
Storage of Transfusion Platelet Concentrates Is Associated with Complement Activation and Reduced Ability of Platelets to Respond to Protease-Activated Receptor-1 and Thromboxane A2 Receptor

 , , , , , , and
, , , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Minor Platelet Consumption during the Storage Period
2.2. Increased Complement Activation during Storage
2.3. Soluble Platelet Activation Markers Increased during Storage
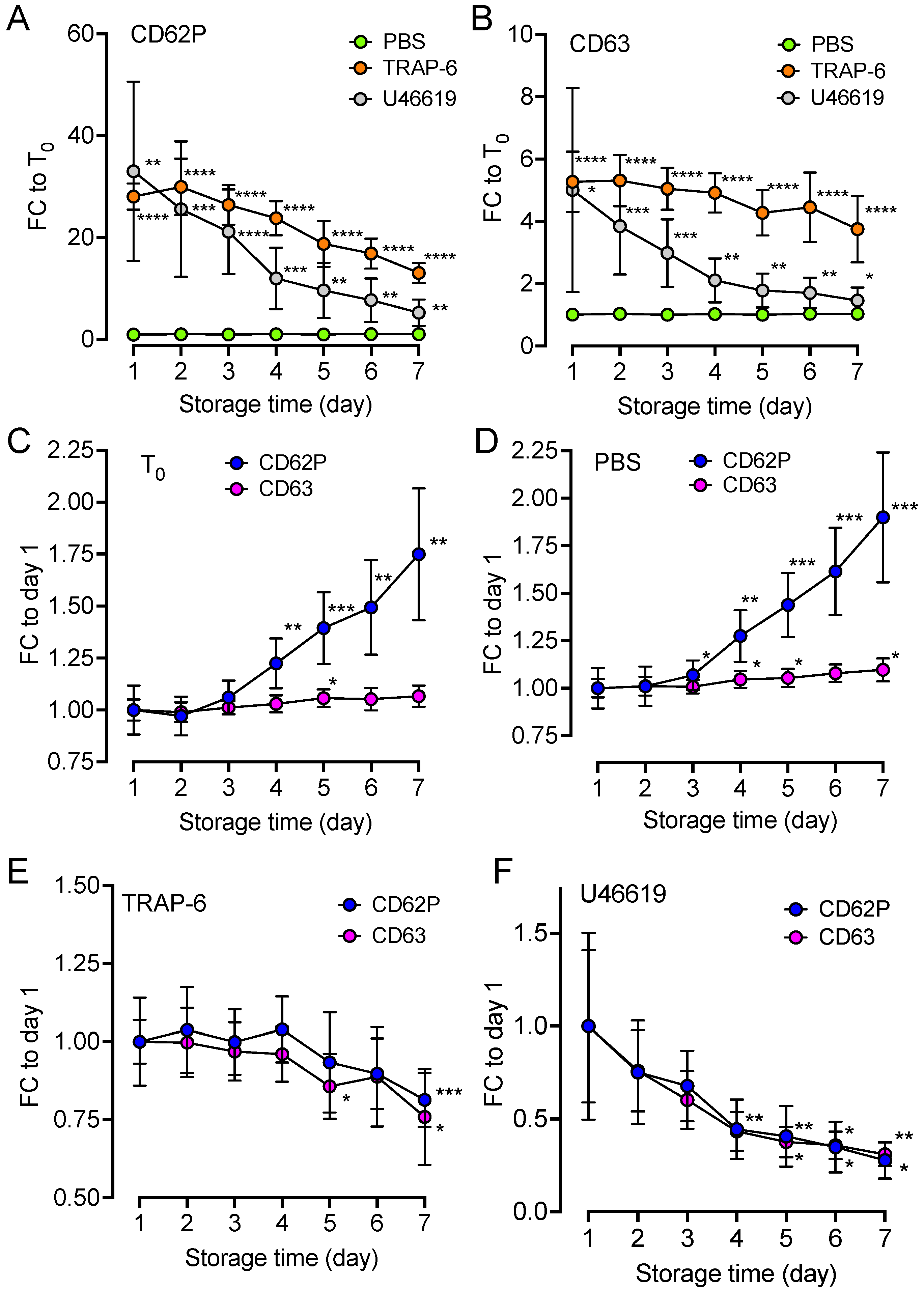
2.4. Upregulation of Platelet Surface Markers
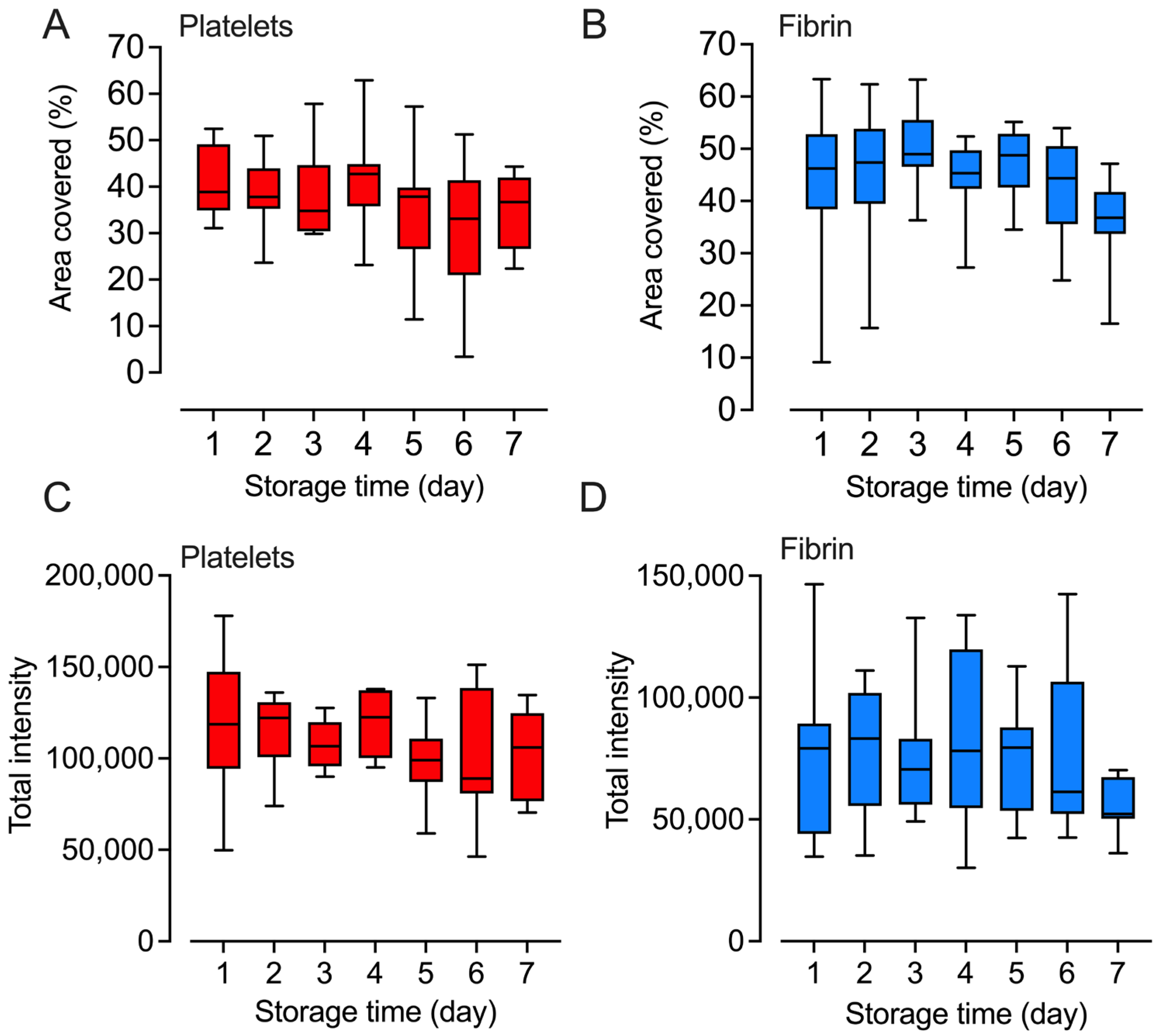
2.5. Platelets Retained Their Adhesion and Fibrin Formation Abilities
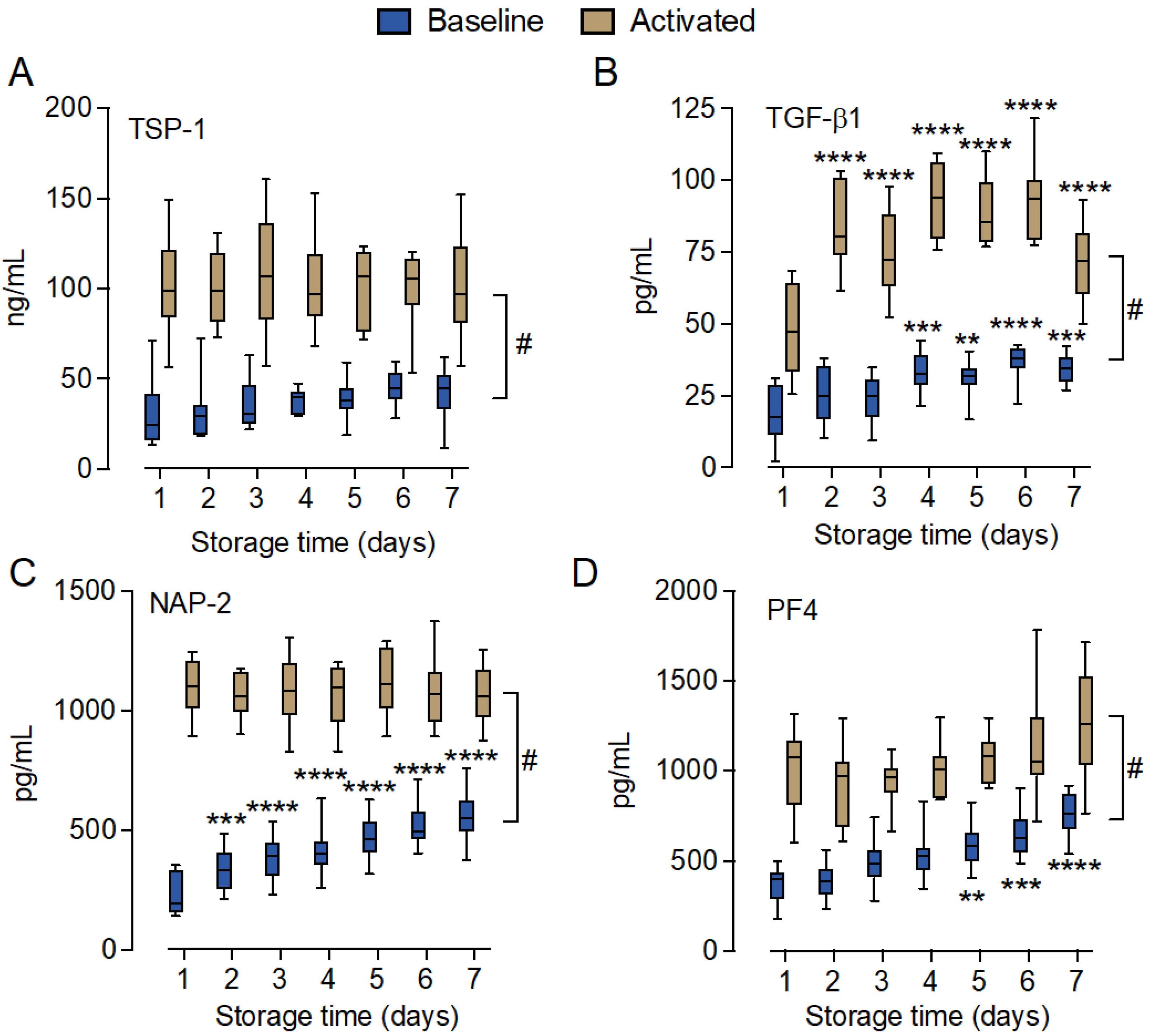
2.6. Marginal Changes in Inflammatory Cytokines and Growth Factors
3. Discussion
4. Materials and Methods
4.1. Preparation of Random Donor Platelet Concentrates
4.2. Storage and Sampling
4.3. Platelet Activation and Control
4.4. Complement Activation Markers
4.5. Soluble Platelet Activation Marker Analysis
4.6. Flow Cytometry
4.7. Platelet Adhesion and Fibrin Deposition
4.8. Cytokine Analysis
4.9. Image Analysis
4.10. Statistical and Data Analyses
4.11. Ethics Statement
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, A.I.; Anwer, F. Platelet Transfusion. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kaufman, R.M.; Djulbegovic, B.; Gernsheimer, T.; Kleinman, S.; Tinmouth, A.T.; Capocelli, K.E.; Cipolle, M.D.; Cohn, C.S.; Fung, M.K.; Grossman, B.J. Platelet transfusion: A clinical practice guideline from the AABB. Ann. Intern. Med. 2015, 162, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Farndale, R.W. Collagen-induced platelet activation. Blood Cells Mol. Dis. 2006, 36, 162–165. [Google Scholar] [CrossRef]
- Schmugge, M.; Rand, M.L.; Freedman, J. Platelets and von Willebrand factor. Transfus. Apher. Sci. 2003, 28, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.M.; Bouchard, B.A.; Tracy, P.B.; Mann, K.G. Prothrombin activation by platelet-associated prothrombinase proceeds through the prethrombin-2 pathway via a concerted mechanism. J. Biol. Chem. 2012, 287, 38647–38655. [Google Scholar] [CrossRef]
