fader, L.D.; Fournier, S.; et al. RP-3500: A Novel, Potent, and Selective ATR Inhibitor That Is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 245–256. [Google Scholar] [CrossRef]
- Restelli, V.; Lupi, M.; Chilà, R.; Vagni, M.; Tarantelli, C.; Spriano, F.; Gaudio, E.; Bertoni, F.; Damia, G.; Carrassa, L. DNA Damage Response Inhibitor Combinations Exert Synergistic Antitumor Activity in Aggressive B-Cell Lymphomas. Mol. Cancer Ther. 2019, 18, 1255–1264. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef] [PubMed]
- Gralewska, P.; Gajek, A.; Marczak, A.; Mikuła, M.; Ostrowski, J.; Śliwińska, A.; Rogalska, A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality-An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. Int. J. Mol. Sci. 2020, 21, 9715. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Hutcherson, R.J.; Kemp, M.G. ATR Kinase Inhibition Sensitizes Quiescent Human Cells to the Lethal Effects of Cisplatin but Increases Mutagenesis. Mutat. Res. 2019, 816–818, 111678. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Frankel, P.H.; Mortazavi, A.; Milowsky, M.; Vaishampayan, U.; Parikh, M.; Lyou, Y.; Weng, P.; Parikh, R.; Teply, B.; et al. Effect of Cisplatin and Gemcitabine with or without Berzosertib in Patients with Advanced Urothelial Carcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.-J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination with Topotecan in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Desai, P.A.; Sciuto, L.; Nichols, S.; Steinberg, S.M.; Thomas, A. Targeting Genomic Instability in Extrapulmonary Small Cell Neuroendocrine Cancers: A Phase II Study with ATR Inhibitor Berzosertib and Topotecan. J. Clin. Orthod. 2022, 40, 8518. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef]
- Angius, G.; Tomao, S.; Stati, V.; Vici, P.; Bianco, V.; Tomao, F. Prexasertib, a Checkpoint Kinase Inhibitor: From Preclinical Data to Clinical Development. Cancer Chemother. Pharmacol. 2020, 85, 9–20. [Google Scholar] [CrossRef]
- Keller, K.M.; Eleveld, T.F.; Schild, L.; van den Handel, K.; van den Boogaard, M.; Amo-Addae, V.; Eising, S.; Ober, K.; Koopmans, B.; Looijenga, L.; et al. Chromosome 11q Loss and MYCN Amplification Demonstrate Synthetic Lethality with Checkpoint Kinase 1 Inhibition in Neuroblastoma. Front. Oncol. 2022, 12, 929123. [Google Scholar] [CrossRef]
- Nair, J.; Huang, T.-T.; Murai, J.; Haynes, B.; Steeg, P.S.; Pommier, Y.; Lee, J.-M. Resistance to the CHK1 Inhibitor Prexasertib Involves Functionally Distinct CHK1 Activities in BRCA Wild-Type Ovarian Cancer. Oncogene 2020, 39, 5520–5535. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Lee, J.-M.; Gao, B.; Miller, R.; Lee, J.-Y.; Colombo, N.; Vergote, I.; Credille, K.M.; Young, S.R.; McNeely, S.; et al. A Phase 2 Study of Prexasertib (LY2606368) in Platinum Resistant or Refractory Recurrent Ovarian Cancer. Gynecol. Oncol. 2022, 167, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.D.; Matthews, T.P.; McHardy, T.; Proisy, N.; Cheung, K.-M.J.; Lainchbury, M.; Brown, N.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; et al. Multiparameter Lead Optimization to Give an Oral Checkpoint Kinase 1 (CHK1) Inhibitor Clinical Candidate: (R)-5-((4-((Morpholin-2-Ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-Yl)amino)pyrazine-2-Carbonitrile (CCT245737). J. Med. Chem. 2016, 59, 5221–5237. [Google Scholar] [CrossRef] [PubMed]
- Walton, M.I.; Eve, P.D.; Hayes, A.; Henley, A.T.; Valenti, M.R.; De Haven Brandon, A.K.; Box, G.; Boxall, K.J.; Tall, M.; Swales, K.; et al. The Clinical Development Candidate CCT245737 Is an Orally Active CHK1 Inhibitor with Preclinical Activity in RAS Mutant NSCLC and Eµ-MYC Driven B-Cell Lymphoma. Oncotarget 2016, 7, 2329–2342. [Google Scholar] [CrossRef]
