
Whole-Exome Screening and Analysis of Signaling Pathways in Multiple Endocrine Neoplasia Type 1 Patients with Different Outcomes: Insights into Cellular Mechanisms and Possible Functional Implications

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results
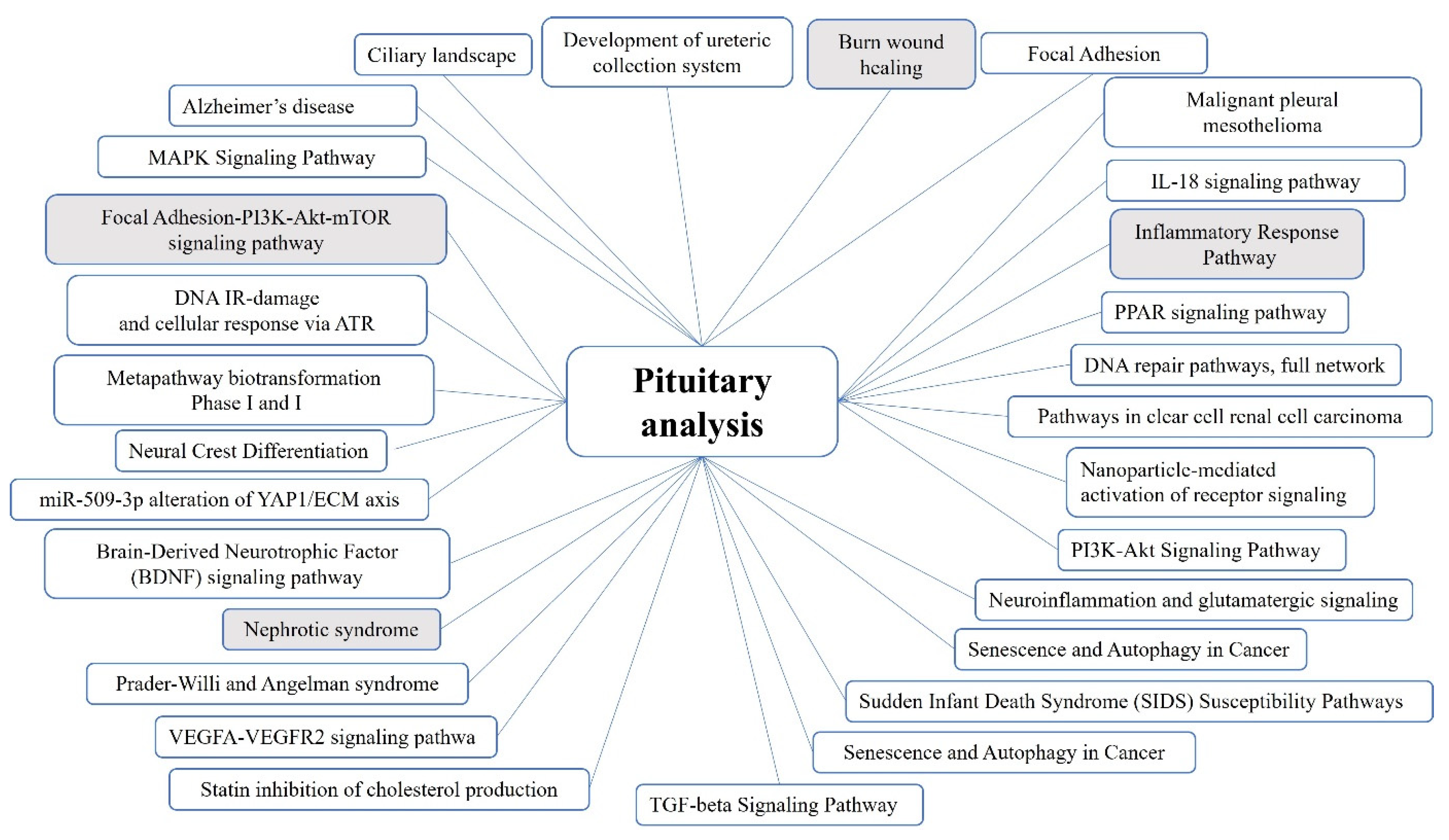
2.1. Pituitary Tumors
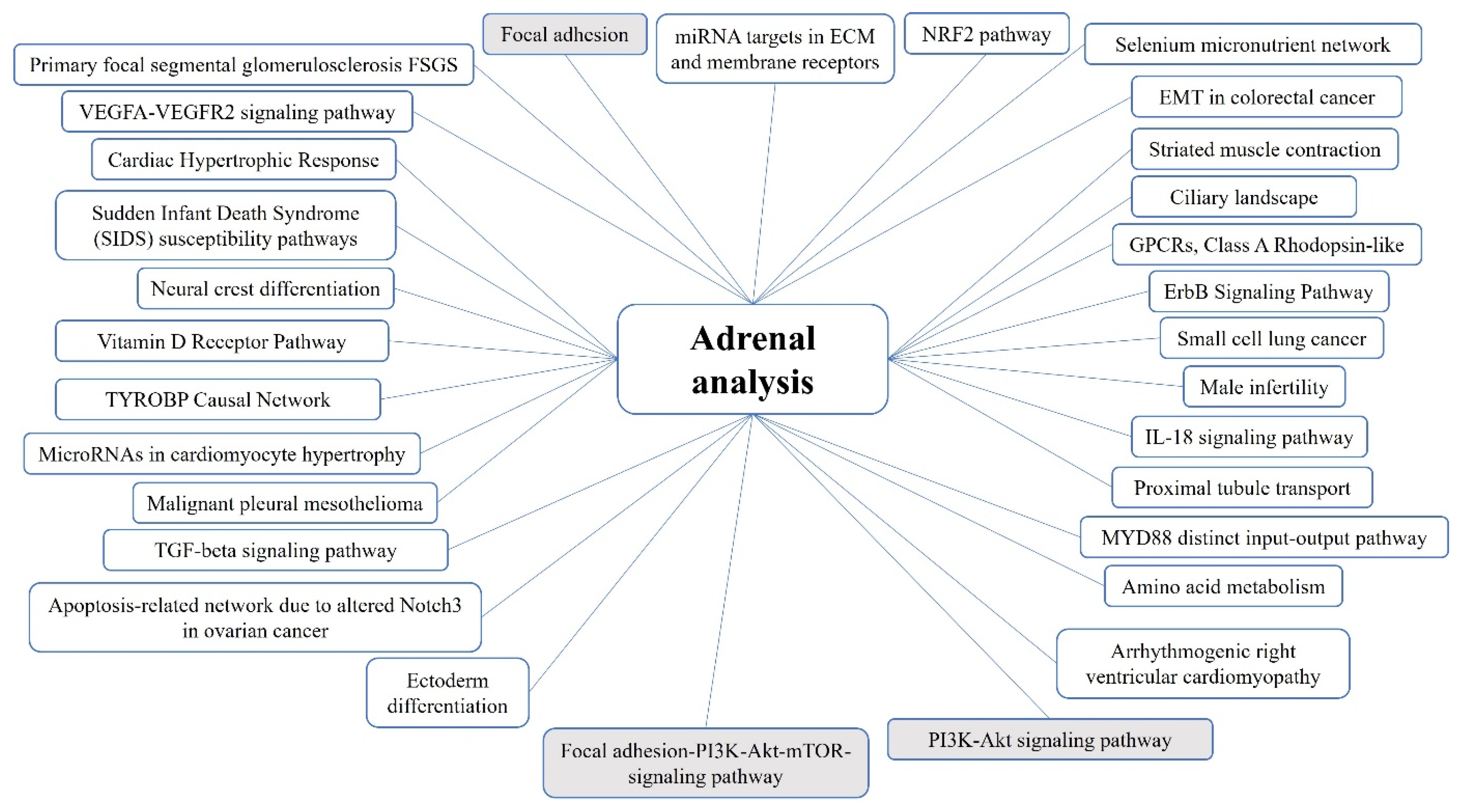
2.2. Adrenocortical Tumors
3. Discussion
3.1. Genes and Pathways Identified in the Pituitary Tumor Patient Groups
3.2. Genes and Pathways Identified in the Adrenocortical Tumor Patient Groups
3.3. Implications for the Future
4. Methods
4.1. Patients
4.2. Ethical Statement
4.3. Genomic DNA Extraction and Whole Exome Library Preparation
4.4. High Throughput Data Analysis
4.5. Downstream Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Mulji, N.J.; Jialal, I. Multiple Endocrine Neoplasia Type 1; StatPearls Publishing LLC: St. Petersburg, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK536980/ (accessed on 12 July 2023).