- Gullriksson, H.; Knutsson, F. Handbok för Blodcentraler. In Kap 4: Blodkomponenter: Framställning och Användning, version 4.0; Svensk Förening för Transfusionsmedicin: Linköping, Sweden, 2015. [Google Scholar]
- Caram-Deelder, C.; Kreuger, A.L.; Jacobse, J.; van der Bom, J.G.; Middelburg, R.A. Effect of platelet storage time on platelet measurements: A systematic review and meta-analyses. Vox Sang. 2016, 111, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Swamy, N.; Thapa, S.; Chakraborthy, S.; Jagannathan, L. Adding to platelet safety and life: Platelet additive solutions. Asian J. Transfus. Sci. 2018, 12, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Hajishengallis, G.; Lambris, J.D. A guide to complement biology, pathology and therapeutic opportunity. Nat. Rev. Immunol. 2023. [Google Scholar] [CrossRef]
- Ekdahl, K.N.; Lambris, J.D.; Elwing, H.; Ricklin, D.; Nilsson, P.H.; Teramura, Y.; Nicholls, I.A.; Nilsson, B. Innate immunity activation on biomaterial surfaces: A mechanistic model and coping strategies. Adv. Drug Deliv. Rev. 2011, 63, 1042–1050. [Google Scholar] [CrossRef]
- Harboe, M.; Ulvund, G.; Vien, L.; Fung, M.; Mollnes, T.E. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 2004, 138, 439–446. [Google Scholar] [CrossRef]
- Nording, H.; Baron, L.; Haberthur, D.; Emschermann, F.; Mezger, M.; Sauter, M.; Sauter, R.; Patzelt, J.; Knoepp, K.; Nording, A.; et al. The C5a/C5a receptor 1 axis controls tissue neovascularization through CXCL4 release from platelets. Nat. Commun. 2021, 12, 3352. [Google Scholar] [CrossRef]
- Sauter, R.J.; Sauter, M.; Reis, E.S.; Emschermann, F.N.; Nording, H.; Ebenhoch, S.; Kraft, P.; Munzer, P.; Mauler, M.; Rheinlaender, J.; et al. Functional Relevance of the Anaphylatoxin Receptor C3aR for Platelet Function and Arterial Thrombus Formation Marks an Intersection Point between Innate Immunity and Thrombosis. Circulation 2018, 138, 1720–1735. [Google Scholar] [CrossRef] [PubMed]