- Rogers, R.F.; Walton, M.I.; Cherry, D.L.; Collins, I.; Clarke, P.A.; Garrett, M.D.; Workman, P. CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells. Cancer Res. 2020, 80, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-R.; Yang, Z.-Z.; Wang, S.-J.; Zhang, L.; Luo, J.-R.; Feng, Y.; Yu, X.-L.; Chen, X.-X.; Guo, X.-M. The Chk1 Inhibitor MK-8776 Increases the Radiosensitivity of Human Triple-Negative Breast Cancer by Inhibiting Autophagy. Acta Pharmacol. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I Dose-Escalation Trial of Checkpoint Kinase 1 Inhibitor MK-8776 as Monotherapy and in Combination with Gemcitabine in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef]
- Cui, Q.; Cai, C.-Y.; Wang, J.-Q.; Zhang, S.; Gupta, P.; Ji, N.; Yang, Y.; Dong, X.; Yang, D.-H.; Chen, Z.-S. Chk1 Inhibitor MK-8776 Restores the Sensitivity of Chemotherapeutics in P-Glycoprotein Overexpressing Cancer Cells. Int. J. Mol. Sci. 2019, 20, 4095. [Google Scholar] [CrossRef]
- Jin, T.; Xu, L.; Wang, P.; Hu, X.; Zhang, R.; Wu, Z.; Du, W.; Kan, W.; Li, K.; Wang, C.; et al. Discovery and Development of a Potent, Selective, and Orally Bioavailable CHK1 Inhibitor Candidate: 5-((4-((3-Amino-3-Methylbutyl)amino)-5-(trifluoromethyl)pyrimidin-2-Yl)amino)picolinonitrile. J. Med. Chem. 2021, 64, 15069–15090. [Google Scholar] [CrossRef]
- Kristeleit, R.; Plummer, R.; Jones, R.; Carter, L.; Blagden, S.; Sarker, D.; Arkenau, T.; Evans, T.R.J.; Danson, S.; Symeonides, S.N.; et al. A Phase 1/2 Trial of SRA737 (a Chk1 Inhibitor) Administered Orally in Patients with Advanced Cancer. Br. J. Cancer 2023, 129, 38–45. [Google Scholar] [CrossRef]
- Karp, J.E.; Thomas, B.M.; Greer, J.M.; Sorge, C.; Gore, S.D.; Pratz, K.W.; Smith, B.D.; Flatten, K.S.; Peterson, K.; Schneider, P.; et al. Phase I and Pharmacologic Trial of Cytosine Arabinoside with the Selective Checkpoint 1 Inhibitor Sch 900776 in Refractory Acute Leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Da Silva, L.A.; Sessa, C.; Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers 2022, 14, 953. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Shacham-Shmueli, E.; Xiao, L.; Varadhachary, G.; Halpern, N.; Fogelman, D.; Boursi, B.; Uruba, S.; Margalit, O.; Wolff, R.A.; et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer with DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings From 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncol. 2021, 7, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.H.A.; Kocherginsky, M.; Agarwal, N.; Zhang, J.; Adra, N.; Paller, C.J.; Picus, J.; Reichert, Z.R.; Szmulewitz, R.Z.; Tagawa, S.T.; et al. BRCAAWAY: A Randomized Phase 2 Trial of Abiraterone, Olaparib, or Abiraterone + Olaparib in Patients with Metastatic Castration-Resistant Prostate Cancer (mCRPC) with DNA Repair Defects. J. Clin. Orthod. 2022, 40, 5018. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.-C.; Choi, Y.-E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of Mutant BRCA1 Protein Confers PARP Inhibitor and Platinum Resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef]
- McMullen, M.; Karakasis, K.; Loembe, B.; Dean, E.; Parr, G.; Oza, A.M. DUETTE: A Phase II Randomized, Multicenter Study to Investigate the Efficacy and Tolerability of a Second Maintenance Treatment in Patients with Platinum-Sensitive Relapsed Epithelial Ovarian Cancer, Who Have Previously Received poly(ADP-Ribose) Polymerase (PARP) Inhibitor Maintenance Treatment. Int. J. Gynecol. Cancer 2020, 30, 1824–1828. [Google Scholar]
- Drewett, L.; Lucey, R.; Pinilla, K.A.; Grybowicz, L.; Wulff, J.; Dayimu, A.; Demiris, N.; Vallier, A.-L.; Qian, W.; Machin, A.; et al. PARTNER: A Randomized, Phase II/III Trial to Evaluate the Safety and Efficacy of the Addition of Olaparib to Platinum-Based Neoadjuvant Chemotherapy in Patients with Triple-Negative And/or Germline BRCA-Mutated Breast Cancer. J. Clin. Orthod. 2022, 40, TPS619. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Rodriguez-Moreno, J.F.; de Velasco, G.; Bravo Fernandez, I.; Alvarez-Fernandez, C.; Fernandez, R.; Vazquez-Estevez, S.; Virizuela, J.A.; Gajate, P.; Font, A.; Lainez, N.; et al. Impact of the Combination of Durvalumab (MEDI4736) plus Olaparib (AZD2281) Administered prior to Surgery in the Molecular Profile of Resectable Urothelial Bladder Cancer: NEODURVARIB Trial. J. Clin. Orthod. 2020, 38, 542. [Google Scholar]