- Lemos, M.C.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1 (MEN1): Analysis of 1336 Mutations Reported in the First Decade Following Identification of the Gene. Hum. Mutat. 2008, 29, 22–32. [Google Scholar] [CrossRef]
- Lips, C.J.M.; Dreijerink, K.M.A.; van der Luijt, R.B.; van Nesselrooij, B.P.M.; Höppener, J.W.M. Multiple Endocrine Neoplasia Type 1 (MEN1). In Genetic Diagnosis of Endocrine Disorders; Weiss, R.E., Refetoff, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 261–270. [Google Scholar]
- Burgess, J.; Shepherd, J.J.; Parameswaran, V.; Hoffman, L.; Greenaway, T.M. Prolactinomas in a Large Kindred with Multiple Endocrine Neoplasia Type 1: Clinical Features and Inheritance Pattern. J. Clin. Endocrinol. Metab. 1996, 81, 1841–1845. [Google Scholar] [PubMed]
- Thevenon, J.; Bourredjem, A.; Faivre, L.; Cardot-Bauters, C.; Calender, A.; le Bras, M.; Giraud, S.; Niccoli, P.; Odou, M.F.; Borson-Chazot, F.; et al. Unraveling the Intrafamilial Correlations and Heritability of Tumor Types in MEN1: A Groupe d’étude Des Tumeurs Endocrines Study. Eur. J. Endocrinol. 2015, 173, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Lemos, M.C.; Harding, B.; Reed, A.A.C.; Jeyabalan, J.; Walls, G.V.; Bowl, M.R.; Sharpe, J.; Wedden, S.; Moss, J.E.; Ross, A.; et al. Genetic Background Influences Embryonic Lethality and the Occurrence of Neural Tube Defects in Men1 Null Mice: Relevance to Genetic Modifiers. J. Endocrinol. 2009, 203, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Mohr, H.; Pellegata, N.S. Animal Models of MEN1. Endocr. Relat. Cancer 2017, 24, T161–T177. [Google Scholar] [CrossRef]
- Lines, K.E.; Javid, M.; Reed, A.A.C.; Walls, G.V.; Stevenson, M.; Simon, M.; Kooblall, K.G.; Piret, S.E.; Christie, P.T.; Newey, P.J.; et al. Genetic Background Influences Tumour Development in Heterozygous Men1 Knockout Mice. Endocr. Connect. 2020, 9, 426–437. [Google Scholar] [CrossRef]
- Scacheri, P.C.; Davis, S.; Odom, D.T.; Crawford, G.E.; Perkins, S.; Halawi, M.J.; Agarwal, S.K.; Marx, S.J.; Spiegel, A.M.; Meltzer, P.S.; et al. Genome-Wide Analysis of Menin Binding Provides Insights into MEN1 Tumorigenesis. PLoS Genet. 2006, 2, e51. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Impey, S.; McWeeney, S.; Scacheri, P.C.; Collins, F.S.; Goodman, R.H.; Spiegel, A.M.; Marx, S.J. Distribution of Menin-Occupied Regions in Chromatin Specifies a Broad Role of Menin in Transcriptional Regulation. Neoplasia 2007, 9, 101–107. [Google Scholar] [CrossRef][Green Version]
- Skalniak, A.; Trofimiuk-Müldner, M.; Jabrocka-Hybel, A.; Totoń-Żurańska, J.; Wołkow, P.; Hubalewska-Dydejczyk, A. Whole-Exome Sequencing as a Tool for Searching for Genetic Background Modifiers in MEN1 Patients with Neuroendocrine Pancreatic Tumours, including Insulinomas. Endokrynol. Pol. 2023, 74, 31–46. [Google Scholar] [CrossRef]
- Chang, M.; Yang, C.; Bao, X.; Wang, R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front. Endocrinol. 2021, 11, 596554. [Google Scholar] [CrossRef]
- Hosseini-Alghaderi, S.; Baron, M. Notch3 in Development, Health and Disease. Biomolecules 2020, 10, 485. [Google Scholar] [CrossRef]
- Kalpachidou, T.; Makrygiannis, A.K.; Pavlakis, E.; Stylianopoulou, F.; Chalepakis, G.; Stamatakis, A. Behavioural Effects of Extracellular Matrix Protein Fras1 Depletion in the Mouse. Eur. J. Neurosci. 2021, 53, 3905–3919. [Google Scholar] [CrossRef]
- Zhuo, L.; Theis, M.; Alvarez-Maya, I.; Brenner, M.; Willecke, K.; Messing, A. HGFAP-cre Transgenic Mice for Manipulation of Glial and Neuronal Function in Vivo. Genesis 2001, 31, 85–94. [Google Scholar] [CrossRef]