- Mannes, M.; Pechtl, V.; Hafner, S.; Dopler, A.; Eriksson, O.; Manivel, V.A.; Wohlgemuth, L.; Messerer, D.A.C.; Schrezenmeier, H.; Ekdahl, K.N.; et al. Complement and platelets: Prothrombotic cell activation requires membrane attack complex-induced release of danger signals. Blood Adv. 2023, 7, 6367–6380. [Google Scholar] [CrossRef]
- Landsem, A.; Emblem, A.; Lau, C.; Christiansen, D.; Gerogianni, A.; Karlsen, B.O.; Mollnes, T.E.; Nilsson, P.H.; Brekke, O.L. Complement C3b contributes to Escherichia coli-induced platelet aggregation in human whole blood. Front. Immunol. 2022, 13, 1020712. [Google Scholar] [CrossRef] [PubMed]
- Hamad, O.A.; Ekdahl, K.N.; Nilsson, P.H.; Andersson, J.; Magotti, P.; Lambris, J.D.; Nilsson, B. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J. Thromb. Haemost. 2008, 6, 1413–1421. [Google Scholar] [CrossRef]
- Peerschke, E.I.; Yin, W.; Grigg, S.E.; Ghebrehiwet, B. Blood platelets activate the classical pathway of human complement. J. Thromb. Haemost. 2006, 4, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Cruz, M.A.; Zhang, H.; Lopez, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef]
- Hamad, O.A.; Nilsson, P.H.; Lasaosa, M.; Ricklin, D.; Lambris, J.D.; Nilsson, B.; Ekdahl, K.N. Contribution of chondroitin sulfate A to the binding of complement proteins to activated platelets. PLoS ONE 2010, 5, e12889. [Google Scholar] [CrossRef]
- Hamad, O.A.; Nilsson, P.H.; Wouters, D.; Lambris, J.D.; Ekdahl, K.N.; Nilsson, B. Complement component C3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J. Immunol. 2010, 184, 2686–2692. [Google Scholar] [CrossRef]
- Saggu, G.; Cortes, C.; Emch, H.N.; Ramirez, G.; Worth, R.G.; Ferreira, V.P. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J. Immunol. 2013, 190, 6457–6467. [Google Scholar] [CrossRef] [PubMed]