- Eder, J.P.; Doroshow, D.B.; Do, K.T.; Keedy, V.L.; Sklar, J.S.; Glazer, P.; Bindra, R.; Shapiro, G.I. Clinical Efficacy of Olaparib in IDH1/IDH2-Mutant Mesenchymal Sarcomas. JCO Precis. Oncol. 2021, 5, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.-H.; Min, A.; Kim, S.; Jang, H.; Kim, S.H.; Kim, H.-J.; Ryu, H.S.; Ku, J.-L.; Lee, K.-H.; Im, S.-A. Antitumor Effect of a WEE1 Inhibitor and Potentiation of Olaparib Sensitivity by DNA Damage Response Modulation in Triple-Negative Breast Cancer. Sci. Rep. 2020, 10, 9930. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity While Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e7. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in Patients with Metastatic Castration-Resistant Prostate Cancer with DNA Repair Gene Aberrations (TOPARP-B): A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidziński, M.; Kim, J.-W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef]
- Kindler, H.L.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall Survival Results from the POLO Trial: A Phase III Study of Active Maintenance Olaparib Versus Placebo for Germline BRCA-Mutated Metastatic Pancreatic Cancer. J. Clin. Oncol. 2022, 40, 3929–3939. [Google Scholar] [CrossRef]
- Senkus, E.; Delaloge, S.; Domchek, S.M.; Conte, P.; Im, S.-A.; Xu, B.; Armstrong, A.; Masuda, N.; Fielding, A.; Robson, M.; et al. Olaparib Efficacy in Patients with Germline BRCA-Mutated, HER2-Negative Metastatic Breast Cancer: Subgroup Analyses from the Phase III OlympiAD Trial. Int. J. Cancer 2023, 153, 803–814. [Google Scholar] [CrossRef]
- Park, J.; Lim, M.C.; Lee, J.-K.; Jeong, D.H.; Kim, S.I.; Choi, M.C.; Kim, B.-G.; Lee, J.-Y. A Single-Arm, Phase II Study of Niraparib and Bevacizumab Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer Patients Previously Treated with a PARP Inhibitor: Korean Gynecologic Oncology Group (KGOG 3056)/NIRVANA-R Trial. J. Gynecol. Oncol. 2022, 33, e12. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N., Jr.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in Patients with Metastatic Castration-Resistant Prostate Cancer and DNA Repair Gene Defects (GALAHAD): A Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- Narayan, V.; Torres, A.; Seger, P.; Aaron, M.; Louie, J.; Lee, D.; Haas, N.B.; Takvorian, S.U.; Maxwell, K.N.; Domchek, S.M. A Phase II Single-Arm Trial of Niraparib in Platinum-Sensitive Metastatic Castration-Resistant Prostate Cancer with DNA Repair Defects (PLATPARP). J. Clin. Orthod. 2023, 41, TPS291. [Google Scholar] [CrossRef]
- Starks, D.; Rojas-Espaillat, L.; Meissner, T.; Elsey, R.; Xu, B.; Koenen, M.; Feng, S.; VanOosbree, A.; Slunecka, J.; Lee, J.; et al. A Phase 1 Evaluation of the Safety and Tolerability of Niraparib in Combination with Everolimus in Advanced Ovarian and Breast Cancers. Cancer Med. 2023, 12, 18654–18665. [Google Scholar] [CrossRef]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-ov45): A Randomized, Phase III Trial to Evaluate Rucaparib as Monotherapy (ATHENA-MONO) and Rucaparib in Combination with Nivolumab (ATHENA-COMBO) as Maintenance Treatment Following Frontline Platinum-Based Chemotherapy in Ovarian Cancer. Int. J. Gynecol. Cancer 2021, 31, 1589–1594. [Google Scholar] [PubMed]
- Patsouris, A.; Diop, K.; Tredan, O.; Nenciu, D.; Gonçalves, A.; Arnedos, M.; Sablin, M.-P.; Jézéquel, P.; Jimenez, M.; Droin, N.; et al. Rucaparib in Patients Presenting a Metastatic Breast Cancer with Homologous Recombination Deficiency, without Germline BRCA1/2 Mutation. Eur. J. Cancer 2021, 159, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Parkinson, C.; Lim, M.C.; O’Malley, D.M.; Oaknin, A.; Wilson, M.K.; Coleman, R.L.; Lorusso, D.; Bessette, P.; Ghamande, S.; et al. A Randomized, Phase III Trial to Evaluate Rucaparib Monotherapy as Maintenance Treatment in Patients with Newly Diagnosed Ovarian Cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J. Clin. Oncol. 2022, 40, 3952–3964. [Google Scholar] [CrossRef]