- Jossin, Y. Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules 2020, 10, 964. [Google Scholar] [CrossRef] [PubMed]
- Schaberg, E.; Götz, M.; Faissner, A. The Extracellular Matrix Molecule Tenascin-C Modulates Cell Cycle Progression and Motility of Adult Neural Stem/Progenitor Cells from the Subependymal Zone. Cell. Mol. Life Sci. 2022, 79, 244. [Google Scholar] [CrossRef]
- Willemsen, M.H.; Ba, W.; Wissink-Lindhout, W.M.; de Brouwer, A.P.M.; Haas, S.A.; Bienek, M.; Hu, H.; Vissers, L.E.L.M.; van Bokhoven, H.; Kalscheuer, V.; et al. Involvement of the Kinesin Family Members KIF4A and KIF5C in Intellectual Disability and Synaptic Function. J. Med. Genet. 2014, 51, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Kong, H.; Ja, Y.; Wang, C.; Qin, L.; Sun, H.; Dai, S. The Relationship between Cancer and Biomechanics. Front. Oncol. 2023, 13, 1273154. [Google Scholar] [CrossRef]
- Berrier, A.L.; Yamada, K.M. Cell–Matrix Adhesion. J. Cell Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef]
- Iyer, S.; Agarwal, S.K. Epigenetic Regulation in the Tumorigenesis of MEN1-associated Endocrine Cell Types. J. Mol. Endocrinol. 2018, 61, R13–R24. [Google Scholar] [CrossRef]
- Feng, Z.; Ma, J.; Hua, X. Epigenetic Regulation by the Menin Pathway. Endocr.-Relat. Cancer 2017, 24, T147–T159. [Google Scholar] [CrossRef]
- Nadeau, J. Modifier Genes and Protective Alleles in Humans and Mice. Curr. Opin. Genet. Dev. 2003, 13, 290–295. [Google Scholar] [CrossRef]
- McCabe, E.R.B. Modifier Genes: Moving from Pathogenesis to Therapy. Mol. Genet. Metab. 2017, 122, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Donati, S.; Ciuffi, S.; Marini, F.; Palmini, G.; Miglietta, F.; Aurilia, C.; Brandi, M.L. Multiple Endocrine Neoplasia Type 1: The Potential Role of microRNAs in the Management of the Syndrome. Int. J. Mol. Sci. 2020, 21, 7592. [Google Scholar] [CrossRef]
- Li, S.-C.; Monazzam, A.; Razmara, M.; Chu, X.; Stålberg, P.; Skogseid, B. MiR-486-3p was Downregulated at microRNA Profiling of Adrenals of Multiple Endocrine Neoplasia Type 1 Mice, and Inhibited Human Adrenocortical Carcinoma Cell Lines. Sci. Rep. 2021, 11, 14722. [Google Scholar] [CrossRef] [PubMed]
- Luzi, E.; Pandolfini, L.; Ciuffi, S.; Marini, F.; Cremisi, F.; Nesi, G.; Brandi, M.L. MicroRNAs Regulatory Networks Governing the Epigenetic Landscape of MEN1 Gastro-Entero-Pancreatic Neuroendocrine Tumor: A Case Report. Clin. Transl. Med. 2021, 11, e351. [Google Scholar] [CrossRef] [PubMed]
- Kooblall, K.G.; Stokes, V.J.; Shariq, O.A.; English, K.A.; Stevenson, M.; Broxholme, J.; Wright, B.; Lockstone, H.E.; Buck, D.; Grozinsky-Glasberg, S.; et al. miR-3156-5p is Downregulated in Serum of MEN1 Patients and Regulates Expression of MORF4L2. Endocr. Relat. Cancer 2022, 29, 557–568. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The Next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Skalniak, A.; Sokołowski, G.; Jabrocka-Hybel, A.; Piątkowski, J.; Białas, M.; Gilis-Januszewska, A.; Pach, D.; Hubalewska-Dydejczyk, A. A Novel In-Frame Deletion in MEN1 (p.Ala416del) Causes Familial Multiple Endocrine Neoplasia Type 1 with an Aggressive Phenotype and Unexpected Inheritance Pattern. Mol. Med. Rep. 2016, 14, 2061–2066. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinform. 2010, 10. [Google Scholar]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ng, P.C. SIFT Indel: Predictions for the Functional Effects of Amino Acid Insertions/Deletions in Proteins. PLoS ONE 2013, 8, e77940. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of Deleterious Mutations within Three Human Genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions Using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and Integration of Deleteriousness Prediction Methods for Nonsynonymous SNVs in Whole Exome Sequencing Studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient ID | Family ID * | Disease-Causing MEN1 Variant | Age at Time of Analysis | Sex | Primary Hyperpara-Thyroidism | Pituitary Tumor | Neuro-Endocrine Neoplasm (NEN) | Pancreatic NEN | Adrenocortical Tumor | Other | Included in Pituitary Analysis (Patient or Group ID) | Included in Adrenal Analysis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A2 | A | c.1246_1248delGCC/p.Ala416del | 59 | F | yes | No | no | yes | no | n/d | neg | - |
| C3 | C | c.945delG/p.Tyr316Profs*57 | 59 | M | yes | No | yes | yes | yes | n/d | - | pos(C) |
| A4 | A | c.1246_1248delGCC/p.Ala416del | 54 | M | yes | Yes | no | yes | yes | n/d | pos(A) | pos(A) |
| X5 | X | c.866C>A/p.Ala289Glu | 59 | F | yes | Yes | no | no | no | nasal cavity cancer | pos(X5) | neg |
| A8 | A | c.1246_1248delGCC/p.Ala416del | 48 | M | yes | yes | no | no | no | n/d | pos(A) | neg |
| C9 | C | c.945delG/p.Tyr316Profs*57 | 33 | M | yes | no | no | no | yes | adrenocortical oncocytoma | - | pos(C) |
| X10 | X | c.945delG/p.Tyr316Profs*57 | 72 | F | yes | n/d | yes | yes (insulinoma) | no | n/d | - | neg |
| X11 | X | c.416A>G/p.His139Arg | 75 | F | yes | no | yes | yes (insulinoma) | yes | n/d | neg | pos(X11) |
| X12 | X | c.796C>T/p.Gln266Ter | 65 | F | yes | no | yes | yes | no | n/d | neg | neg |
| X15 | X | c.1256delTGC/p.Leu419del | 75 | M | yes | yes | yes | no | yes | n/d | pos(X15) | pos(X15) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skalniak, A.; Trofimiuk-Müldner, M.; Surmiak, M.; Totoń-Żurańska, J.; Jabrocka-Hybel, A.; Hubalewska-Dydejczyk, A. Whole-Exome Screening and Analysis of Signaling Pathways in Multiple Endocrine Neoplasia Type 1 Patients with Different Outcomes: Insights into Cellular Mechanisms and Possible Functional Implications. Int. J. Mol. Sci. 2024, 25, 1065. https://doi.org/10.3390/ijms25021065
Skalniak A, Trofimiuk-Müldner M, Surmiak M, Totoń-Żurańska J, Jabrocka-Hybel A, Hubalewska-Dydejczyk A. Whole-Exome Screening and Analysis of Signaling Pathways in Multiple Endocrine Neoplasia Type 1 Patients with Different Outcomes: Insights into Cellular Mechanisms and Possible Functional Implications. International Journal of Molecular Sciences. 2024; 25(2):1065. https://doi.org/10.3390/ijms25021065
Chicago/Turabian StyleSkalniak, Anna, Małgorzata Trofimiuk-Müldner, Marcin Surmiak, Justyna Totoń-Żurańska, Agata Jabrocka-Hybel, and Alicja Hubalewska-Dydejczyk. 2024. "Whole-Exome Screening and Analysis of Signaling Pathways in Multiple Endocrine Neoplasia Type 1 Patients with Different Outcomes: Insights into Cellular Mechanisms and Possible Functional Implications" International Journal of Molecular Sciences 25, no. 2: 1065. https://doi.org/10.3390/ijms25021065
APA StyleSkalniak, A., Trofimiuk-Müldner, M., Surmiak, M., Totoń-Żurańska, J., Jabrocka-Hybel, A., & Hubalewska-Dydejczyk, A. (2024). Whole-Exome Screening and Analysis of Signaling Pathways in Multiple Endocrine Neoplasia Type 1 Patients with Different Outcomes: Insights into Cellular Mechanisms and Possible Functional Implications. International Journal of Molecular Sciences, 25(2), 1065. https://doi.org/10.3390/ijms25021065