- De Wit, Y.E.S.; Hamzeh-Cognasse, H.; Cognasse, F.; Ten Brinke, A.; Zeerleder, S.S. DAMPS and complement activation in platelet concentrates that induce adverse reactions in patients. Transfusion 2022, 62, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.; Mitchell, D.A.; Schmid, R.; Schwaeble, W.J.; Keeble, A.H. Paths reunited: Initiation of the classical and lectin pathways of complement activation. Immunobiology 2010, 215, 1–11. [Google Scholar] [CrossRef]
- Mortensen, S.; Jensen, J.K.; Andersen, G.R. Solution Structures of Complement C2 and Its C4 Complexes Propose Pathway-specific Mechanisms for Control and Activation of the Complement Proconvertases. J. Biol. Chem. 2016, 291, 16494–16507. [Google Scholar] [CrossRef] [PubMed]
- Torreira, E.; Tortajada, A.; Montes, T.; Rodriguez de Cordoba, S.; Llorca, O. 3D structure of the C3bB complex provides insights into the activation and regulation of the complement alternative pathway convertase. Proc. Natl. Acad. Sci. USA 2009, 106, 882–887. [Google Scholar] [CrossRef]
- Brandwijk, R.; Michels, M.; van Rossum, M.; de Nooijer, A.H.; Nilsson, P.H.; de Bruin, W.C.C.; Toonen, E.J.M. Pitfalls in complement analysis: A systematic literature review of assessing complement activation. Front. Immunol. 2022, 13, 1007102. [Google Scholar] [CrossRef] [PubMed]
- Hurler, L.; Toonen, E.J.M.; Kajdacsi, E.; van Bree, B.; Brandwijk, R.; de Bruin, W.; Lyons, P.A.; Bergamaschi, L.; Cambridge Institute of Therapeutic Immunolog; Infectious Disease-National Institute of Health Research (CITIID-NIHR) COVID BioResource Collaboration. Distinction of early complement classical and lectin pathway activation via quantification of C1s/C1-INH and MASP-1/C1-INH complexes using novel ELISAs. Front. Immunol. 2022, 13, 1039765. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Sandholm, K.; Jonsson, N.; Nilsson, A.; Wieslander, A.; Grundstrom, G.; Hancock, V.; Ekdahl, K.N. Low concentrations of citrate reduce complement and granulocyte activation in vitro in human blood. Clin. Kidney J. 2015, 8, 31–37. [Google Scholar] [CrossRef]
- Cardigan, R.; Sutherland, J.; Wadhwa, M.; Dilger, P.; Thorpe, R. The influence of platelet additive solutions on cytokine levels and complement activation in platelet concentrates during storage. Vox Sang. 2003, 84, 28–35. [Google Scholar] [CrossRef]