- Fizazi, K.; Piulats, J.M.; Reaume, M.N.; Ostler, P.; McDermott, R.; Gingerich, J.R.; Pintus, E.; Sridhar, S.S.; Bambury, R.M.; Emmenegger, U.; et al. Rucaparib or Physician’s Choice in Metastatic Prostate Cancer. N. Engl. J. Med. 2023, 388, 719–732. [Google Scholar] [CrossRef]
- De Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib Monotherapy in Metastatic Castration-Resistant Prostate Cancer with DNA Repair Alterations (TALAPRO-1): An Open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Gruber, J.J.; Gross, W.; McMillan, A.; Ford, J.M.; Telli, M.L. A Phase II Clinical Trial of Talazoparib Monotherapy for PALB2 Mutation-Associated Advanced Breast Cancer. J. Clin. Orthod. 2021, 39, TPS1109. [Google Scholar] [CrossRef]
- Gruber, J.J.; Afghahi, A.; Timms, K.; DeWees, A.; Gross, W.; Aushev, V.N.; Wu, H.-T.; Balcioglu, M.; Sethi, H.; Scott, D.; et al. A Phase II Study of Talazoparib Monotherapy in Patients with Wild-Type BRCA1 and BRCA2 with a Mutation in Other Homologous Recombination Genes. Nat Cancer 2022, 3, 1181–1191. [Google Scholar] [CrossRef]
- Yap, T.A.; Bardia, A.; Dvorkin, M.; Galsky, M.D.; Beck, J.T.; Wise, D.R.; Karyakin, O.; Rubovszky, G.; Kislov, N.; Rohrberg, K.; et al. Avelumab Plus Talazoparib in Patients with Advanced Solid Tumors: The JAVELIN PARP Medley Nonrandomized Controlled Trial. JAMA Oncol. 2023, 9, 40–50. [Google Scholar] [CrossRef]
- Ghelli Luserna di Rorà, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 Family Business: Regulation of Mitosis, Cancer Progression, and Therapeutic Target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.A.; Pal, S.; Kudgus, R.A.; Ijaz, H.; Liu, X.; Minard, C.G.; Pawel, B.R.; Maris, J.M.; Haas-Kogan, D.A.; Voss, S.D.; et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors: A COG Phase I Consortium Report (ADVL1312). Clin. Cancer Res. 2020, 26, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients with Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.J.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus Gemcitabine for Platinum-Resistant or Platinum-Refractory Recurrent Ovarian Cancer: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Hamilton, E.P.; Wang, J.S.; Falchook, G.S.; Oza, A.M.; Rodrigo Imedio, E.; Kumar, S.; Mugundu, G.M.; De Bruin, E.; Spigel, D.R.; et al. 1785P Phase Ib Expansion Study of Adavosertib plus Olaparib in Patients with Extensive-Stage or Relapsed Small Cell Lung Cancer. Ann. Oncol. 2020, 31, S1035. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Tang, L.; Tanaka, N.; Gleber-Netto, F.O.; Bartels, M.D.; Wang, L.; McGrail, D.J.; Lin, S.-Y.; Frank, S.J.; et al. Combined Inhibition of Rad51 and Wee1 Enhances Cell Killing in HNSCC Through Induction of Apoptosis Associated With Excessive DNA Damage and Replication Stress. Mol. Cancer Ther. 2021, 20, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Patel, A.A.; Tang, L.; Silver, N.L.; Lindemann, A.; Takahashi, H.; Jaksik, R.; Rao, X.; Kalu, N.N.; Chen, T.-C.; et al. Replication Stress Leading to Apoptosis within the S-Phase Contributes to Synergism between Vorinostat and AZD1775 in HNSCC Harboring High-Risk TP53 Mutation. Clin. Cancer Res. 2017, 23, 6541–6554. [Google Scholar] [CrossRef]
- Rampias, T.; Sasaki, C.; Weinberger, P.; Psyrri, A. E6 and e7 Gene Silencing and Transformed Phenotype of Human Papillomavirus 16-Positive Oropharyngeal Cancer Cells. J. Natl. Cancer Inst. 2009, 101, 412–423. [Google Scholar] [CrossRef]
- Qiao, G.-B.; Wu, Y.-L.; Yang, X.-N.; Zhong, W.-Z.; Xie, D.; Guan, X.-Y.; Fischer, D.; Kolberg, H.-C.; Kruger, S.; Stuerzbecher, H.-W. High-Level Expression of Rad51 Is an Independent Prognostic Marker of Survival in Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef]
- Diab, A.; Kao, M.; Kehrli, K.; Kim, H.Y.; Sidorova, J.; Mendez, E. Multiple Defects Sensitize p53-Deficient Head and Neck Cancer Cells to the WEE1 Kinase Inhibition. Mol. Cancer Res. 2019, 17, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Moser, R.; Xu, C.; Kao, M.; Annis, J.; Lerma, L.A.; Schaupp, C.M.; Gurley, K.E.; Jang, I.S.; Biktasova, A.; Yarbrough, W.G.; et al. Functional Kinomics Identifies Candidate Therapeutic Targets in Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 4274–4288. [Google Scholar] [CrossRef] [PubMed]
- Chayka, O.; D’Acunto, C.W.; Middleton, O.; Arab, M.; Sala, A. Identification and Pharmacological Inactivation of the MYCN Gene Network as a Therapeutic Strategy for Neuroblastic Tumor Cells. J. Biol. Chem. 2015, 290, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Z.-H.; Wang, H.-P.; Zhang, X.-N.; Chen, Z.-X.; Zhang, H.-Y.; Gu, M.-M. PKMYT1 Aggravates the Progression of Ovarian Cancer by Targeting SIRT3. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5259–5266. [Google Scholar]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-Wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef]
- Benada, J.; Bulanova, D.; Azzoni, V.; Petrosius, V.; Ghazanfar, S.; Wennerberg, K.; Sørensen, C.S. Synthetic Lethal Interaction between WEE1 and PKMYT1 Is a Target for Multiple Low-Dose Treatment of High-Grade Serous Ovarian Carcinoma. NAR Cancer 2023, 5, zcad029. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.P.H.; Poon, R.Y.C. The CDK1 Inhibitory Kinase MYT1 in DNA Damage Checkpoint Recovery. Oncogene 2013, 32, 4778–4788. [Google Scholar] [CrossRef]
- Oza, A.M.; Estevez-Diz, M.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.; Uyar, D.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-Enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-Sensitive TP53-Mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) with Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef]
- Takebe, N.; Naqash, A.R.; O’Sullivan Coyne, G.; Kummar, S.; Do, K.; Bruns, A.; Juwara, L.; Zlott, J.; Rubinstein, L.; Piekarz, R.; et al. Safety, Antitumor Activity, and Biomarker Analysis in a Phase I Trial of the Once-Daily Wee1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Madariaga, A.; Mitchell, S.A.; Pittman, T.; Wang, L.; Bowering, V.; Kavak, N.; Quintos, J.; Chang, K.; Ramsahai, J.; Karakasis, K.; et al. Patient Self-Reporting of Tolerability Using PRO-CTCAE in a Randomized Double-Blind, Placebo-Controlled Phase II Trial Comparing Gemcitabine in Combination with Adavosertib or Placebo in Patients with Platinum Resistant or Refractory Epithelial Ovarian Carcinoma. Gynecol. Oncol. 2022, 167, 226–233. [Google Scholar] [PubMed]
- Cole, K.A.; Ijaz, H.; Surrey, L.F.; Santi, M.; Liu, X.; Minard, C.G.; Maris, J.M.; Voss, S.; Reid, J.M.; Fox, E.; et al. Pediatric Phase 2 Trial of a WEE1 Inhibitor, Adavosertib (AZD1775), and Irinotecan for Relapsed Neuroblastoma, Medulloblastoma, and Rhabdomyosarcoma. Cancer 2023, 129, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Embaby, A.; Kutzera, J.; Geenen, J.J.; Pluim, D.; Hofland, I.; Sanders, J.; Lopez-Yurda, M.; Beijnen, J.H.; Huitema, A.D.R.; Witteveen, P.O.; et al. WEE1 Inhibitor Adavosertib in Combination with Carboplatin in Advanced TP53 Mutated Ovarian Cancer: A Biomarker-Enriched Phase II Study. Gynecol. Oncol. 2023, 174, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yao, S.; Yuan, Y.; Previs, R.A.; Elias, A.D.; Carvajal, R.D.; George, T.J.; Yuan, Y.; Yu, L.; Westin, S.N.; et al. Multicenter Phase II Trial of the WEE1 Inhibitor Adavosertib in Refractory Solid Tumors Harboring CCNE1 Amplification. J. Clin. Oncol. 2023, 41, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.J.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination with Gemcitabine, Cisplatin, or Carboplatin in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, J.; Bao, X.; Honea, N.; Xie, Y.; Kim, S.; Sparreboom, A.; Sanai, N. Quantitative and Mechanistic Understanding of AZD1775 Penetration across Human Blood-Brain Barrier in Glioblastoma Patients Using an IVIVE-PBPK Modeling Approach. Clin. Cancer Res. 2017, 23, 7454–7466. [Google Scholar] [CrossRef]