- Bode, A.P.; Miller, D.T.; Newman, S.L.; Castellani, W.J.; Norris, H.T. Plasmin activity and complement activation during storage of citrated platelet concentrates. J. Lab. Clin. Med. 1989, 113, 94–102. [Google Scholar] [PubMed]
- Chen, J.; Losos, M.; Yang, S.; Li, J.; Wu, H.; Cataland, S. Increased complement activation during platelet storage. Transfusion 2017, 57, 2182–2188. [Google Scholar] [CrossRef]
- Gyongyossy-Issa, M.I.; McLeod, E.; Devine, D.V. Complement activation in platelet concentrates is surface-dependent and modulated by the platelets. J. Lab. Clin. Med. 1994, 123, 859–868. [Google Scholar]
- De Wit, Y.E.S.; Vlaar, R.; Gouwerok, E.; Hamzeh-Cognasse, H.; van Mierlo, G.; Bulder, I.; Lagerberg, J.W.M.; de Korte, D.; Cognasse, F.; Ten Brinke, A.; et al. Platelet concentrates in platelet additive solutions generate less complement activation products during storage than platelets stored in plasma. Blood Transfus. 2023, 21, 157–167. [Google Scholar] [CrossRef]
- Quach, H.Q.; Johnson, C.; Ekholt, K.; Islam, R.; Mollnes, T.E.; Nilsson, P.H. Platelet-Depletion of Whole Blood Reveals That Platelets Potentiate the Release of IL-8 from Leukocytes into Plasma in a Thrombin-Dependent Manner. Front. Immunol. 2022, 13, 865386. [Google Scholar] [CrossRef]
- Zhong, R.; Wang, H.; Wu, X.; Cao, Y.; He, Z.; He, Y.; Liu, J. In vitro investigation of the effect of plasticizers on the blood compatibility of medical grade plasticized poly (vinyl chloride). J. Mater. Sci. Mater. Med. 2013, 24, 1985–1992. [Google Scholar] [CrossRef]
- Lachmann, P.J.; Lay, E.; Seilly, D.J. Experimental confirmation of the C3 tickover hypothesis by studies with an Ab (S77) that inhibits tickover in whole serum. FASEB J. 2018, 32, 123–129. [Google Scholar] [CrossRef]
- Elvington, M.; Liszewski, M.K.; Liszewski, A.R.; Kulkarni, H.S.; Hachem, R.R.; Mohanakumar, T.; Kim, A.H.J.; Atkinson, J.P. Development and Optimization of an ELISA to Quantitate C3(H2O) as a Marker of Human Disease. Front. Immunol. 2019, 10, 703. [Google Scholar] [CrossRef]