- Méndez, E.; Rodriguez, C.P.; Kao, M.C.; Raju, S.; Diab, A.; Harbison, R.A.; Konnick, E.Q.; Mugundu, G.M.; Santana-Davila, R.; Martins, R.; et al. A Phase I Clinical Trial of AZD1775 in Combination with Neoadjuvant Weekly Docetaxel and Cisplatin before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2740–2748. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination with Gemcitabine and Radiation for Patients with Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Kato, H.; de Souza, P.; Kim, S.-W.; Lickliter, J.D.; Naito, Y.; Park, K.; Kumar, S.; Mugundu, G.M.; Bang, Y.-J. Safety, Pharmacokinetics, and Clinical Activity of Adavosertib in Combination with Chemotherapy in Asian Patients with Advanced Solid Tumors: Phase Ib Study. Target. Oncol. 2020, 15, 75–84. [Google Scholar] [CrossRef]
- Kong, A.; Good, J.; Kirkham, A.; Savage, J.; Mant, R.; Llewellyn, L.; Parish, J.; Spruce, R.; Forster, M.; Schipani, S.; et al. Phase I Trial of WEE1 Inhibition with Chemotherapy and Radiotherapy as Adjuvant Treatment, and a Window of Opportunity Trial with Cisplatin in Patients with Head and Neck Cancer: The WISTERIA Trial Protocol. BMJ Open 2020, 10, e033009. [Google Scholar] [CrossRef] [PubMed]
- Någård, M.; Ah-See, M.-L.; So, K.; Vermunt, M.; Thistlethwaite, F.; Labots, M.; Roxburgh, P.; Ravaud, A.; Campone, M.; Valkenburg-van Iersel, L.; et al. Effect of Food on the Pharmacokinetics of the WEE1 Inhibitor Adavosertib (AZD1775) in Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2020, 86, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chera, B.S.; Sheth, S.H.; Patel, S.A.; Goldin, D.; Douglas, K.E.; Green, R.L.; Shen, C.J.; Gupta, G.P.; Moore, D.T.; Grilley Olson, J.E.; et al. Phase 1 Trial of Adavosertib (AZD1775) in Combination with Concurrent Radiation and Cisplatin for Intermediate-Risk and High-Risk Head and Neck Squamous Cell Carcinoma. Cancer 2021, 127, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Li, T.; Vallius, T.; Guerriero, J.L.; Tayob, N.; Kochupurakkal, B.; Davis, J.; Pastorello, R.; Tahara, R.K.; Anderson, L.; et al. Clinical Efficacy and Molecular Response Correlates of the WEE1 Inhibitor Adavosertib Combined with Cisplatin in Patients with Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2021, 27, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Moore, K.N.; Rader, J.S.; Simpkins, F.; Mita, A.C.; Beck, J.T.; Hart, L.; Chu, Q.; Oza, A.; Tinker, A.V.; et al. A Phase Ib Study Assessing the Safety, Tolerability, and Efficacy of the First-in-Class Wee1 Inhibitor Adavosertib (AZD1775) as Monotherapy in Patients with Advanced Solid Tumors. Target. Oncol. 2023, 18, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Sachdev, J.; Imedio, E.R.; Kumar, S.; Mugundu, G.M.; Jenkins, S.; Chmielecki, J.; Jones, S.; Spigel, D.R.; Johnson, M. A Phase Ib Study of Adavosertib, a Selective Wee1 Inhibitor, in Patients with Locally Advanced or Metastatic Solid Tumors. Investig. New Drugs 2023, 41, 493–502. [Google Scholar] [CrossRef]
- Gonzalez-Ochoa, E.; Milosevic, M.; Corr, B.; Abbruzzese, J.L.; Girda, E.; Miller, R.W.; Croke, J.; Mackay, H.; Lee, Y.C.; Bowering, V.; et al. A Phase I Study of the Wee1 Kinase Inhibitor Adavosertib (AZD1775) in Combination with Chemoradiation in Cervical, Upper Vaginal, and Uterine Cancers. Int. J. Gynecol. Cancer 2023, 33, 1208–1214. [Google Scholar] [CrossRef]
- Shafer, D.; Kagan, A.B.; Rudek, M.A.; Kmieciak, M.; Tombes, M.B.; Shrader, E.; Bandyopadhyay, D.; Hudson, D.; Sankala, H.; Weir, C.; et al. Phase 1 Study of Belinostat and Adavosertib in Patients with Relapsed or Refractory Myeloid Malignancies. Cancer Chemother. Pharmacol. 2023, 91, 281–290. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-Dependent Kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Johnson, N.; Cai, D.; Kennedy, R.D.; Pathania, S.; Arora, M.; Li, Y.-C.; D’Andrea, A.D.; Parvin, J.D.; Shapiro, G.I. Cdk1 Participates in BRCA1-Dependent S Phase Checkpoint Control in Response to DNA Damage. Mol. Cell 2009, 35, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Li, Y.