- De Boer, E.; Sokolova, M.; Quach, H.Q.; McAdam, K.E.; Gotz, M.P.; Chaban, V.; Vaage, J.; Fagerang, B.; Woodruff, T.M.; Garred, P.; et al. Synthetic Oligodeoxynucleotide CpG Motifs Activate Human Complement through Their Backbone Structure and Induce Complement-Dependent Cytokine Release. J. Immunol. 2022, 209, 1760–1767. [Google Scholar] [CrossRef]
- Chaban, V.; de Boer, E.; McAdam, K.E.; Vaage, J.; Mollnes, T.E.; Nilsson, P.H.; Pischke, S.E.; Islam, R. Escherichia coli-induced inflammatory responses are temperature-dependent in human whole blood ex vivo. Mol. Immunol. 2023, 157, 70–77. [Google Scholar] [CrossRef]
- Senzel, L.; Gnatenko, D.V.; Bahou, W.F. The platelet proteome. Curr. Opin. Hematol. 2009, 16, 329–333. [Google Scholar] [CrossRef]
- Damas, J.K.; Waehre, T.; Yndestad, A.; Otterdal, K.; Hognestad, A.; Solum, N.O.; Gullestad, L.; Froland, S.S.; Aukrust, P. Interleukin-7-mediated inflammation in unstable angina: Possible role of chemokines and platelets. Circulation 2003, 107, 2670–2676. [Google Scholar] [CrossRef] [PubMed]
- Battegay, E.J.; Rupp, J.; Iruela-Arispe, L.; Sage, E.H.; Pech, M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J. Cell Biol. 1994, 125, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Weber, K.S.; Huo, Y.; Proudfoot, A.E.; Nelson, P.J.; Ley, K.; Weber, C. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef]
- Lord, M.S.; Cheng, B.; Farrugia, B.L.; McCarthy, S.; Whitelock, J.M. Platelet Factor 4 Binds to Vascular Proteoglycans and Controls Both Growth Factor Activities and Platelet Activation. J. Biol. Chem. 2017, 292, 4054–4063. [Google Scholar] [CrossRef]
- Wu, Q.; Tu, H.; Li, J. Multifaceted Roles of Chemokine C-X-C Motif Ligand 7 in Inflammatory Diseases and Cancer. Front. Pharmacol. 2022, 13, 914730. [Google Scholar] [CrossRef]
- Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFbeta. Int. J. Mol. Sci. 2021, 22, 4492. [Google Scholar] [CrossRef]
- Prud’homme, G.J. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Investig. 2007, 87, 1077–1091. [Google Scholar] [CrossRef]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef]
- Rink, T.J.; Smith, S.W.; Tsien, R.Y. Cytoplasmic free Ca2+ in human platelets: Ca2+ thresholds and Ca-independent activation for shape-change and secretion. FEBS Lett. 1982, 148, 21–26. [Google Scholar] [CrossRef]
- Kiefel, V. Reactions Induced by Platelet Transfusions. Transfus. Med. Hemother. 2008, 35, 354–358. [Google Scholar] [CrossRef]
- Cognasse, F.; Aloui, C.; Anh Nguyen, K.; Hamzeh-Cognasse, H.; Fagan, J.; Arthaud, C.A.; Eyraud, M.A.; Sebban, M.; Fromont, E.; Pozzetto, B.; et al. Platelet components associated with adverse reactions: Predictive value of mitochondrial DNA relative to biological response modifiers. Transfusion 2016, 56, 497–504. [Google Scholar] [CrossRef]
- Cognasse, F.; Sut, C.; Hamzeh-Cognasse, H.; Garraud, O. Platelet-derived HMGB1: Critical mediator of SARs related to transfusion. Ann. Transl. Med. 2020, 8, 140. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundback, P.; Long, W.; Valdes-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef]
- Mangsbo, S.M.; Sanchez, J.; Anger, K.; Lambris, J.D.; Ekdahl, K.N.; Loskog, A.S.; Nilsson, B.; Totterman, T.H. Complement activation by CpG in a human whole blood loop system: Mechanisms and immunomodulatory effects. J. Immunol. 2009, 183, 6724–6732. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Blumberg, N.; Gettings, K.F.; Turner, C.; Heal, J.M.; Phipps, R.P. An association of soluble CD40 ligand (CD154) with adverse reactions to platelet transfusions. Transfusion 2006, 46, 1813–1821. [Google Scholar] [CrossRef]
- Kluter, H.; Bubel, S.; Kirchner, H.; Wilhelm, D. Febrile and allergic transfusion reactions after the transfusion of white cell-poor platelet preparations. Transfusion 1999, 39, 1179–1184. [Google Scholar] [CrossRef]