-C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 Activity Sensitizes BRCA-Proficient Cancers to PARP Inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Enders, G.H. Cdk Inhibition in Human Cells Compromises chk1 Function and Activates a DNA Damage Response. Cancer Res. 2005, 65, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; Katuwal, N.B.; Park, N.; Hur, J.; Cho, Y.B.; Kim, S.K.; Lee, S.A.; Kim, I.; Lee, S.-R.; Moon, Y.W. Combination of Abemaciclib Following Eribulin Overcomes Palbociclib-Resistant Breast Cancer by Inhibiting the G2/M Cell Cycle Phase. Cancers 2022, 14, 210. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, J.; Zhang, L.; Wang, L.; Cheng, C.; Ji, H.; Altilia, S.; Ding, X.; Cai, G.; Altomare, D.; et al. CDK8 and CDK19: Positive Regulators of Signal-Induced Transcription and Negative Regulators of Mediator Complex Proteins. Nucleic Acids Res. 2023, 51, 7288–7313. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Urban, V.; Muñoz-Martínez, F.; Ayestaran, I.; Thomas, J.C.; de Renty, C.; O’Connor, M.J.; Forment, J.V.; Galanty, Y.; Jackson, S.P. Loss of Cyclin C or CDK8 Provides ATR Inhibitor Resistance by Suppressing Transcription-Associated Replication Stress. Nucleic Acids Res. 2021, 49, 8665–8683. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Shamby, R.; Stansbury, N.; Schenken, R.; de la Pena Avalos, B.; Javanmardi, S.; Dray, E.; Sung, P.; Boyer, T.G. Aberrant R-Loop-Induced Replication Stress in MED12-Mutant Uterine Fibroids. Sci. Rep. 2022, 12, 6169. [Google Scholar] [CrossRef]
- Khamidullina, A.I.; Yastrebova, M.A.; Bruter, A.V.; Nuzhina, J.V.; Vorobyeva, N.E.; Khrustaleva, A.M.; Varlamova, E.A.; Tyakht, A.V.; Abramenko, Y.E.; Ivanova, E.S.; et al. CDK8/19 Inhibition Attenuates G1 Arrest Induced by BCR-ABL Antagonists and Accelerates Death of Chronic Myelogenous Leukemia Cells. BioRxiv 2023, 2023.09.25.559286. [Google Scholar] [CrossRef]
- Köhler, K.; Sanchez-Pulido, L.; Höfer, V.; Marko, A.; Ponting, C.P.; Snijders, A.P.; Feederle, R.; Schepers, A.; Boos, D. The Cdk8/19-Cyclin C Transcription Regulator Functions in Genome Replication through Metazoan Sld7. PLoS Biol. 2019, 17, e2006767. [Google Scholar] [CrossRef]
- Poss, Z.C.; Ebmeier, C.C.; Odell, A.T.; Tangpeerachaikul, A.; Lee, T.; Pelish, H.E.; Shair, M.D.; Dowell, R.D.; Old, W.M.; Taatjes, D.J. Identification of Mediator Kinase Substrates in Human Cells Using Cortistatin A and Quantitative Phosphoproteomics. Cell Rep. 2016, 15, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wen, P.; Hu, G.; Gao, Y.; Qi, X.; Zhu, K.; Chen, S.; Wu, L.; Xu, A.; Zhao, G. Antagonizing CDK8 Sensitizes Colorectal Cancer to Radiation Through Potentiating the Transcription of e2f1 Target Gene apaf1. Front. Cell Dev. Biol. 2020, 8, 408. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 Complex Maintains Genomic Stability via Regulation of Expression of DNA Damage Response Genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [PubMed]
- Chirackal Manavalan, A.P.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 Controls G1/S Progression by Regulating RNAPII Processivity at Core DNA Replication Genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef] [PubMed]
- Coelho, R.; Tozzi, A.; Disler, M.; Lombardo, F.; Fedier, A.; López, M.N.; Freuler, F.; Jacob, F.; Heinzelmann-Schwarz, V. Overlapping Gene Dependencies for PARP Inhibitors and Carboplatin Response Identified by Functional CRISPR-Cas9 Screening in Ovarian Cancer. Cell Death Dis. 2022, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- Ragupathi, A.; Singh, M.; Perez, A.M.; Zhang, D. Targeting the BRCA1/2 Deficient Cancer with PARP Inhibitors: Clinical Outcomes and Mechanistic Insights. Front. Cell Dev. Biol. 2023, 11, 1133472. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Juric, D.; Henick, B.S.; Duska, L.R.; Wu, R.; Guo, J.; Zhang, H.; Newberry, K.; Rinne, M.; Yap, T.A. BLU-222, an Oral, Potent, and Selective CDK2 Inhibitor, in Patients with Advanced Solid Tumors: Phase 1 Monotherapy Dose Escalation. J. Clin. Orthod. 2023, 41, 3095. [Google Scholar] [CrossRef]
- Lulla, A.; Nguyen, T.D.; Li, M.; Mastoraki, S.; Wang, Y.; Bui, T.; Pina, M.; Tsavachidis, S.; Marshall, G.; Hunt, K.K.; et al. Abstract 950: Targeting PKMYT1 Kinase Is an Effective Treatment Strategy in Triple Negative Breast Cancers with Low Molecular Weight Cyclin E (LMW-E) Expression. Cancer Res. 2023, 83, 950. [Google Scholar] [CrossRef]
- Durinikova, E.; Reilly, N.M.; Buzo, K.; Mariella, E.; Chilà, R.; Lorenzato, A.; Dias, J.M.L.; Grasso, G.; Pisati, F.; Lamba, S.; et al. Targeting the DNA Damage Response Pathways and Replication Stress in Colorectal Cancer. Clin. Cancer Res. 2022, 28, 3874–3889. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef]
- Cash, T.; Fox, E.; Liu, X.; Minard, C.G.; Reid, J.M.; Scheck, A.C.; Weigel, B.J.; Wetmore, C. A Phase 1 Study of Prexasertib (LY2606368), a CHK1/2 Inhibitor, in Pediatric Patients with Recurrent or Refractory Solid Tumors, Including CNS Tumors: A Report from the Children’s Oncology Group Pediatric Early Phase Clinical Trials Network (ADVL1515). Pediatr. Blood Cancer 2021, 68, e29065. [Google Scholar] [CrossRef] [PubMed]
- Guerrero Llobet, S.; van der Vegt, B.; Jongeneel, E.; Bense, R.D.; Zwager, M.C.; Schröder, C.P.; Everts, M.; Fehrmann, R.S.N.; de Bock, G.H.; van Vugt, M.A.T.M. Cyclin E Expression Is Associated with High Levels of Replication Stress in Triple-Negative Breast Cancer. NPJ Breast Cancer 2020, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kim, S.; Schultz, C.W.; Rajapakse, V.N.; Zhang, Y.; Redon, C.E.; Fu, H.; Pongor, L.; Kumar, S.; Pommier, Y.; et al. Replication Stress Defines Distinct Molecular Subtypes across Cancers. Cancer Res. Commun. 2022, 2, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-H.; Hong, Y.-K.; Du, H.; Ke, W.-Q.; Lin, B.-B.; Li, Y.-L. A Machine Learning Framework Develops a DNA Replication Stress Model for Predicting Clinical Outcomes and Therapeutic Vulnerability in Primary Prostate Cancer. J. Transl. Med. 2023, 21, 20. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Mehanna, H. WEE1 Inhibitor: Clinical Development. Curr. Oncol. Rep. 2021, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- Shkundina, I.S.; Gall, A.A.; Dick, A.; Cocklin, S.; Mazin, A.V. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes 2021, 12, 920. [Google Scholar] [CrossRef] [PubMed]
- Guerrero Llobet, S.; Bhattacharya, A.; Everts, M.; Kok, K.; van der Vegt, B.; Fehrmann, R.S.N.; van Vugt, M.A.T.M. An mRNA Expression-Based Signature for Oncogene-Induced Replication-Stress. Oncogene 2022, 41, 1216–1224. [Google Scholar] [CrossRef]
- Dreyer, S.B.; Upstill-Goddard, R.; Paulus-Hock, V.; Paris, C.; Lampraki, E.-M.; Dray, E.; Serrels, B.; Caligiuri, G.; Rebus, S.; Plenker, D.; et al. Targeting DNA Damage Response and Replication Stress in Pancreatic Cancer. Gastroenterology 2021, 160, 362–377.e13. [Google Scholar] [CrossRef]
- Benada, J.; Ejlertsen, B.; Sørensen, C.S. Overcoming Treatment Toxicity through Sequential Therapy. Cancer Cell 2019, 35, 821–822. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khamidullina, A.I.; Abramenko, Y.E.; Bruter, A.V.; Tatarskiy, V.V. Key Proteins of Replication Stress Response and Cell Cycle Control as Cancer Therapy Targets. Int. J. Mol. Sci. 2024, 25, 1263. https://doi.org/10.3390/ijms25021263
Khamidullina AI, Abramenko YE, Bruter AV, Tatarskiy VV. Key Proteins of Replication Stress Response and Cell Cycle Control as Cancer Therapy Targets. International Journal of Molecular Sciences. 2024; 25(2):1263. https://doi.org/10.3390/ijms25021263
Chicago/Turabian StyleKhamidullina, Alvina I., Yaroslav E. Abramenko, Alexandra V. Bruter, and Victor V. Tatarskiy. 2024. "Key Proteins of Replication Stress Response and Cell Cycle Control as Cancer Therapy Targets" International Journal of Molecular Sciences 25, no. 2: 1263. https://doi.org/10.3390/ijms25021263
APA StyleKhamidullina, A. I., Abramenko, Y. E., Bruter, A. V., & Tatarskiy, V. V. (2024). Key Proteins of Replication Stress Response and Cell Cycle Control as Cancer Therapy Targets. International Journal of Molecular Sciences, 25(2), 1263. https://doi.org/10.3390/ijms25021263