- Bergseth, G.; Ludviksen, J.K.; Kirschfink, M.; Giclas, P.C.; Nilsson, B.; Mollnes, T.E. An international serum standard for application in assays to detect human complement activation products. Mol. Immunol. 2013, 56, 232–239. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersson, L.I.; Sjöström, D.J.; Quach, H.Q.; Hägerström, K.; Hurler, L.; Kajdácsi, E.; Cervenak, L.; Prohászka, Z.; Toonen, E.J.M.; Mohlin, C.; et al. Storage of Transfusion Platelet Concentrates Is Associated with Complement Activation and Reduced Ability of Platelets to Respond to Protease-Activated Receptor-1 and Thromboxane A2 Receptor. Int. J. Mol. Sci. 2024, 25, 1091. https://doi.org/10.3390/ijms25021091
Andersson LI, Sjöström DJ, Quach HQ, Hägerström K, Hurler L, Kajdácsi E, Cervenak L, Prohászka Z, Toonen EJM, Mohlin C, et al. Storage of Transfusion Platelet Concentrates Is Associated with Complement Activation and Reduced Ability of Platelets to Respond to Protease-Activated Receptor-1 and Thromboxane A2 Receptor. International Journal of Molecular Sciences. 2024; 25(2):1091. https://doi.org/10.3390/ijms25021091
Chicago/Turabian StyleAndersson, Linnea I., Dick J. Sjöström, Huy Quang Quach, Kim Hägerström, Lisa Hurler, Erika Kajdácsi, László Cervenak, Zoltán Prohászka, Erik J. M. Toonen, Camilla Mohlin, and et al. 2024. "Storage of Transfusion Platelet Concentrates Is Associated with Complement Activation and Reduced Ability of Platelets to Respond to Protease-Activated Receptor-1 and Thromboxane A2 Receptor" International Journal of Molecular Sciences 25, no. 2: 1091. https://doi.org/10.3390/ijms25021091
APA StyleAndersson, L. I., Sjöström, D. J., Quach, H. Q., Hägerström, K., Hurler, L., Kajdácsi, E., Cervenak, L., Prohászka, Z., Toonen, E. J. M., Mohlin, C., Mollnes, T. E., Sandgren, P., Tjernberg, I., & Nilsson, P. H. (2024). Storage of Transfusion Platelet Concentrates Is Associated with Complement Activation and Reduced Ability of Platelets to Respond to Protease-Activated Receptor-1 and Thromboxane A2 Receptor. International Journal of Molecular Sciences, 25(2), 1091. https://doi.org/10.3390/ijms25021091